MOE Key Laboratory of Macromolecular Synthesis and Functionalization, International Research Center for X Polymers, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou 310027, China
Received Date:
06 July 2025 Accepted Date:
20 October 2025 Revised Date:
16 October 2025 Available Online:
15 February 2026
Abstract:
In 2024, the MOE Key Laboratory of Macromolecular Synthesis and Functionalization at Zhejiang University continued its impactful researches across five core areas. In controllable catalytic polymerization, organoboron catalysts were developed for CO2 copolymerization and novel photoresist materials. Studies in microstructure and rheology elucidated universal deformation modes in graphene-based 2D membranes and improved graphene fiber properties through shear alignment engineering, defect control, and enhanced interlayer entanglement. For separating functional polymers, Janus membranes and channels were created for multiphase separation, liquid-phase molecular layer-by-layer deposition technique was developed to fabricate aromatic polyamide nanofilms, and the harmonic amide bond density was established as a valuable parameter for polyamide structural analysis. In biomedical functional polymers, a sustainable carboxyl-ester transesterification strategy was proposed for upcycling poly(ethylene terephthalate) (PET) waste into biodegradable plastics. Additionally, immunocompatible biomaterials were designed utilizing zwitterionic polypeptides and albumin-derived coatings, and Cu2+-phenolic nanoflower was designed to combat fungal infections by combining cuproptosis and cell wall digestion. Further, the researchers developed a gelatin-DOPA-knob/fibrinogen hydrogel to achieve rapid and robust hemostatic sealing, utilized a double-network polyelectrolyte-coated hydrogel for enhancing endothelialization of left atrial appendage (LAA) occluders, and the researchers also demonstrated that image-guided high-intensity focused ultrasound enables manipulation of shape-memory polymers. Finally, in the realm of photo-electro-magnetic functional polymers, precise control of through-space conjugation was shown to enhance organic luminescence. Topologically structured hydrogels were revealed to exhibit autonomous actuation. Also, solar-driven photothermal ion pumps were developed for selective lithium extraction from seawater, and high-performance non-solvated C60 single-crystal films were prepared via facile bar coating. Lastly, the researchers demonstrated outstanding dielectric properties of polyethylene (PE) lamellar single crystals. The relevant works are reviewed in this paper.
Founded in 2005, the MOE Key Laboratory of Macromolecular Synthesis and Functionalization serves as a prominent research center dedicated to advancing both fundamental and applied knowledge in polymer science. Supported by diverse disciplines, including chemistry, materials science, and biology, at Zhejiang University, the laboratory pursues cutting-edge investigations to address critical challenges across diverse sectors, including sustainable resources, carbon utilization, new energy technologies, and human health. To this end, the laboratory has established five key research directions: controllable catalytic polymerization, microstructure and rheology, biomedical functional polymers, separation functional polymers and photo-electro-magnetic functional polymers [1-4].
In 2024, the laboratory achieved substantial research progress in all five thematic areas spanning from polymer synthesis to the investigation of structure-property relationships and culminating in the development of advanced functional materials, the performance of which, in turn, informs and refines future polymer synthesis (Fig. 1). This review highlights a selection of these high-impact studies, providing a concise overview of the laboratory's contributions to the broader polymer science community over the past year. The review begins with an exposition of recent progress in organoboron catalysis for polymerization, focusing on its applications in developing sustainable materials, facilitating carbon utilization, and expanding applications within high-technology industries. Subsequently, the focus shifts to structural investigations elucidating formation mechanisms in two-dimensional materials, including studies on shear-assisted wet-spinning, synergistic alignment engineering, and graphene aerogels, contributing to the evolution of advanced materials design methodologies. A detailed discussion of innovative separation technologies follows, covering Janus membranes and channels, polyamide nanofilms, and refined techniques for organic solvent nanofiltration. Shifting focus to biomedical applications, the review elucidates new progresses including studies of the upcycling of PET, immunocompatible biomaterials, anti-fungal strategies, hydrogel hemostatic sealants, modification of LAA occluders, and shape-memory polymers. Finally, we will discuss research in photo-electro-magnetic functional polymers, multi-aryl alkanes with through-space conjugation, architectures and controls of hydrogels, innovative lithium extraction, high-performance non-solvated C60 single-crystal films, and refined control for growth and dielectric properties of PE lamellar single crystals. This review ultimately serves as a testament to the laboratory's unwavering commitment to excellence in polymer research and its dedication to translating fundamental scientific discoveries into real-world innovations with the long-term goal to contribute to the sustainable development.
Figure 1
Figure 1.
Schematic illustration about the key progresses of MOE Key Laboratory of Macromolecular Synthesis and Functionalization in 2024. Adapted with permission [10,12,23,27,66,103,140,161]. Copyright 2024, Wiley Publishing Group; Copyright 2024, American Association for the Advancement of Science; Copyright, 2024, Elsevier; Copyright 2024, Springer Nature; Copyright 2024, Wiley Publishing Group.
In 2024, Wu’s group advanced organoboron catalysis by elucidating structure-activity relationships, optimizing polymerization methodologies, and expanding applications in sustainable materials (CO2-PCs) and high-tech industries (photoresists). Key innovations include catalyst design for stereoselective and alternating copolymerizations, mechanistic insights via computational tools, and strategic evaluations of polymerization techniques (Fig. 2). These contributions collectively position organoboron chemistry as a pivotal tool for addressing challenges in polymer synthesis, carbon utilization, and advanced material development. The following summarizes recent key contributions across six publications. The review systematically reviewed the evolution of organoboron compounds as versatile catalysts and mediators for diverse polymerization modes, including controlled radical, Lewis pair, ionic, and polyhomologation processes. Emphasis was placed on their role in synthesizing linear, block, cyclic, and graft polymers. This work highlighted mechanistic insights and structure-property relationships, offering a foundation for designing advanced organoboron catalysts [5]. The review detailed the progress in converting CO2 and epoxides into aliphatic polycarbonates (CO2-PCs), emphasizing the transition from metal-based to organoborane catalysts. Innovations in catalyst design enabled precise control over polymer selectivity, sequence, and architecture. Computational studies elucidated mechanistic pathways, while developments in closed-loop recycling and industrialization efforts underscored the sustainability potential of CO2-PCs [6]. By optimizing mononuclear organoborane catalysts with α-H-containing cations, the group achieved >99% selectivity for alternating poly(propylene carbonate) (PPC), overcoming challenges of cyclic carbonate byproduct formation. Phosphonium-based catalysts exhibited superior activity due to enhanced Lewis acidity, while mechanistic studies revealed α-H atoms suppress backbiting, favoring chain propagation [7]. A direct comparison between ring-opening copolymerization (ROCOP) of ethylene oxide/succinic anhydride and traditional polycondensation demonstrated ROCOP’s advantages in atom economy and mild conditions, albeit with lower molecular weights (~3.7 kDa). The researchers proposed ROCOP as a complementary method for high-value polyesters (e.g., block copolymers) rather than a replacement for industrial-scale polycondensation [8]. Bifunctional organoborane catalysts outperformed binary systems in terpolymerization of epoxides, CO2, and anhydrides, producing gradient polyester-polycarbonates via enhanced CO2 incorporation. Density functional theory (DFT) calculations attributed this to intramolecular synergistic effects, including optimized charge distribution and electrostatic interactions, guiding future catalyst design [9]. The group developed aqueous-developable CO2-PCs bearing acid-cleavable acetal groups, serving as high-performance photoresists. These materials achieved exceptional sensitivity (1.9 mJ/cm2), resolution (750 nm), and etch resistance, surpassing commercial KrF/ArF resists. This work bridges sustainable polymer chemistry with semiconductor manufacturing, showcasing CO2-PCs’ potential in deep ultraviolet (DUV) and extreme ultraviolet (EUV) lithography [10].
Figure 2
Figure 2.
(a) Progress in converting CO2 and epoxides into aliphatic polycarbonates. Reproduced with permission [6]. Copyright 2024, American Chemical Society. (b) Organoboron compounds as versatile catalysts and mediators for diverse polymerization modes. Reproduced with permission [5]. Copyright 2024, The Royal Society of Chemistry. (c) Aqueous-developable CO-PCs bearing acid-cleavable acetal groups, serving as high-performance photoresists. Reproduced with permission [10]. Copyright 2024, Wiley Publishing Group.
Progresses have been made in the research of two-dimensional (2D) macromolecular conformations represented by graphene oxide (GO) and graphene-based macroscopic assemblies.
For 2D macromolecular conformations and their condensed structures, the researchers deciphered two universal deformation modes of 2D membranes under 2D shrinkage fields, that are scrolling and folding. They unveiled a determinative relation between folding numbers (N), Föppl-von Kármán number (γ) and shrinkage ratio (ϕ) as N ~ ln(γ/ϕ2). The revealed rules are validated to be general for the deformation of membranes, spanning from the microscale to atomic thickness [11].
In the realm of high-performance graphene fibers, the group proposed a multiple shear-flow assisted wet-spinning technology, and obtain graphene fibers with ultrahigh modulus and thermal conductivity. During the wet-spinning process, a radial rotating shear-flow field is introduced to yield a concentric liquid crystal texture. This process greatly enlarges the three-dimensional crystallite sizes in graphene fibers and achieves the super-high modulus and excellent thermal conductivity. Young’s modulus and the thermal conductivity of graphene fiber reach 901 GPa and 1660 W m−1 K−1, respectively. The graphene fiber with a concentric circular texture would be an ideal skeleton model for carbonaceous fibers with extraordinary thermal conductivity and high modulus (Fig. 3) [12].
Figure 3
Figure 3.
Schematic of the concept of bidirectionally promoting assembly order of graphene. (a) GO fibers prepared by multiple shear-flow fields. (b-f) Structural illustration of the sheet-order in the whole spinning tube under multiple flow fields (c), where Plane Ⅰ (d, e) shows the aligned sheets along the flow direction but wrinkling conformation in perpendicular direction under the unidirectionally flow field, Plane Ⅱ (f) presents the aligned slices in two directions under multiple flow fields, and Plane Ⅲ (b) depicts the optimally concentric structure by bidirectionally propelling assembly order. Copied with permission [12]. Licensed under CC-BY.
The researchers investigated the effect of atomic defects of restored graphene on the electrical conductivity of fibers and electromagnetic interference (EMI) shielding properties of textiles. They prepared highly conductive graphene fiber fabrics that can be applied in EMI shielding applications. By weaving of graphene fiber tows with a high conductivity of 8.5 × 105 S/m, the graphene fiber fabric with a thickness of 0.41 mm achieves an X-band EMI shielding effectiveness up of 96 dB [13].
A dual-field synergistic alignment engineering strategy was developed to produce a graphene fiber array with a high orientation degree (0.88) and excellent array density (33.5 mg/cm2). The flexible graphene fiber-based thermal interface material exhibits metal-level thermal conductivity (82.4 W m−1 K−1) and outstanding elastic compliance, accompanying with excellent structure and stability of thermal conductivity [14].
For high-performance graphene composite fibers, the researchers firstly tailored hydrogen bonds to strengthen the entanglement network for stronger interlayer energy dissipation. The strength and toughness of resulting materials were improved simultaneously to 1.58 GPa and 52 MJ/m3. They also modulated hydrogen bonds and ionic coordinating bonding simultaneously to enhance the entanglement network, realizing the high strength and modulus of biomimetic graphene composite fibers, finally up to 2.3 and 253 GPa, respectively. This advance provides an alternative method to overcome the performance limitations of graphene-based composite fibers in terms of the strength-toughness conflict [15].
The group fabricated a high-performance graphene aerogel served as wearable AI smart devices. A strategy based on the topological cellular hierarchy design was proposed to design graphene aerogels with ultra-stiffness (>10 MPa modulus) yet super-elasticity (>90% recoverable strain). This structure relieves the conflict between high stiffness and superior recoverability of traditional aerogel materials. The hierarchically topological cellular graphene aerogel also exhibited exceptional ballistic resistance to block high-speed projectiles (200 m/s) [16].
The researchers presented a trans-scale porosity design of graphene nanofibrous aerogels (GNFAs), achieving the high flexibility yet superelasticity of the one-dimensional fiber aerogel. GNFAs can fully recover after 1000 compression cycles under 60% compressive strain, and keep unexceptionable structural integrity after 10,000 fatigue cycles under 90% folding strain. As a wearable sensor, GNFA enables the intelligent recognition of sign language with high accuracy via a multi-layer artificial neural network [17].
4.
Separating functional polymers
4.1
Janus membrane and channel of membranes
Janus membranes have garnered significant attention in multiphase processes in recent years. These membranes enable unidirectional liquid transport, facilitating lyophobic-to-lyophilic transport while preventing the reverse process. This unique transport behavior provides the foundation for various applications, including fog harvesting, demulsification, and simultaneous filtration, among others. Despite numerous hypotheses, the underlying mechanism remains elusive [18]. In their recent work, the researchers proposed a three-stage model: comprising liquid intrusion, capillary wetting, and liquid transport, to describe this process (Fig. 4a) [19]. Through finite element analysis, they identified two key mechanisms: the direct contact mechanism and the surface-energy-driven mechanism for liquid intrusion in bi-layer and gradient Janus membranes, respectively. The predicted critical thickness of the lyophobic layer for liquid intrusion aligns with previous reports. During the capillary wetting stage, the Laplace pressure at the concave interface within the pores is significantly higher than that of the droplet on the membrane surface, resulting in capillary forces dominating the rapid liquid transport at this stage. Once the hydrophilic layer becomes saturated with liquid, the Laplace pressure of the droplet governs the slower transport.
Figure 4
Figure 4.
(a) Three-stage mode of unidirectional liquid transport through Janus membrane. Reproduced with permission [19]. Licensed under CC-BY. (b) Janus substrate to regulate interfacial polymerization. Reproduced with permission [20]. Licensed under CC-BY. (c) Janus seesaw evaporator for self-descaling during hypersaline brine evaporation. Reproduced with permission [21]. Copyright 2024, Wiley Publishing Group. (d) Janus channel of membranes for concurrent recovery of oil and water from emulsions. Reproduced with permission [23]. Copyright 2024, American Association for the Advancement of Science.
In addition to these fundamental mechanisms, recent advancements in Janus structures have expanded their applications. For instance, Janus membranes can act as substrates for regulating interfacial polymerization (Fig. 4b) [20]. By adjusting the thickness of the hydrophilic layer, the solution reserved within the substrate can be precisely tuned. This allows for the decoupling of monomer concentration and total quantity, enabling the formation of thin, dense films with high rejection and permeate flux simultaneously. Notably, this strategy offers a novel method for substrate regulation in interfacial polymerization, eliminating the need for an additional interlayer. In the field of solar-driven evaporation, the researchers have demonstrated a Janus seesaw evaporator capable of mitigating salt scaling during evaporation (Fig. 4c) [21]. The salt accumulates at the upper end of the seesaw, and the evaporator self-flips once the salt reaches a critical threshold. It is worth noting that conventional Janus evaporators normally leverage the hydrophilic layer to supply water while the hydrophobic layer to prevent scaling [22]. This innovation offers a novel approach to achieving durable evaporation, even in hypersaline solutions.
The Janus configuration can be further extended from a material to an apparatus. Oil/water emulsions are prevalent as waste or by-products in industries such as petroleum, metallurgy, textiles, and food processing. Conventional separation technologies typically separate one phase from the emulsion, leaving a residual product that requires further treatment. Most recently, the researchers introduced a novel concept of Janus Channel of Membranes (JCMs), which consists of a narrow channel between a pair of hydrophilic and hydrophobic membranes (Fig. 4d) [23]. This design facilitates the concurrent recovery of both oil and water from surfactant-stabilized emulsions. The recoveries of oil and water improve significantly as the channel width decreases. The confined space enhances demulsification through a local enrichment effect and increased collision probability. Furthermore, the interplay between the hydrophilic and hydrophobic membranes introduces a novel mechanism in membrane separation. As water permeates through the hydrophilic membrane, the emulsion within the channel becomes concentrated, leading to concentration polarization that compromises water flux. The enriched emulsion promotes collision and demulsification of oil droplets, which in turn enhances removal efficiency by the hydrophobic membrane. The reduced oil content further accelerates water permeation. This feedback mechanism offers insights for the future design of membrane modules to address concentration polarization issues.
4.2
Polyimide membranes for organic solvent nanofiltration
The application of membrane technology in water treatment has achieved significant success, and the technology is becoming increasingly mature. However, its application in chemical and pharmaceutical is still in an early developing stage. Organic solvent nanofiltration (OSN) and reverse osmosis (RO) are the pressure-driven membrane processes developed from traditional aqueous membrane technology. They have great application prospects in drug concentration, solvent recovery, natural product extraction, and electronic chemical preparation etc. Membrane materials are the core of membrane technology. From a materials science angle of view, the membrane with an ultra-thin selective layer, uniform pore size, and high porosity are ideal structural characteristics for these membrane processes. In addition, the separation in organic solvent systems also requires membrane materials to have high solvent resistance.
Recently the researchers developed a liquid-phase molecular layer-by-layer deposition (MLD) technique to fabricate aromatic polyamide (PA) nanofilms, which served as selective layers of thin-film composite (TFC) membranes used in OSN process (Fig. 5). The resultant nanofilms exhibited tunable thickness within the range from 10 nm to 50 nm, significantly thinner than that achieved by traditional interfacial polymerization [24]. This technique allows precise design and control of membrane microstructures at molecular level. They further demonstrated the use of MLD to synthesize PA nanofilms with controlled microporosity, achieving a thickness of 25 nm and exceptional solvent permeance by introducing amine monomers with aromatic rings and non-planar configuration into reaction system [25]. To extend the application of this technique, covalent organic polymer (COP) nanofilms were synthesized through the Schiff-base reaction with a slower reaction rate. The ultrathin nanofilms exhibited precisely tunable solute rejection profiles in molecular weight range of 200–400 Da, which was significant for fine separations in pharmaceutical molecules [26]. The researchers further incorporated cardo-pendant groups into amine monomers and developed PA nanofilms with enhanced ultramicroporosity and a thickness of 23–36 nm [27]. These studies collectively highlight the importance of monomer design and MLD in creating membranes with high solvent permeability, precise molecular sieving, and robust stability, and offers a promising pathway for developing next-generation OSN membranes with improved performance in energy-efficient separations. To achieve green manufacturing of polymeric OSN membranes, the research team proposed a novel strategy to prepare integrally-skin asymmetric (ISA) polyimide (PI) membranes by the combination of non-solvent induced phase separation (NIPS) and thermal-induced surface densification [28,29]. They further extended this approach to poly(ether sulfone) (PES) membranes, demonstrating that sub-Tg thermal treatment can generate acid-resistant NF membranes with a ultra-thin skin layer and high water permeance [30]. In addition, tris(hydroxymethyl) aminomethane (Tris) was chemically grafted onto PI membranes followed by thermal annealing. It was found that the required annealing temperature was significantly lowered, while the solvent resistance and separation accuracy were improved [31]. These studies highlight the significant progress made in membrane science, emphasizing the importance of material design, fabrication techniques to achieve high-performance solvent-resistant polymeric membranes for precise molecular separations.
Figure 5
Figure 5.
Preparation of PA nanofilms with highly intrinsic microporosity for efficient organic solvent nanofiltration. (a) Schematic of PA nanofilm preparation by tailoring the monomer structure in molecular layer deposition. 4,4′-Methylenedianiline (MDA), 1,1-bis(4-aminophenyl)cyclohexane (BACH), and 9,9-bis(4-aminophenyl)fluorene (BAF) were used as amino monomers, and trimesoyl chloride (TMC) was used as the acyl chloride monomer. (b) PA-BAF amorphous cell (50 Å), where the Connolly surface area is colored gray/blue with a probe radius of 1 Å, as described in the simulation method. (c) Photograph of a typical free-standing PA nanofilm from the reaction between BAF and TMC. (d) Schematic of PA nanofilms with controlled chemical and morphological structures used for molecular separation in organic solvents. Reproduced with permission [27]. Copyright 2024, Elsevier.
4.3
Harmonic amide bond density (HABD) in polyamide network structure
Polyamide nanofilms, stemmed from a convenient preparation method and highly adjustable crosslinking structure, offer the opportunity for customizable properties to fulfill specific separation requirements. This makes them valuable for a wide range of applications, including desalination, gas purification, and drug concentration. Despite their practical application value, there has always been a mystery surrounding the structural analysis of crosslinked polyamide networks and their relation to separation performance.
To address this gap, the researchers introduce a novel structural parameter called the harmonic amide bond density (HABD, mmol/g) as a replacement for the degree of network crosslinking (DNC) in describing the compactness and structural characteristics of polyamide networks (Fig. 6) [32]. While conventional DNC overlooks unreacted amino groups and hydrolyzed carboxyl groups present in polyamide networks, these groups significantly deviate the calculated DNC from the actual situation. By reclassifying crosslinked polyamide networks based on their functional groups and defining the number of amide bonds per unit mass of polyamide as HABD, the researchers create a segmentation method that accounts for all terminal groups and provides a more comprehensive depiction of the crosslinked polyamide networks.
Figure 6
Figure 6.
(a) Comparison of harmonic amide bond density (HABD) and the degree of network crosslinking (DNC) in analyzing structures and performances of desalination polyamide membranes. (b) HABD for analyzing complex crosslinked polyamide networks. Reproduced with permission [32]. Licensed under CC-BY.
To ensure accurate calculation of HABD, the research group has designed an optimized X-ray photoelectron spectroscopy data processing protocol and calculation method that employ the cross-peak self-consistency of atoms in diverse chemical environments. These designs enable HABD to accurately describe the network structure of desalination-oriented polyamide membranes, specifically those developed through the reaction of trimesoyl chloride with piperazine and m-phenylenediamine, for reverse osmosis and nanofiltration polyamides. Furthermore, HABD exhibits a theoretically expected correlation with water/salt separation performance in desalination membranes, where increasing HABD results in a consistent decrease in water permeance and a significant increase in salt rejection. Additionally, HABD can be extended to complex polyamides, formed by highly functional monomers, to design customized crosslinked polyamide networks that go beyond the scope and ability of DNC.
5.
Biomedical functional polymers
5.1
Upcycling PET waste into biodegradable copolyesters
The researchers proposed a sustainable strategy for upcycling PET waste to biodegradable plastics through carboxyl-ester transesterification [33,34], effectuating the process from polymer to polymer without degradation to monomer. PET is one of the most popular plastics in the world mainly applied to food packaging and textiles [35,36]. The annually manufactured PET is almost 70 million tons worldwide [37,38]. As the consumption of PET plastics increases, the amount of discarded PET becomes significant, accounting for nearly 12% of total solid waste [39]. PET waste is accumulating in the environment and becoming one of the main sources of “white pollution” [40]. Notably, the treatment of waste PET is increasingly becoming a challenge. “Upcycling” refers to improving the performance and value of recycled materials through physical and chemical means [41]. Nevertheless, current physical and chemical recycling strategies of PET may not meet the requirements of upcycling [42]. Mechanical recycling of PET can be described as a “downcycling”, owing to relatively low intrinsic viscosity values of recycled PET [43]. High recycling cost limits large-scale application of chemical recycling [44]. Thus, those strategies cannot be the main pathway to upcycle PET waste.
Copolyesters polymerized from aromatic diacids, aliphatic diacids and diols, e.g. polybutylene adipate/terephthalate (PBAT), are widely used biodegradable plastics with high added value [45]. Currently, the main strategy to prepare terephthalate-based degradable copolyesters from PET waste is to first degrade it into monomers, and then perform copolycondensation with aliphatic dicarboxylic acids. The price of monomers obtained by PET waste decomposition is even higher than that of virgin monomers, and copolyesters obtained by this route are disadvantageous in terms of cost [46]. Thus, it is urgent to make a breakthrough in the principle of PET macromolecular reaction and efficiently prepare products with high added value and wide applications.
The group utilized PET waste and bio-based hydrogenated dimer acid (HDA) as feedstocks, to synthesize biodegradable poly(ethylene hydrogenated dimerate-co-terephthalate) (PEHT) plastics through melt reaction without any additional catalyst (Fig. 7) [47]. This carboxyl-ester transesterification was based on the principle of replacing terephthalic acid (TPA) with nonvolatile HDA in the polymer chain. TPA removal is the key to producing high molecular weight PEHT. Various PEHTs with different chemical compositions were synthesized by simply tuning the feed ratio of HDA to PET. Without additional catalyst and solvent, this macromolecular reaction can be facilely carried out on current equipment of polyester industry, thus will enable low-cost and large-scale production. This process achieves 100% atomic utilization, due to the repeated application of by-product TPA in the polyester industry. With excellent mechanical properties, regulable flexibility and great biodegradability, PEHT is expected to become a competitive candidate of high value-added biodegradable material. Moreover, with good film-forming performance, PEHT is promising to be applied as biodegradable mulch films on a large scale. The research group is looking forward to solving the ecological environment sustainability issues associated with end-of-life PET by this feasible upcycling approach.
Figure 7
Figure 7.
Bio-based HDA enables the upcycling of PET waste to biodegradable PEHT. Copied with permission [47]. Copyright 2024, Wiley Publishing Group.
Implantable biomaterials and devices are fundamental to contemporary medical practice, yet their functionality and long-term stability are often compromised by the host immune response triggered upon implantation [48,49]. The use of synthetic materials in medical devices can induce varying degrees of immune-mediated foreign body response (FBR), forming a dense fibrous capsule around the implant, which serves as a barrier to electrical, chemical, and optical communication [50]. Molecular design of materials has emerged as a promising strategy to attenuate the immune response to implants, offering sustained effects and design flexibility [51,52]. Building on this concept, the research group has developed a series of anti-FBR materials and strategies, integrating mechanisms such as anti-protein non-specific adsorption and specific immunomodulation to address the diverse application requirements of different materials.
Non-specific protein adsorption at the material interface is considered the initial step of the immune cascade upon implantation [53]. To address this, ultra-low-fouling materials, such as zwitterionic polymers with equally and evenly distributed cationic and anionic functional groups at the molecular level, have demonstrated effectiveness in resisting the FBR [54-57]. EK peptide-based materials, a pseudo zwitterionic material, have alternating glutamic acid and lysine sequences. These materials exhibit enzymatic degradability while maintaining their inherent zwitterionic properties to resist non-specific protein adsorption [58]. In a recent study, the researchers prepared a class of zwitterionic polypeptide (ZIP) hydrogels with alternating E and K sequences for the first time [59]. When implanted subcutaneously, the ZIP hydrogels elicited minimal inflammation and no significant collagen capsulation after six months in mice. Furthermore, the enzymatic degradability of the hydrogels could be controlled by adjusting the crosslinking density or the optical isomerism of the amino acid monomers. The long-term FBR resistance and controlled degradability of ZIP hydrogels open new avenues for various biomedical applications [59]. This material is immunocompatible, controllably degradable, and highly amenable to further functionalization. Next, the team extend the strategy of eliminating non-specific protein adsorption to design implantable elastomers. The group developed an elastomer material capable of generating a super-hydrophilic, anti-fouling zwitterionic layer in situ on its surface through a simple chemical trigger [60]. This hydrolyzed elastomer effectively repels protein adsorption and the adhesion of cells, platelets, and diverse microbes. The hydrolyzed elastomer elicited negligible inflammatory responses and mitigated fibrotic reactions in rodents. This elastomer is designed without the need for coating construction and features a stable, in-situ-generated hydrophilic surface, providing a new material option for implantable medical devices.
Another approach to evade immune system recognition involves utilizing the body's components. For example, biomaterials derived from natural mucin glycoproteins have demonstrated the ability to inhibit immune cascades and evade the FBR for up to 21 days in mice [61]. Albumin, the most abundant protein in plasma, has been extensively studied for its biomedical applications. The researchers developed a simple and scalable strategy to covalently graft human serum albumin (HSA) onto the surface of silicone implants [62]. This covalently grafted albumin coating remains stable and resistant to displacement by other proteins. Notably, the polydimethylsiloxane (PDMS) implants with covalently grafted HSA strongly resist the fibrotic capsule formation following a 180-day subcutaneous implantation in mice. This albumin-based immunocompatible coating method is simple and practical, with potential for industrial-scale production.
In addition to the rational design of anti-FBR materials, high-throughput screening methods utilizing combinatorial material libraries offer a promising approach to discovering novel biomaterials with immunomodulatory properties [63]. Recent studies have shown that a coating derived from tetrahydropyran phenyl triazole (THPT) effectively mitigated capsule formation around silicone devices and prostheses [64,65]. Inspired by the THPT group’s efficacy in modulating the FBR of elastomers, the research group hypothesize that the more straightforward structure of the tetrahydropyran ring may play a critical role in its effectiveness. Based on this, they designed easy-to-synthesize vinyl-based anti-FBR dense elastomer (EVADE) materials containing the tetrahydropyran ring, synthesized via free radical polymerization of methacrylate monomers (Fig. 8a) [66]. EVADE materials effectively suppress inflammation and capsule formation in subcutaneous models of rodents and non-human primates for at least one year and two months, respectively (Fig. 8b). Implantation of EVADE materials significantly reduces the expression of inflammation-related proteins S100A8/A9 in adjacent tissues compared to silicone elastomers. Additionally, they demonstrate that inhibition or knockout of S100A8/A9 results in substantial attenuation of fibrosis in mice, suggesting a target for fibrosis inhibition (Fig. 8c). Continuous subcutaneous insulin infusion (CSII) catheters made from EVADE elastomers show significantly improved longevity and performance compared to commercial catheters (Fig. 8d). The EVADE materials reported here may enhance and extend function in various medical devices by resisting the local immune responses.
Figure 8
Figure 8.
(a) Photograph showing the flexible sheet of EVADE material and its corresponding chemical structure. (b) Digital photos, H&E-, and M&T-stained histological sections of excised 1-year post-implantation in mice (representative for n = 6 biologically independent samples). (c) The construction and experimental overview of the S100A8-KO mice model and digital photos and M&T-stained histological sections of excised 4-week post-implantation in S100A8-KO mice. (d) Schematic representation of CSII catheter made of EVADE elastomer enabling the long-term use of infusion catheter and faster insulin absorption. Reproduced with permission [66]. Copyright 2024, Springer Nature.
5.3
Cuproptosis-inducing copper nanoflowers for antifungal applications
Fungal infection has become a serious public health problem in the world due to the very limited antifungal drugs in clinic and increasing drug resistance [67-69]. Especially, invasive fungal infections can result in high morbidity. It is urgently needed to develop innovative non-antibiotic antifungal strategies for the treatment of fungal infections [70-72]. Transition metals such as silver, copper, and zinc are well-known antifungal drugs [73,74]. However, the in vivo antimicrobial applications of transition metal ions are strongly restricted owing to the deactivation by proteins [75]. In order to improve the fungicidal efficacy of metal ions in vivo, the researchers prepared Cu2+-containing nanoflowers by supramolecular self-assembly (Fig. 9) [76]. Protocatechuic acid (PA)-Cu2+ (PC) nanopetals were synthesized via coordination self-assembly of Cu2+ and PA. Lywallzyme (Lyw)-loaded PC (PCW) nanoflowers were then synthesized by Lyw-induced assembly of PC nanopetals. PCW nanoflowers could effectively adhere onto fungal cells. Lyw was then released from PCW nanoflower to digest the fungal cell walls, which facilitated the internalization of Cu2+ by fungal cells. Therefore, PCW nanoflowers exhibited much stronger fungicidal activity than dissociative Cu2+. It should be emphasized that PCW nanoflowers exhibited strong fungicidal activity in protein-rich environment, while dissociative Cu2+ was completely deactivated with very limited fungicidal effect. Furthermore, the internalization of abundant Cu2+ by fungal cells, led to a decrease of pyruvate dehydrogenase (PDH) activity, a restricted tricarboxylic acid (TCA) cycle, reduced Fe-S cluster proteins, and significant lipid peroxides (LPO) accumulation, which implied that PCW nanoflowers could induce fungicidal cuproptosis. It is the first example about the occurrence of cuproptosis in fungal cells, which provides a new strategy for the treatment of fungal infections.
Figure 9
Figure 9.
Schematic illustration of the synthesis and fungicidal mechanism of PCW nanoflowers. Copied with permission [76]. Copyright 2024, Springer Nature.
5.4
Gelatin-DOPA-knob/fibrinogen hydrogel for rapid and robust hemostatic sealing
The researchers achieved an efficient hemostatic seal by developing a novel hydrogel adhesive that characterized by rapid gelation, strong adhesion and excellent biocompatibility. Uncontrolled bleeding from surgical incisions and traumatic injuries is a leading cause of global mortality [77,78]. Rapid bleeding control can significantly improve patients’ survival [79,80]. Tissue adhesives have emerged as a promising alternative for wound closure and surgical hemostasis, providing adhesive sealing of bleeding tissues [81,82]. However, their clinical applications are limited by inadequate tissue adhesion and biocompatibility [83]. To address these issues, common strategies for improving the properties of tissue adhesives include removing the interfacial water [84], enhancing cohesion and tissue interface adhesion [85,86], and incorporating hemostatic functions [87]. One novel example involves enhancing adhesion performance on the wet tissue through a dry cross-linking method combined with robust intra/inter-molecular interactions [84].
Herein, drawing inspiration from the mechanisms of fibrin polymerization and mussel adhesion, a gelatin-DOPA-knob/fibrinogen (GDK/Fg) hydrogel is designed for rapid and robust hemostatic sealing (Fig. 10) [88]. The strategy involves using knob peptide and catechol groups to modify gelatin (GDK), which enables crosslinking with fibrinogen (Fg) through knob-hole interactions. Additionally, the cohesion of the hydrogel is enhanced through non-covalent interactions between the catechol groups. This crosslinking mechanism facilitates the rapid formation of the hydrogel and provides a robust physical barrier. When applied to hemostasis, the GDK/Fg hydrogel interacts with tissue surface through catechol groups, achieving firm adhesion and effectively sealing the wound. Additionally, the knobs in the GDK/Fg hydrogel can bind to Fg in the blood-covered tissue surface via knob-hole interactions, accelerating the coagulation cascade and forming a tight fibrin gel layer on the tissue surface, thereby enhancing interfacial adhesion. Compared with commercial fibrin glue, GDK/Fg hydrogel shows similar gelation time and biocompatibility, as well as stronger adhesive properties and storage stability. This hydrogel exhibits excellent hemostatic sealing effects in both rat severe liver injury and heart puncture injury. The results show that GDK/Fg is a safe and effective hemostatic adhesive with significant prospect in medical settings.
Figure 10
Figure 10.
Schematic illustration of the preparation, crosslinking, and mechanism of tissue adhesion and hemostasis for the GDK/Fg hydrogel. Reproduced with permission [88]. Copyright 2025, Elsevier.
5.5
Hydrogel coatings to promote endothelialization of LAA occluders
Atrial fibrillation (AF) is a common arrhythmia affecting 2%–4% adult population worldwide [89]. It is estimated that approximately 25% of ischemic strokes are caused by AF, with a substantial proportion of these cases being caused by cardiogenic embolism originating from the left atrial appendage (LAA) [90]. Acute ischemic stroke patients with AF have higher mortality, poorer functional outcomes, greater risk of intracerebral hemorrhage [91]. Percutaneous closure of the LAA using occluders has become a commonly adopted preventive strategy for AF patients at risk of stroke, given that it can considerably reduce the incidence of stroke and systemic embolism [92]. Commercial LAA occluders are typically composed of a nitinol metal framework and a polyethylene terephthalate (PET) fiber membrane [93]. The occluder disk diameter is approximately 2–4 cm [89]. The poor biocompatibility of these materials and the large surface area may lead to delayed endothelialization and thrombogenesis on the surface [94,95], resulting in device-related thrombus (DRT) [96]. Endothelialization of the disk surface typically requires 1–3 months [97]. Some patients have not yet endothelialized even 2–3 years after the occluder is implanted [98,99]. Under this circumstance, the risk of DRT accumulates continuously when the device comes into long-term contact with blood. DRT occurs in 4%–7% of patients and is associated with higher rates of ischemic stroke and systemic embolism [96,100]. Therefore, addressing the issue of DRT when using LAA occluders for stroke prevention in patients with AF is crucial. The common approach is to coat the surface of medical devices with biocompatible coatings to inhibit the thrombosis and promote endothelialization. However, due to the complex surface structure of the occluder and the severe folding and friction it encounters during delivery, the stability of the coating on the surface of the occluder faces great challenges.
To address the above-mentioned challenge, the researchers first reported a hydrogel coating by the in situ UV-triggered polymerization of double-network polyelectrolytes (Figs. 11a and b) [101]. Their findings revealed that the double network and electrostatic interactions between the networks resulted in excellent mechanical properties of the hydrogel coating. The sulfonate and Arg-Gly-Asp (RGD) groups in the coating promoted hemocompatibility and endothelial growth of the occluder, respectively. The group further demonstrated that the coating significantly accelerated the endothelialization of the LAA occluder in a canine model.
Figure 11
Figure 11.
(a) Preparation process of hemocompatible coatings on the surface of the LAA occluder for enhanced endothelialization. (b) Schematic illustration of the polyelectrolyte coating on LAA occluder. Reproduced with permission [101]. Licensed under CC-BY. (c) Schematic diagram of drop-shaped microgrooves and their influence on cell migration. Reproduced with permission [102]. Copyright 2024, Springer Nature.
In addition, to further combine the stability and functionality of the coating, they have developed a special surface microstructure to promote the endothelialization of the occluder surface (Fig. 11c) [102]. The group hypothesize that guiding the directional migration of cells on the surface through local microtopography can accelerate endothelialization. Inspired by the ratchet-shaped grooves, here the research group design microgrooves composed of a continuous series of drop shapes. They observed that cells tend to migrate from the wider part of the drop towards the narrower part, crossing the ‘neck’ to enter the wider section, and repeating this process. Reverse migration of cells is limited. In this microgroove, there is only one direction for cell migration. By arranging these drop-shaped microgrooves on specific surfaces, they found that this configuration can enhance the collective migration efficiency of cells, accelerating the coverage of the surface by cells and tissue. The research group further apply this kind of microgroove to the surface of occluder discs, confirming its ability to expedite endothelialization. Their research presents a surface microstructure design that effectively accelerates endothelialization on the surfaces of interventional medical devices.
5.6
Image-guided HIFU for triggering shape recovery in deep tissue-implanted shape-memory polymers
Shape-memory polymer materials offer significant potential for enhancing medical devices with functionalities for implantation, locomotion, drug delivery, and removal. However, their clinical translation is hindered by the lack of non-invasive and precise methods to trigger and control shape recovery, particularly for devices implanted in deep tissues. This year, the researchers evaluated the application of image-guided high-intensity focused ultrasound (HIFU) heating (Fig. 12) [103]. Magnetic resonance-guided HIFU successfully triggers shape recovery in a polyurethane urea device while enabling real-time temperature monitoring through magnetic resonance thermometry. In a live canine bladder (5 cm deep), deformation of the polyurethane urea is achieved within 8 s using ultrasound-guided HIFU with millimeter-scale energy focus. Histological analysis confirms the absence of hyperthermic tissue injury. A conceptual application in ureteral stent shape recovery demonstrates reduced removal resistance. In conclusion, image-guided HIFU enables deep energy penetration, precise control, and rapid actuation, offering a promising approach for the clinical translation of shape-memory polymeric medical devices.
Figure 12
Figure 12.
(a) Equipment layout of image-guided high-intensity focused ultrasound (HIFU). Image-guided HIFU facilitates precise, non-invasive, and real-time monitored heating of shape memory medical devices in vivo. This technology has potential applications in medical device implantation, functional activation, including drug release, and device extraction. (b) Concept of shape memory ureteral stent removal. Copied with permission [103]. Licensed under CC-BY.
6.1
Organic luminescent materials based on through-space conjugation
The regulation of electronic structure based on through-bond conjugation (TBC) has been widely applied to traditional planar conjugated molecules, enabling numerous organic functional materials to exhibit exceptional optical properties [104]. However, the mechanisms underlying through-space conjugation (TSC), which involves spatial electron delocalization, and its effects on electronic structures and material performance remain insufficiently understood. Materials exhibiting this weak interaction-based TSC are referred to as weak interaction-based organic luminescent materials [105]. Previous studies have partially elucidated the mechanisms of TSC in multiaryl-substituted alkanes (MAAs), such as diphenylmethane [106,107], triphenylmethane [108], tri(biphenyl)methane [109], and tetraphenylethane [110]. Nonetheless, the roles played by their intrinsic molecular skeletons and extrinsic aggregated states are still unclear [111].
In order to explore the influence of single molecular skeletons and aggregates on TSC, a systematic study of the trinaphthylmethane isomers was conducted [112]. As illustrated in Fig. 13a, four trinaphthylmethane isomers (222-TNM, 122-TNM, 112-TNM, and 111-TNM) were successfully synthesized. The photophysical properties of these molecules revealed that altering the connection sites of the naphthyl groups significantly impacts the TSC in molecular aggregates. In pure acetonitrile (ACN) solution, all four molecules exhibit a naphthyl ring emission peak at 339 nm. However, upon forming aggregates, these molecules can be classified into two types: (ⅰ) For 222-TNM and 122-TNM, a weak shoulder peak extending to 500 nm is observed, indicating the presence of weak and unstable TSC, as previously reported (Fig. 13b). (ⅱ) In contrast, 112-TNM and 111-TNM display strong and broad emission peaks at 384 and 395 nm, respectively. These findings suggest that aggregates of 112-TNM and 111-TNM are capable of producing strong TSC.
Figure 13
Figure 13.
(a) Single-crystal structures of TNM isomers and the manipulating strategies presented in this work. (b) Photoluminescence (PL) spectra of 222-TNM, 122-TNM, 112-TNM, and 111-TNM in ACN/water mixtures with different water fractions (fw), concentration (c) = 10 µmol/L. (c) Schematic plot of the relationship between intramolecular and intermolecular interactions and the strength of TSC, and the schematic plot of the relationship between the aggregate flexibility and TSC-emission wavelength of TNM isomers. Reproduced with permission [112]. Copyright 2024, Springer Nature.
Subsequently, theoretical calculations revealed that from 222-TNM to 111-TNM, the intermolecular interactions gradually weaken, while the intramolecular interactions progressively strengthen (Fig. 13c). Combining experimental and theoretical analyses, the pronounced TSC exhibited by 111-TNM is attributed to its strong intramolecular interactions, with intermolecular interactions serving to stabilize these intramolecular interactions. Therefore, this work not only unveils the intrinsic and extrinsic aspects of manipulating TSC, but also provides a novel strategy for achieving ultra-strong TSC and highly efficient organic emitters. The mechanistic investigation may further contribute to understanding the processes and mechanisms involved in modulating electronic structure and photophysical properties.
6.2
Architectured hydrogels as autonomous soft robots
Hydrogels serving as a typical class of soft and smart materials have garnered substantial interest in the development of soft actuators and robots capable of executing tasks in biomedical and engineering fields [113-121]. The continuous motions of these soft robots rely on spatially heterogeneous, dynamic stimulations or complex control modules for cyclic shape-morphing [119-124]. However, biological systems could exhibit autonomous motion by harvesting energy from the ambient environment and built-in feedback loops for self-regulation [125]. The underlying mechanisms have inspired many researchers to design intelligent soft actuators and robots to emulate the adaptive behaviors of living organisms. In recent years, researchers have developed several soft robots with self-sustained oscillations based on rhythmic chemical reactions or self-shadowing effects in responsive materials [126,127]. However, these soft robots exhibit intermittent motions due to the cyclic shape morphing with a delay. There are few reports on continuum soft robots with constant and autonomous motions under time-invariant conditions. Topological geometry features with an unchanged configuration with time upon external stimuli (i.e., self-similarity) [128,129], and the resulting topological intelligence may favor the actuation and self-control of shape morphing, which should be promising for the achievement of autonomous motions.
Recently, the researchers designed a series of anisotropic hydrogel-based soft robots with different topologies and achieved various autonomous motions under constant conditions [130,131]. The poly(N-isopropylacrylamide) (PNIPAm) hydrogel with shear-aligned nanosheets (NSs) and randomly dispersed gold nanoparticles is obtained by photopolymerization (Fig. 14a), which exhibits fast and isochoric deformation upon heating or light irradiation [121,122]. Due to the gradient photothermal strain, the hydrogel exhibits fast and reversible bending deformation upon light stimulations, which breaks the symmetry of the gel in the through-thickness direction (Fig. 14b). The trefoil knotbot (i.e., soft robot with a knotted structure) is fabricated by tangling the cylindrical hydrogel and then jointing the two ends (Fig. 14c). The topological geometry endows the soft robots with self-shadowing effect, topological constrains, and invariant morphology with time. Under static light irradiation, the additional hoop stress drives the knotbot to roll inward, which spontaneously exhibits continuous and autonomous rolling and braid rotation, owing to the synergistic contributions of topological intelligence and fast shape morphing (Figs. 14d and e).
Figure 14
Figure 14.
(a) Schematic for the fabrication of anisotropic hydrogel with shear-induced alignment of nanosheets. (b) Photothermal bending of the anisotropic gel upon light stimulus. (c) Schematic for the fabrication of the right-handed trefoil knotbot. (d) Simulated strain energy of the knotbot under static light irradiation. (e) Autonomous rolling and braid rotation of the trefoil knotbot under constant light irradiation. θ and γ represent the rotation and rolling angles, respectively. Reproduced with permission [130]. Licensed under CC-BY. (f) Schematic for the fabrication of the Möbius strip robot. (g) Simulated strain energy of the Möbius strip under static light irradiation. (h) Autonomous rolling and braid rotation of the Möbius strip robot under constant light irradiation. β and γ represent the rotation and flipping angles, respectively. Reproduced with permission [131]. Copyright 2024, Wiley Publishing Group.
Base on the similar mechanism, the research team also devised a Möbius strip robot by twisting a slender anisotropic gel sheet and jointing the two ends, which shows autonomous rotation and flipping motions upon constant irradiation (Fig. 14f). The Möbius strip is a classical topologic structure of a twisted ribbon with distributed torsional stress [123]. Under static light irradiation, the exposed regions near the twist locus bends towards the light source, breaking the local balance of internal stresses and driving the twist locus anticlockwise spreading along the Möbius strip. Owing to geometry-related refresh of the exposed regions and fast response of the anisotropic gel, the motions are continuous and autonomous (Figs. 14g and h). Such hydrogel robots with autonomous motions under constant condition should be promising in diverse fields, especially when the controlled dynamic stimulations are inaccessible.
6.3
Lithium extraction via functional polymers
Extracting lithium from seawater is highly promising to resolve the conflict between ever-growing lithium shortage and increasing need for electric devices, owing to the abundant yet untapped lithium resources of >230 billion tons in seawater [132]. However, existing lithium extraction strategies suffer from some fundamental challenges, such as limited Li+ trapping performance, severe concentration polarization and scaling, as well as high energy consumption. To overcome these challenges, the researchers proposed a novel design of photothermal “ion pumps” (PIPs) that enable solar-driven spontaneous, continuous Li+ transport and enrichment, allowing for efficient and durable lithium extraction from seawater [133]. The PIPs were elaborated by the function fusion and spatial configuration manipulation of a hydrophilic Li+-trapping nanofibrous core and a hydrophobic photothermal shell for governing gravity-driven water flow and solar-driven water evaporation. Briefly, the gravity-driven water flow can increase mass transfer within core-shell structured fiber for offering abundant ions replenishing as well as timely removing the interfering ions, which minimizes the impact of concentration polarization as well as eliminates the electrostatic repulsion of interfering ions to the freshly-replenished Li+. On the other hand, solar-driven water evaporation allows for rapid and persistent diffusion and enrichment of Li+ within core-shell structured fiber, ensuring Li+ with strengthened interaction to Li+-trapping sites of the Li+-trapping nanofibrous core in PIPs. Such a design endows PIPs with continuous and augmented Li+ replenishment-diffusion-enrichment, circumventing concentration polarization and scaling of interfering ions. The concurrent attainment of reinforced Li+ replenishment-diffusion-enrichment is not achievable with the conventional strategies. With this design, the PIPs exhibit improved Li+ trapping rate by nearly 80%, and outstanding Li+ separation factor (Li+/Na+ of ~53,747 and Li+/Mg2+ of ~13,151) toward seawater only consuming ultralow electric energy consuming several orders of magnitude lower than other reported methods. Moreover, the research team show that the PIPs deliver ultra-stable Li+ trapping performance without scaling even under high-concentration interfering ions for 140 h operation, as opposed to the significant decrease of nearly 56% in conventional photothermal configuration. To manifest the potential of practical application, the research team further manufactured multiple-PIPs systems with an array of 18 PIPs, exhibiting a Li+ production rate of up to 308.6 mg m−2 d−1.
In addition to the adsorption-based lithium extraction, the research group also developed high-performance polyamide membranes for effective lithium extraction. Normally, current polyamide membranes are negatively charged once using commercial piperazine and trimesoyl chloride as monomers, as imposed by their insufficient diffusion. Moreover, these polyamide membranes fabricated by piperazine and trimesoyl chloride are largely limited by narrow charge tailoring window, owing to their synchronous change of bulk and interfacial diffusion. Last year, the group developed a series of thermodynamic and kinetic regulation strategies to orchestrate the bulk and interfacial diffusion during interfacial polymerization [134-139], aiming at optimizing the structure and property of polyamide membranes and eventually boosting their separation performance. Particularly, the research team presented a facile ionic liquid-decoupled strategy that enables double charge flips of polyamide membranes (Fig. 15) [140]. Distinct form conventional interfacial polymerization, their design leverages unique physicochemical properties of ionic liquid as co-solvent to decouple bulk and interfacial diffusion during interfacial polymerization, achieving enhanced interfacial diffusion and inhibited bulk diffusion simultaneously. Such an ionic liquid-manipulated platform not only offers many open possibilities to construct charged polyamide membranes with broad charge tailoring window, but also is able to dramatically boost positive surface charges with zeta potential of +34 mV, as opposed to the conventional polyamide membranes with zeta potential of −25 mV. As such, this polyamide membrane yields an ultrahigh Mg2+/Li+ selectivity up to 68, which are >13.7 times than that of conventional polyamide membranes.
Figure 15
Figure 15.
(a) Schematic illustration of negatively charged polyamide membranes fabricated by conventional interfacial polymerization at the alkane-water interface. (b) Schematic illustration of double charge flips of polyamide membranes by ionic liquid-decoupled bulk/interfacial diffusion during interfacial polymerization. Copied with permission [140]. Licensed under CC-BY.
6.4
Solution-processed non-solvated C60 single-crystal films for high-performance n-type OFETs
The development of high-performance n-type organic semiconductors generally lagged the p-type ones. C60 has been one of the star n-type materials in organic field-effect transistors (OFETs) [141-143]. But the solution processing of C60 into large-area continuous single-crystal films remains challenging [144]. In the current literature, regular ribbon-shaped C60 crystals that are advantageous in OFETs are deposited either from mixed solvents or low-boiling-point toxic solvents such as CS2, which hinders the steady and continuous crystal growth [141,145,146]. Moreover, C60 tends to form various solvated crystals when crystallized from different solvents. Such high concentration of residual solvent molecules is known to be harmful to the mobility and stability of n-type organic semiconductors [142,147]. On the other hand, it is difficult to explicitly study the performance of C60 in n-type OFETs with such a number of different solvated structures. It is thus critical to develop methods that directly obtain non-solvated C60 single crystals, which will certainly boost the potential of C60 in high-performance n-type OFETs.
Herein, centimeter-scale non-solvated C60 single-crystal films were successfully grown by a facile bar coating method without the aid of extra growth-promoting materials (Fig. 16) [148]. With appropriate solvent selection and temperature control, non-solvated hexagonal close packed (HCP) C60 crystals were directly grown. Uniformly aligned regular ribbon-shaped crystals could be obtained by controlling the coating speed and concentration. The n-type OFETs exhibited average electron mobility as high as 2.28 cm2 V−1 s−1 and excellent uniformity with the coefficient of variance (CV) of mobility and threshold voltage as low as 13.6% and 16.5%, respectively. The non-solvated nature also gave raised to the enhanced operational and temporal stability, which would bring advantages in the further applications of C60 in various optoelectronic devices.
Figure 16
Figure 16.
(a-h) The optical microscopy (OM) images of C60 crystals grown from different solvents. Molecular structure of solvents was shown at the top right corner of each figure. The scale bars in all figures are 50 µm. (i, j) Typical transfer and output characteristics curve of the C60-crystal-based devices. (k) Transfer characteristics curves of 80 OFETs on a 19 mm × 10 mm substrate. Histogram of the mobility (l), threshold voltage (m), and on-state current (n) based on the 80 OFETs. Reproduced with permission [148]. Copyright 2024, Wiley Publishing Group.
6.5
Enabling polymer single crystals to be high-performance dielectrics
Polymer lamellar single crystals (PLSCs), represent not only the most highly ordered polymer structure achievable under mild conditions but also fundamental building blocks of more complex aggregates, such as spherulites and shish-kebabs, providing insights into solid-state physics [149]. Since the discovery of polyethylene (PE) PLSCs in the mid-20th century [150], sustained research has enabled the establishment of fundamental physical models and theories for understanding polymer crystallization. Furthermore, the well-defined aggregated structure and long-range order serves as an ideal platform for studying polymer structure-property relationships at the molecular scale, as well as elucidating performance limits and application potential in ordered polymer materials. For instance, Semicrystalline polymer dielectrics (SPDs) are widely used in electric power transmission, energy storage, and advanced microelectronic devices due to their lightweight, low-cost, flexible nature, facile processability, and exceptional dielectric properties [151-155]. These advantageous properties stem from the unique characteristics of polymers, including their elemental composition, long and flexible chains, and chain-folding crystallization behavior. However, these same characteristics also lead to complex aggregating microstructures, such as different crystal phases and mixtures of crystalline and amorphous regions, hindering a complete understanding of structure-property relationships in SPDs [156-158]. PLSCs characterized by long-range order and well-defined chain orientation, offer a pathway to investigate and potentially mitigate the detrimental effects of these disordered regions on material dielectric properties. However, investigating the macroscopic physical properties of PLSCs remains challenging due to difficulties in material preparation and the scarcity of appropriate testing methods, thereby restricting research progress in this field [159,160].
Recently, the researchers devised a strategy for preparing large-area PLSCs by controlling the crystallization kinetics inherent in the solution crystallization process, achieving controllable growth of polyethylene lamellar single crystals with optimized defect density, reaching sizes on the order of a hundred micrometers (Figs. 17a and b) [161]. In addition, to address the inherent fragility of the resulting lamellae, the research group proposed a “flat-template lift-off” (FTL) process to prepare planarized electrodes. This approach effectively avoids interference from lamellar damage during the construction of parallel-plate capacitors, consequently enabling the successful fabrication of vertical capacitors incorporating polyethylene lamellar dielectrics (Fig. 17c). Characterization of these vertical capacitors, realized through non-destructive processing, demonstrated markedly improved dielectric properties, evidenced by a high Weibull breakdown strength of 6.95 MV/cm and a low dielectric constant of 2.14, exceeding the performance characteristics of most currently available polyethylene-based insulating materials. This exceptional dielectric performance is attributed to the long-range ordered structure and molecular chain orientation inherent within the polyethylene PLSCs. Beyond demonstrating the exceptional dielectric performance conferred by the long-range ordered structure of PLSCs and their promising application within electronic devices, this work fosters a more fundamental understanding conducive to the development of advanced high-performance insulating materials.
Figure 17
Figure 17.
(a) Illustration of the fabrication process and the crystal structure of PE PLSCs. (b) An optical microscope (OM) image of PE PLSC. (c) The fabrication process of MIM capacitor device. Reproduced with permission [161]. Copyright 2024, Wiley Publishing Group.
In conclusion, this review has highlighted the breadth and depth of research advances made by the MOE Key Laboratory of Macromolecular Synthesis and Functionalization during 2024, showcasing significant progress across its five core research areas. In controllable catalytic polymerization, innovative organoboron catalysts have achieved >99% selectivity for alternating PPC, while CO2-PCs are emerging as high-performance developed photoresists. This achievement paves the way for greater control over polymer synthesis as well as bridging polymer chemistry with semiconductor manufacturing. The efforts in microstructure and rheology have elucidated deformation mechanisms in graphene-based 2D membranes, leading to superhigh modulus and excellent thermal conductivity in graphene with topological cellular structures. These findings provide a foundation for future efforts focused on translating these findings into scalable fabrication techniques for high-performance graphene materials, controlling the assembly/orientation of graphene sheets to optimize properties across diverse applications. Regarding separating functional polymers, novel Janus membranes and channels, refined techniques for organic solvent nanofiltration, and HABD models have offered excellent results, providing a platform for prioritizing the design of adaptive membranes responding to changing conditions and developing advanced separating membranes for concentration polarization issues and precise molecular separations. In the domain of biomedical functional polymers, studies explored the upcycling of PET waste to biodegradable PEHT plastics, creation of immunocompatible surfaces, and treatment of fungal infections via induced fungal cuproptosis. Success was also achieved in fast hemostatic sealing using GDK/Fg hydrogels, promoting endothelialization of LAA occluders by introducing double-network polyelectrolyte hydrogels, and designing deep-tissue activation for shape-memory polymers. These studies offer a compelling trajectory for continued development, emphasizing the translation of these materials into clinically relevant medical devices, enhancing biocompatibility, and integrating diverse functionalities. Finally, control of through-space conjugation enhanced organic luminescence, topologically structured hydrogels demonstrated autonomous performance, photothermal ion pump successfully transported ions, high-performance C60 single crystals was manufactured for electronics and extraordinary dielectric properties of PE PLSCs was demonstrated, illustrating the broad potential of polymer materials with rationally designed structures across fields as varied as electronics, photonics, and energy. These advances, ranging from polymer synthesis to investigations of structure-property relationships and the development of advanced functional materials, are expected to stimulate significant research in both fundamental science and practical industrial applications.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Kangyuan Xie: Writing – review & editing, Writing – original draft. Tianxiang Fang: Writing – original draft. Qingli Zhu: Writing – original draft. Qingyang Xu: Writing – original draft. Boyu Peng: Writing – review & editing, Writing – original draft. Guangpeng Wu: Writing – review & editing, Writing – original draft. Chao Gao: Writing – review & editing, Writing – original draft. Haocheng Yang: Writing – review & editing, Writing – original draft. Liping Zhu: Writing – review & editing, Writing – original draft. Hongqing Liang: Writing – review & editing, Writing – original draft. Weipu Zhu: Writing – review & editing, Writing – original draft. Peng Zhang: Writing – review & editing, Writing – original draft. Qiao Jin: Writing – review & editing, Writing – original draft. Zhengwei Mao: Writing – review & editing, Writing – original draft. Kefeng Ren: Writing – review & editing, Writing – original draft. Yang Zhu: Writing – review & editing, Writing – original draft. Haoke Zhang: Writing – review & editing, Writing – original draft. Ziliang Wu: Writing – review & editing, Writing – original draft. Chao Zhang: Writing – review & editing, Writing – original draft. Hanying Li: Writing – review & editing, Writing – original draft, Supervision, Project administration, Conceptualization.
Acknowledgment
This work was supported by the Fundamental Research Funds for the Central Universities (No. 226-2025-00031).
[1]
J. Ren, X. Shu, Y. Wang, et al., Chin. Chem. Lett. 33 (2022) 1650–1658.
[2]
Q. Wen, Q. Cai, P. Fu, et al., Chin. Chem. Lett. 34 (2023) 107592.
[3]
X. Deng, K. Chen, K. Pang, et al., Chin. Chem. Lett. 35 (2024) 108861.
[4]
G. Yu, C. Xu, H. Ju, et al., Chin. Chem. Lett. 35 (2024) 109893.
[5]
Y.Y. Zhang, G.W. Yang, C. Lu, et al., Chem. Soc. Rev. 53 (2024) 3384–3456. doi: 10.1039/d3cs00115f
Y. Miyoshi, K. Chino, Jpn. J. Appl. Phys. 6 (1967) 181.
[161]
M. Chen, W.L. Ong, B. Peng, et al., Angew. Chem. Int. Ed. 63 (2024) e202314685.
Figure 1
Schematic illustration about the key progresses of MOE Key Laboratory of Macromolecular Synthesis and Functionalization in 2024. Adapted with permission [10,12,23,27,66,103,140,161]. Copyright 2024, Wiley Publishing Group; Copyright 2024, American Association for the Advancement of Science; Copyright, 2024, Elsevier; Copyright 2024, Springer Nature; Copyright 2024, Wiley Publishing Group.
Figure 2
(a) Progress in converting CO2 and epoxides into aliphatic polycarbonates. Reproduced with permission [6]. Copyright 2024, American Chemical Society. (b) Organoboron compounds as versatile catalysts and mediators for diverse polymerization modes. Reproduced with permission [5]. Copyright 2024, The Royal Society of Chemistry. (c) Aqueous-developable CO-PCs bearing acid-cleavable acetal groups, serving as high-performance photoresists. Reproduced with permission [10]. Copyright 2024, Wiley Publishing Group.
Figure 3
Schematic of the concept of bidirectionally promoting assembly order of graphene. (a) GO fibers prepared by multiple shear-flow fields. (b-f) Structural illustration of the sheet-order in the whole spinning tube under multiple flow fields (c), where Plane Ⅰ (d, e) shows the aligned sheets along the flow direction but wrinkling conformation in perpendicular direction under the unidirectionally flow field, Plane Ⅱ (f) presents the aligned slices in two directions under multiple flow fields, and Plane Ⅲ (b) depicts the optimally concentric structure by bidirectionally propelling assembly order. Copied with permission [12]. Licensed under CC-BY.
Figure 4
(a) Three-stage mode of unidirectional liquid transport through Janus membrane. Reproduced with permission [19]. Licensed under CC-BY. (b) Janus substrate to regulate interfacial polymerization. Reproduced with permission [20]. Licensed under CC-BY. (c) Janus seesaw evaporator for self-descaling during hypersaline brine evaporation. Reproduced with permission [21]. Copyright 2024, Wiley Publishing Group. (d) Janus channel of membranes for concurrent recovery of oil and water from emulsions. Reproduced with permission [23]. Copyright 2024, American Association for the Advancement of Science.
Figure 5
Preparation of PA nanofilms with highly intrinsic microporosity for efficient organic solvent nanofiltration. (a) Schematic of PA nanofilm preparation by tailoring the monomer structure in molecular layer deposition. 4,4′-Methylenedianiline (MDA), 1,1-bis(4-aminophenyl)cyclohexane (BACH), and 9,9-bis(4-aminophenyl)fluorene (BAF) were used as amino monomers, and trimesoyl chloride (TMC) was used as the acyl chloride monomer. (b) PA-BAF amorphous cell (50 Å), where the Connolly surface area is colored gray/blue with a probe radius of 1 Å, as described in the simulation method. (c) Photograph of a typical free-standing PA nanofilm from the reaction between BAF and TMC. (d) Schematic of PA nanofilms with controlled chemical and morphological structures used for molecular separation in organic solvents. Reproduced with permission [27]. Copyright 2024, Elsevier.
Figure 6
(a) Comparison of harmonic amide bond density (HABD) and the degree of network crosslinking (DNC) in analyzing structures and performances of desalination polyamide membranes. (b) HABD for analyzing complex crosslinked polyamide networks. Reproduced with permission [32]. Licensed under CC-BY.
Figure 8
(a) Photograph showing the flexible sheet of EVADE material and its corresponding chemical structure. (b) Digital photos, H&E-, and M&T-stained histological sections of excised 1-year post-implantation in mice (representative for n = 6 biologically independent samples). (c) The construction and experimental overview of the S100A8-KO mice model and digital photos and M&T-stained histological sections of excised 4-week post-implantation in S100A8-KO mice. (d) Schematic representation of CSII catheter made of EVADE elastomer enabling the long-term use of infusion catheter and faster insulin absorption. Reproduced with permission [66]. Copyright 2024, Springer Nature.
Figure 9
Schematic illustration of the synthesis and fungicidal mechanism of PCW nanoflowers. Copied with permission [76]. Copyright 2024, Springer Nature.
Figure 10
Schematic illustration of the preparation, crosslinking, and mechanism of tissue adhesion and hemostasis for the GDK/Fg hydrogel. Reproduced with permission [88]. Copyright 2025, Elsevier.
Figure 11
(a) Preparation process of hemocompatible coatings on the surface of the LAA occluder for enhanced endothelialization. (b) Schematic illustration of the polyelectrolyte coating on LAA occluder. Reproduced with permission [101]. Licensed under CC-BY. (c) Schematic diagram of drop-shaped microgrooves and their influence on cell migration. Reproduced with permission [102]. Copyright 2024, Springer Nature.
Figure 12
(a) Equipment layout of image-guided high-intensity focused ultrasound (HIFU). Image-guided HIFU facilitates precise, non-invasive, and real-time monitored heating of shape memory medical devices in vivo. This technology has potential applications in medical device implantation, functional activation, including drug release, and device extraction. (b) Concept of shape memory ureteral stent removal. Copied with permission [103]. Licensed under CC-BY.
Figure 13
(a) Single-crystal structures of TNM isomers and the manipulating strategies presented in this work. (b) Photoluminescence (PL) spectra of 222-TNM, 122-TNM, 112-TNM, and 111-TNM in ACN/water mixtures with different water fractions (fw), concentration (c) = 10 µmol/L. (c) Schematic plot of the relationship between intramolecular and intermolecular interactions and the strength of TSC, and the schematic plot of the relationship between the aggregate flexibility and TSC-emission wavelength of TNM isomers. Reproduced with permission [112]. Copyright 2024, Springer Nature.
Figure 14
(a) Schematic for the fabrication of anisotropic hydrogel with shear-induced alignment of nanosheets. (b) Photothermal bending of the anisotropic gel upon light stimulus. (c) Schematic for the fabrication of the right-handed trefoil knotbot. (d) Simulated strain energy of the knotbot under static light irradiation. (e) Autonomous rolling and braid rotation of the trefoil knotbot under constant light irradiation. θ and γ represent the rotation and rolling angles, respectively. Reproduced with permission [130]. Licensed under CC-BY. (f) Schematic for the fabrication of the Möbius strip robot. (g) Simulated strain energy of the Möbius strip under static light irradiation. (h) Autonomous rolling and braid rotation of the Möbius strip robot under constant light irradiation. β and γ represent the rotation and flipping angles, respectively. Reproduced with permission [131]. Copyright 2024, Wiley Publishing Group.
Figure 15
(a) Schematic illustration of negatively charged polyamide membranes fabricated by conventional interfacial polymerization at the alkane-water interface. (b) Schematic illustration of double charge flips of polyamide membranes by ionic liquid-decoupled bulk/interfacial diffusion during interfacial polymerization. Copied with permission [140]. Licensed under CC-BY.
Figure 16
(a-h) The optical microscopy (OM) images of C60 crystals grown from different solvents. Molecular structure of solvents was shown at the top right corner of each figure. The scale bars in all figures are 50 µm. (i, j) Typical transfer and output characteristics curve of the C60-crystal-based devices. (k) Transfer characteristics curves of 80 OFETs on a 19 mm × 10 mm substrate. Histogram of the mobility (l), threshold voltage (m), and on-state current (n) based on the 80 OFETs. Reproduced with permission [148]. Copyright 2024, Wiley Publishing Group.
Figure 17
(a) Illustration of the fabrication process and the crystal structure of PE PLSCs. (b) An optical microscope (OM) image of PE PLSC. (c) The fabrication process of MIM capacitor device. Reproduced with permission [161]. Copyright 2024, Wiley Publishing Group.


 DownLoad:
DownLoad:

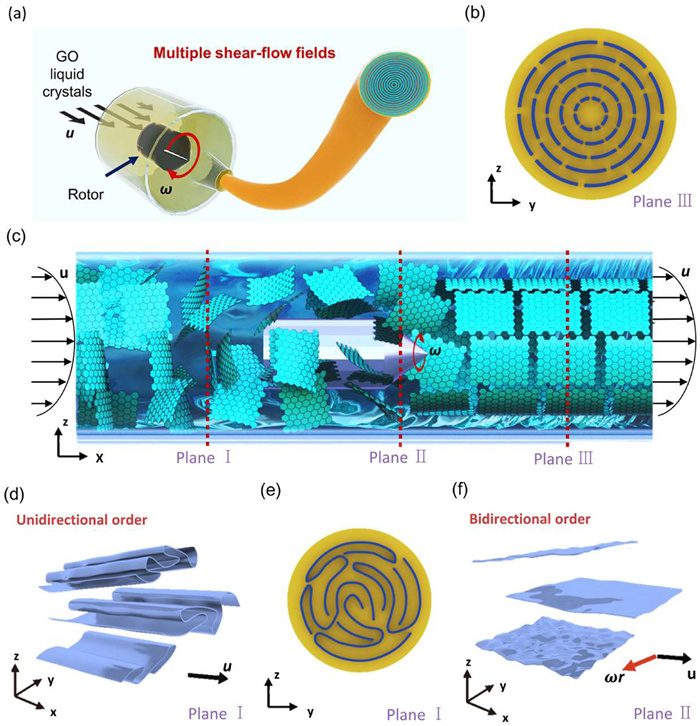
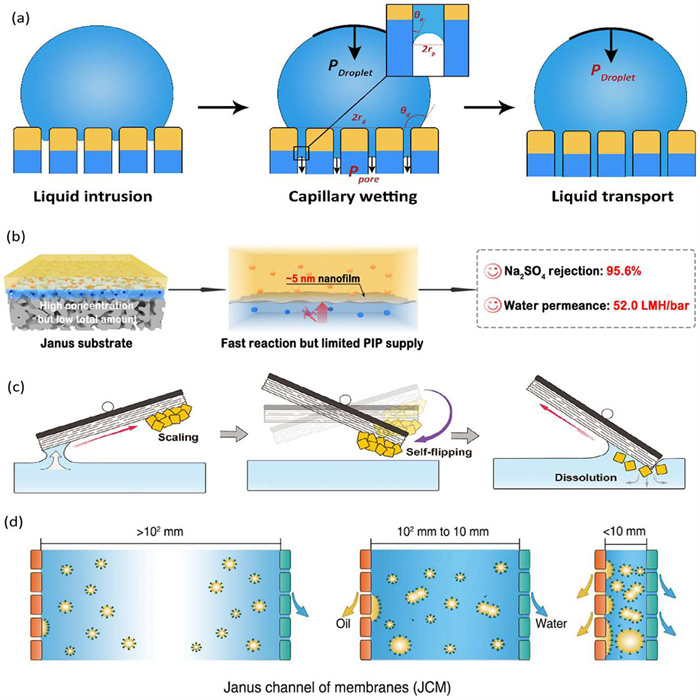
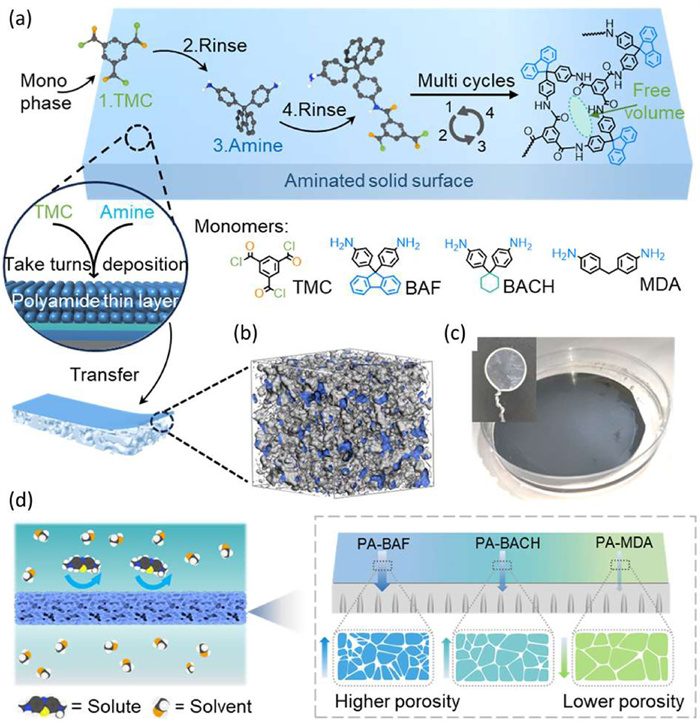
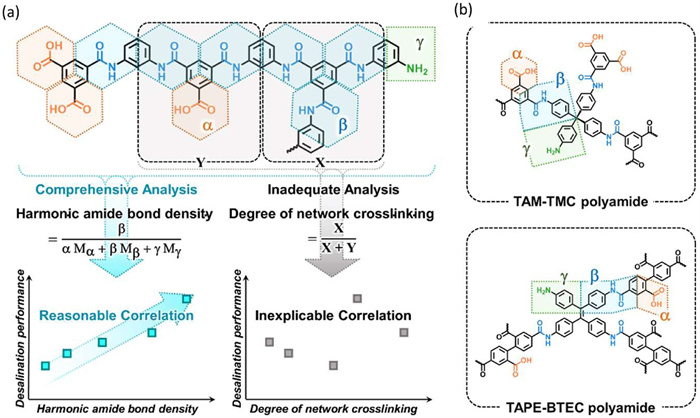

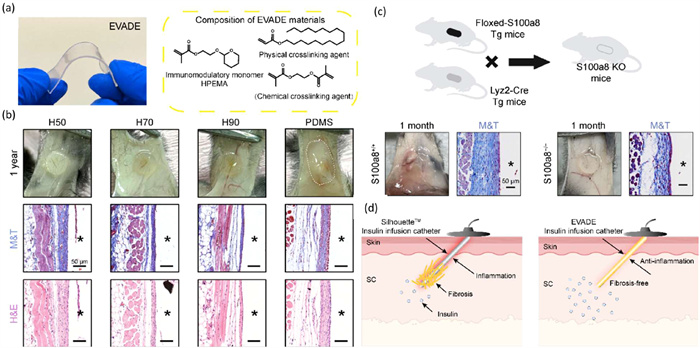
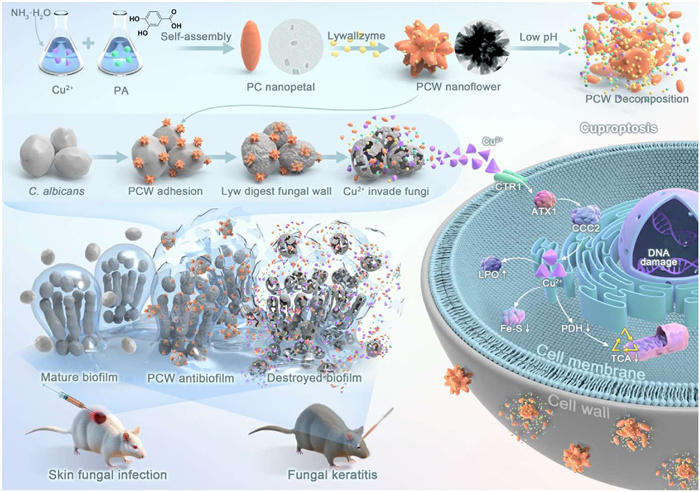
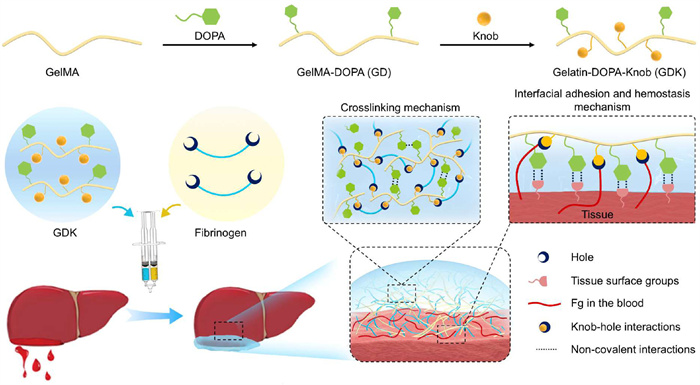

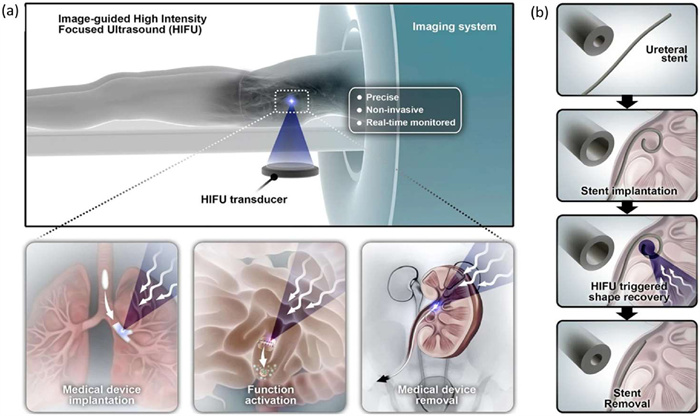
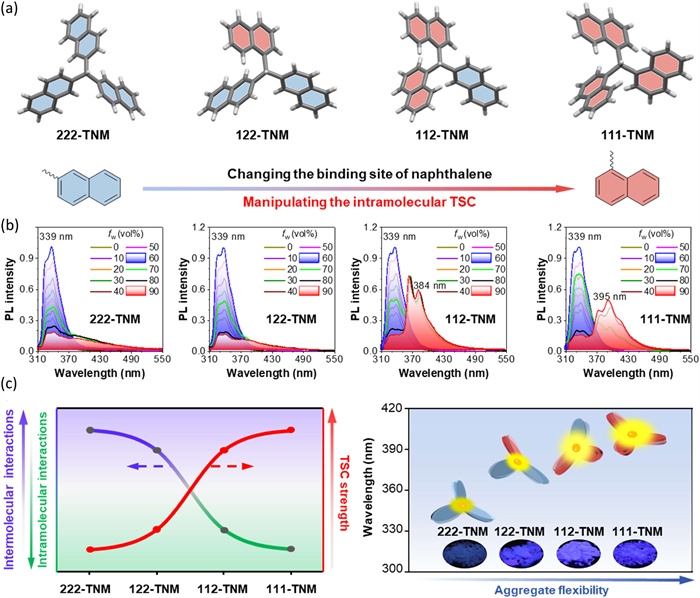
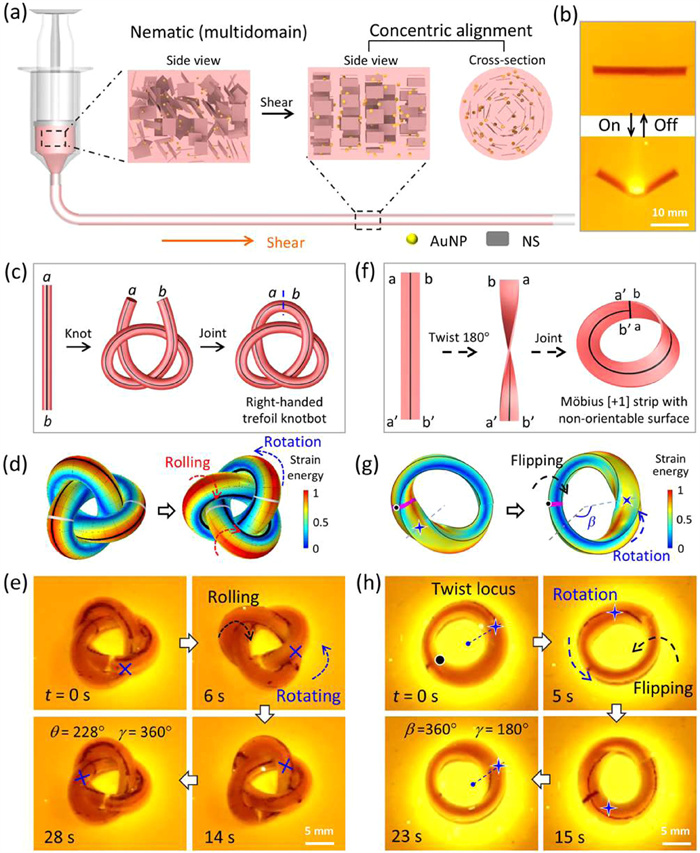
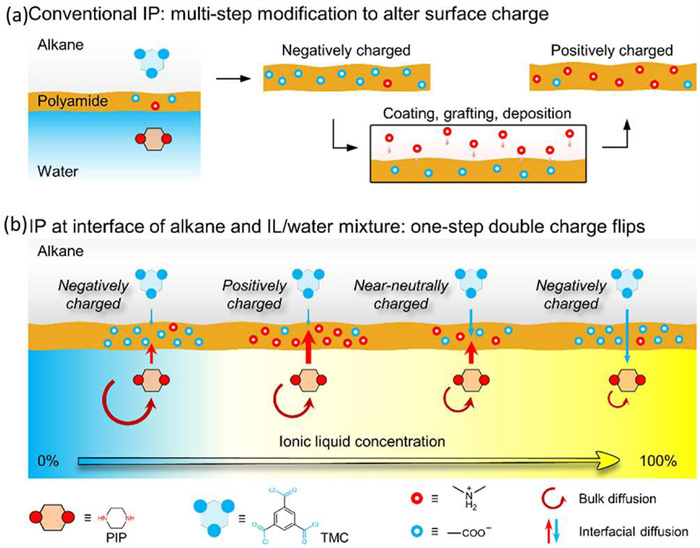
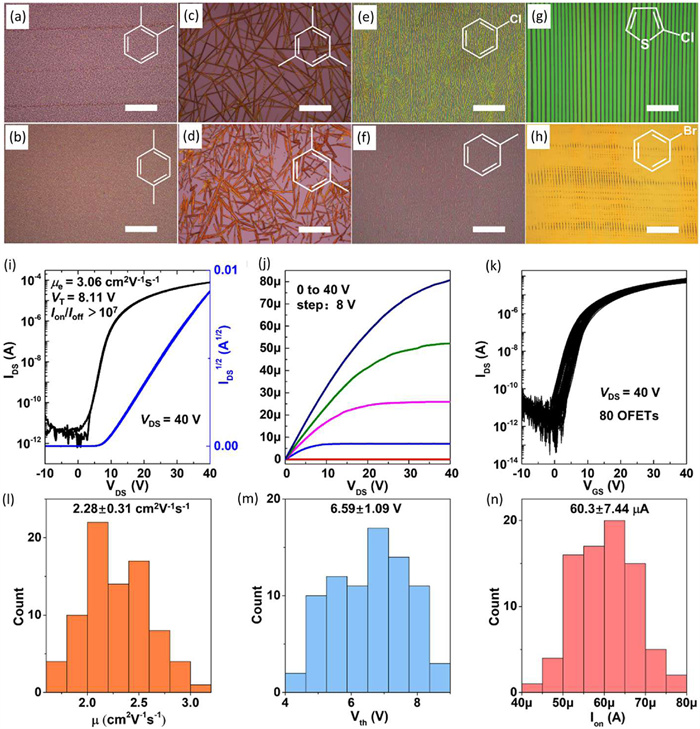
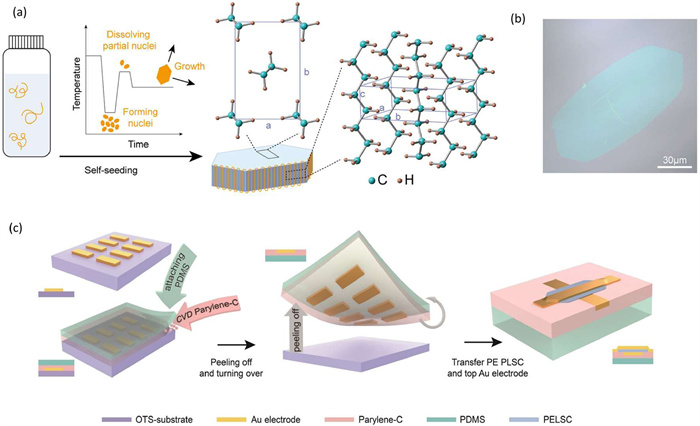

 下载:
下载:


















