Key Laboratory for Advanced Materials and Joint International Research Laboratory of Precision Chemistry and Molecular Engineering, Feringa Nobel Prize Scientist Joint Research Center, Frontiers Science Center for Materiobiology and Dynamic Chemistry, Institute of Fine Chemicals, School of Chemistry and Molecular Engineering, East China University of Science and Technology, Shanghai 200237, China
b.
Strait Institute of Flexible Electronics (SIFE, Future Technologies), Fujian Key Laboratory of Flexible Electronics, Fujian Normal University and Strait Laboratory of Flexible Electronics (SLoFE), Fuzhou 350117, China
feitong@ecust.edu.cn (F. Tong). 1 These authors contributed equally to this work.
Received Date:
12 June 2025 Accepted Date:
12 October 2025 Revised Date:
08 October 2025 Available Online:
15 February 2026
Abstract:
Developing advanced polymeric materials with enhanced mechanical properties and functionalities has been a long-standing goal in materials science. Recently, supramolecular polymeric materials (SPMs) have drawn increased attention due to their unique properties and potential applications in self-healing, shape memory, sensors, and flexible electronics. Here, we develop an ionic cluster-optimized microphase separation strategy to enhance the toughening and energy dissipation capabilities of polydisulfide-based supramolecular polymers. The mechanical properties, including Young’s modulus and toughness, are significantly improved by integrating the quadruple H-bonding 2-ureido-4-pyrimidone (UPy) induced microphase separation with iron(Ⅲ)-to-carboxylate ionic clusters. By combining established chemical approaches with adjustable polymer phase ratios, it is revealed that the synergistic effect of these factors expands the interchain spacing, facilitates the formation of microphase domains, and enhances the tolerance of polythioctic acid-based polymers to external mechanical and thermal stimuli, meeting the practical requirements for industrial plastic applications. Moreover, the UPy-functionalized polymers incorporating iron carboxylate clusters exhibit good one-way shape memory behavior with practical applicability at a relatively low recovery temperature. Our work demonstrates a novel strategy for constructing industrially viable shape memory dynamic SPMs and paves the way for future innovations in developing SPMs.
Nature enables life to adapt to environmental stimuli, including mechanical and thermal stressors, through sophisticated, synergistic, yet highly functional systems and to maintain the balance of living organisms by dissipating energy through increased entropy [1,2]. Comprehending and leveraging these architectures brings valuable insights and significant benefits to various innovative composite materials [3,4]. Integrating multiple stimuli-responsive mechanisms, such as hydrogen bonds [5–7], microphase separation [8–10], metal-ligand interactions [11,12], ionic bonds [13,14], and host-guest interactions [15,16], is desirable to fulfill novel properties in traditional plastics. However, a trade-off relationship is usually generated between tunable designed functions and high processing difficulties for the inherently fixed networks. Supramolecular polymeric materials (SPMs) bond distinct polymer blocks together through reversible interactions, allowing for dynamic repair, reuse, and recycling [17–21]. Among these, disulfide bonds have garnered promising and growing attention in polymer science, endowing polydisulfide materials with various intriguing functions [22–24]. In recent years, the research focus has gradually shifted from molecular-level chemical modifications and property regulation [25,26] to studies oriented toward industrial applications [27–30]. However, despite their exceptional strain capabilities, polythioctic acid networks exhibit inadequate mechanical properties and high environmental sensitivity, which limits broader industrial applications. The critical challenge in the field of polydisulfide materials lies in strain control, necessitating a delicate balance between network strength optimization while maintaining structural simplicity and minimizing auxiliary components. On the other hand, introducing robust dynamic chemical bonds into the microstructure design opens up new possibilities for enhancing the performance of SPMs and developing high-energy dissipation capabilities [10,13,31]. In polymeric materials, typical approaches include microphase separation and the electrostatic interaction induced by ionic clusters. For microphase separation, the hard segments require appropriate block lengths and sufficient steric hindrance to aggregate together and form crystalline domains. Therefore, it prevents them from being squeezed out by the soft segments with higher conformational flexibility, endowing the polymer with the ability to absorb energy without fracturing, resulting in high toughness. The quadruple hydrogen bonding 2-ureido-4-pyrimidone (UPy) motifs have gradually emerged as a prominent functional group in supramolecular chemistry and advanced materials design, owing to the straightforward synthetic methodology [32], robust binding constants [33], and efficient energy dissipation mechanisms [34]. Moreover, the presence of ionic clusters leads to a higher crosslink density, resulting in an increased glass transition temperature and higher stiffness (higher Young’s modulus). However, previous literature suggested that the assistance of ionic clusters in enhancing the restoring force and energy dissipation of polymers is limited to low-strain conditions [35,36]. As a result, achieving both reinforcement and toughening of SPMs while improving their ability to dissipate energy under external mechanical and thermal stimuli remains challenging.
Here, we develop an ionic cluster-optimized microphase separation to achieve both toughening and enhanced energy dissipation capabilities for designing potential engineering plastics. The polydisulfides serve as flexible polymeric backbones, while the dynamically bonded UPy motifs limit the high-strain properties and increase the mechanical strength. Due to the rigid structure of the UPy dimerization, the UPy motifs, which serve as tertiary clusters, often exhibit coplanar stacking, imparting multifaceted thermal effects on the polymeric network [10,34,37]. The incorporation of iron(Ⅲ)-to-carboxylate ionic clusters (serving as secondary clusters) into the polymer backbone provides electrostatic interactions while simultaneously replacing a portion of the carboxylic acid pairs (serving as primary clusters). As a result, it expands the interchain spacing by reducing the entanglement of the polymer chains and endows more space to facilitate the microphase separation induced by UPy motifs. The low strain caused by the UPy motifs also allows the ionic clusters to undergo facile ligand exchange, thereby enabling the dissipation of the input energy. The synergistic interplay of these factors has resulted in a multiplicative improvement in the strength and toughness of the polymeric materials, concomitantly endowing them with a wide range of mechanical and thermal resistance properties. Capitalizing on this, we have engineered a class of polydisulfide-based shape-memory SPMs that can undergo rapid, unidirectional shape recovery at moderately low temperatures alongside notable performance repeatability. These advantages render polydisulfide-based SPMs with practical applications in intelligent materials.
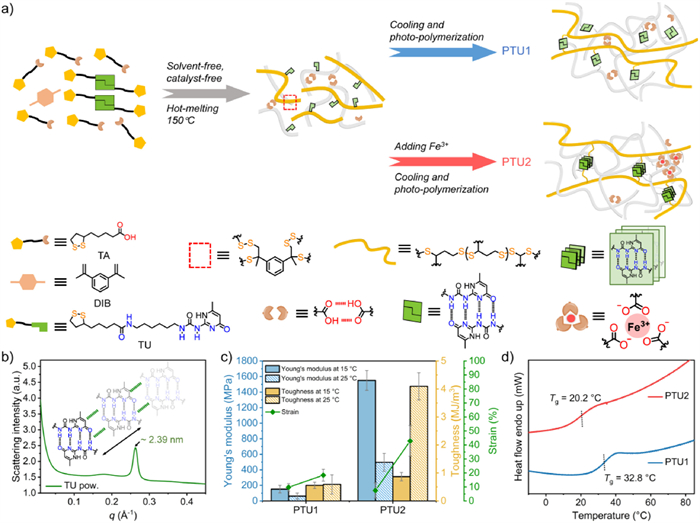
As depicted in Fig. 1a, polymer networks are prepared by the hot-melt mixing and photo-polymerization of monomer molecules, which is based on an optimization of our previous procedure involving 1,3-diisopropenylbenzene (DIB, 20 wt%) as the cross-linker [38,39]. The carbon-carbon double bonds in DIB molecules can undergo rapid click reactions with sulfur radicals, efficiently terminating terminal sulfur radicals along polymer chains. This dual-functional process facilitates both inverse vulcanization and molecular-scale network toughening. Pre-polymerization can be achieved through a solvent-free, catalyst-free hot-melting method, while further cooling and photo-polymerization result in denser and more stable polymeric backbones [40]. To better harness the quadruple hydrogen bonding interactions (quadruple H-bonds) of the UPy motifs as hard segments and increase the compatibility, we synthesized the monomer molecule TU with an alkyl chain length twice that of TA (Fig. 1a). The powder X-ray diffraction (XRD) patterns of TU exhibited a significant baseline rise (Fig. S5 in Supporting information), indicating potential molecular clustering between the monomers and a concomitant decrease in long-range crystalline order. However, the small-angle X-ray scattering (SAXS) pattern of monomer TU featured one distinct sharp peak, corresponding to a length of 2.39 nm (Fig. 1b). We deduced that the unique dimerization of UPy and its one-dimensional (1D) stacking of the dimerized motifs are responsible for the presence of this peak [32,41].
Figure 1
Figure 1.
Synthesis and the mechanical properties of polymeric materials. (a) Cartoon diagram of the preparation of polydisulfide-based SPMs: PTU1 and PTU2. (b) The SAXS curve of monomer TU with the proposed interstack interaction between UPy motifs. (c) Comparison of Young’s modulus, toughness, and strain rates of PTU1 and PTU2. Error bars represent standard deviations. (d) The DSC spectrum of PTU1 and PTU2 with their Tg.
The optimal doping ratio, achieved by varying the ratios of TU (with a weight ratio of TA to TU of 3:1) and varying ratios of Fe3+, was characterized by the tensile stress curve (Figs. S6 and S7 in Supporting information). The experimental groups without doped Fe3+ are denoted as PTU1. Those with Fe3+ (1/4500 molar ratio to carboxyl group) are denoted as PTU2 (Fig. 1a). The incorporation of Fe3+ leads to the formation of iron(Ⅲ)-to-carboxylate ionic clusters, introducing additional supramolecular interactions, while simultaneously enhancing the network strength, toughness, and energy dissipation efficiency of the system [35,38,39]. The optimal ratio of DIB to Fe3+ was determined through established optimization protocols to maximize the efficacy of design modulation of the polymer phase composition [38]. The controlled hot-melting temperature, below the monomer TU’s melting point (Tm, Fig. S4 in Supporting information), facilitated processing by reducing the mixture viscosity while preserving the UPy motifs’ aggregation capability [34,42]. Thermal gravimetric analysis (TGA) of the obtained polymeric materials revealed that the decomposition temperature at 95% residue weight is ~230 ℃ (Fig. S8 in Supporting information), indicating impressive thermal stability. The variations in color and infrared spectra of the copolymer indicate the formation of iron(Ⅲ)-to-carboxylate ionic clusters (Figs. S9 and S10 in Supporting information) [38].
Compared to the original poly(TA-DIB-Fe) [38,39] without doping monomer TU, both PTU1 and PTU2 achieve significant improvements in mechanical performance. By introducing TU with the proper proportion, Young’s modulus increased from 0.086 MPa (poly(TA-DIB-Fe), 1/18000 ratio of Fe3+) or 0.83 MPa (poly(TA-DIB-Fe), 1/1000 ratio of Fe3+) to 151.81 MPa (PTU1, Table S1 in Supporting information), showing nearly three orders of magnitude improvement. This significant reinforcement was ascribed to the robust and stiff quadruple H-bonds-induced microphase separation. Moreover, for poly(TA-DIB-Fe), the inherent soft and dynamic nature of polydisulfides enables them to maintain a high strain even after doping with ionic clusters, which is unfavorable for energy dissipation and polymeric toughening (>8000% strain) as the restoring force is insufficient to drive ligand exchange [35,36]. In contrast, PTU1 successfully reduced the strain of polymeric materials to a suitable level (9.75%), which favors ligand exchange and energy dissipation via ionic clusters. Successfully, the Fe3+-doped PTU2 furnishes a substantial enhancement in modulus, approaching a ten-fold increase in its glassy state (Fig. 1c, Fig. S13 and Table S1 in Supporting information). According to differential scanning calorimetry (DSC, Fig. 1d) results, PTU2 exhibited a glass transition temperature (Tg) of 20 ℃, which is near ambient temperature and lower than the Tg of PTU1 (33 ℃). We attribute this phenomenon to the incorporation of Fe3+, which increases the interchain spacing and reduces the degree of polymer chain entanglement, as will be elaborated in subsequent sections. Therefore, mechanical performance comparisons were conducted at 15 ℃ and 25 ℃ to ensure equal ΔT from the glass transition. Notably, at room temperature (25 ℃), PTU2 exhibited a remarkable enhancement in toughness (Fig. 1c), showing approximately seven-fold improvement compared to PTU1 at relatively low strain levels. This significant enhancement can be attributed to the efficient energy dissipation mechanism arising from the synergistic effect of UPy motifs and iron(Ⅲ)-to-carboxylate ionic clusters.
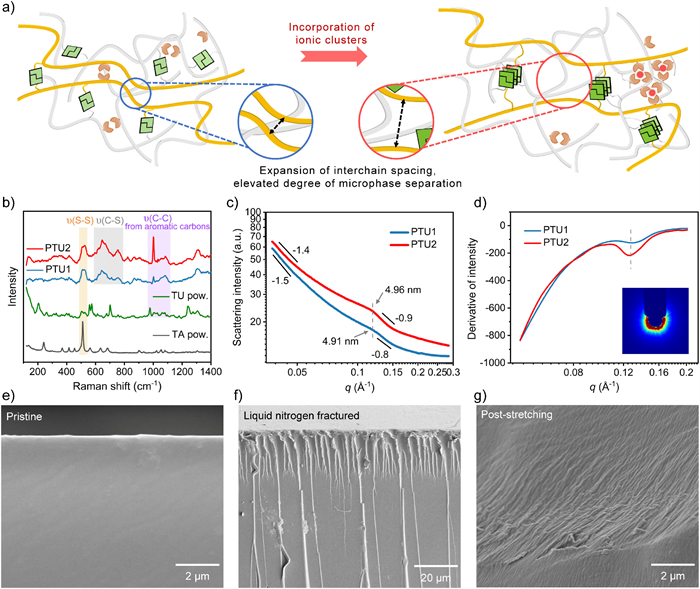
The obtained high toughness and low strain of PTU2 have reconciled the potential connection between microphase separation and ionic clusters, where we next proceed to the hypothesis that ionic clusters-optimized microphase separation toughening mechanism (Fig. 2a). The pristine amorphous polymer backbone lacks the long-range order and the densely packed network hinders further aggregation of the UPy motifs (Fig. S5). Adding Fe3+ replaces part interactions between polymer chains formed by carboxylic acid dimers with iron(Ⅲ)-to-carboxylate ionic clusters, thereby expanding the interchain distances. As shown by Raman measurements (Fig. 2b), both PTU1 and PTU2 exhibited characteristic peaks attributed to the disulfide (S–S) bond and carbon-sulfur (C–S) bonds, along with distinctive Raman peaks corresponding to the C–C bonds in the aromatic rings that differed from those of the monomers. These can be attributed to the crosslinker DIB, which connects the polymer backbone. It is noticeable that PTU2 displays more intense peaks than PTU1 on aromatic C–C bonds, indicating that it has more exposed aromatic rings. The initially dense polymer network affected the peak intensities, while the subsequent increase in interchain spacing enabled enhanced peak heights. Our results suggest that ionic clusters play a role in expanding the chain distances within the polymer matrix.
Figure 2
Figure 2.
The toughening mechanism of PTU2 and the microstructural characterization. (a) Cartoon diagram of the mechanism of ionic cluster-optimized microphase separation. (b) The Raman spectra of monomer TA (grey curve), monomer TU (green curve), polymer PTU1 (blue curve, the same as follows), and polymer PTU2 (red curve, the same as follows). (c) The experimental SAXS curve of PTU1 and PTU2. (d) The first derivative of SAXS intensity. Inset: 2D image of PTU2. (e-g) SEM cross-section images of pristine, liquid-nitrogen-fractured, and tensile-fractured PTU2 samples, respectively.
The resulting sufficient steric hindrance provides the UPy motifs with greater ease in forming aggregation micro-domains. As indicated by the SAXS patterns and the derivative of scattering intensity value (Figs. 2c and d), one new broad envelope peak emerged at q ≈ 0.128 Å-1 both in PTU1 and PTU2, which is distinct from the single sharp peak observed in the TU monomer spectrum. This characteristic peak indicates the emergence of a newly generated phase within the polymer network, induced by UPy motifs [41]. Comparative analysis reveals that while the peak positions remain invariant between PTU1 and PTU2, the observed differences in peak intensity suggest that the emergent microphase does not originate from alterations in interchain distances but can be attributed to an enhanced density of identical aggregated structures. The incorporation of Fe3+ facilitates the substitution of interchain carboxylic acid dimers with larger ionic clusters, thereby expanding the interchain spacing. The resultant reduced spatial constraints enable the emergence of additional UPy-induced microphases, which are evidenced by the markedly intensified diffraction peaks observed in the SAXS profile of PTU2. The insert in Fig. 2d suggests that no long-range order or structural orientation exists, indicating that the ionic clusters favor more microphase domain formation originating from the consequent stacking of the UPy motifs rather than the formation or crystallization of clusters themselves [10,32,43,44].
To further elucidate the network toughening mechanism, we analyzed a pristine PTU2 sheet sample, a non-stretched sheet fractured in liquid nitrogen, and another sheet stretched until fracture, along with their cross sections, using scanning electron microscopy (SEM, Figs. 2e-g) [8,45]. The pristine one exhibits a smooth and flat cross-section free from visible defects due to the dynamic and high compatibility of the polymer. After liquid nitrogen treatment, relatively high internal stress was induced due to the high aggregation tendency of the hard segment quadruple hydrogen-bonded UPy motifs within the polymer. The high internal stress also leads to the large-scale formation of distinct longitudinal clustering patterns observed in the cross-section. Upon stretching, numerous nanoscale-level wrinkles were observed on the tensile fracture surface. These wrinkles and the pronounced uneven multi-layered block section (Fig. S15 in Supporting information) further substantiate the role of UPy motifs-induced microphase separation. Tension-induced wrinkles can promote stress dissipation well, but at the same time, the tightly packed hard segment crystalline domains also lead to a sharp decline in strain. As a result, the uniformly distributed microphase structures in the material’s bulk phase can serve as dissipation points for external mechanical stimuli [8], thereby achieving toughening effects for the integral polymer and improving the ability to dissipate external energy.
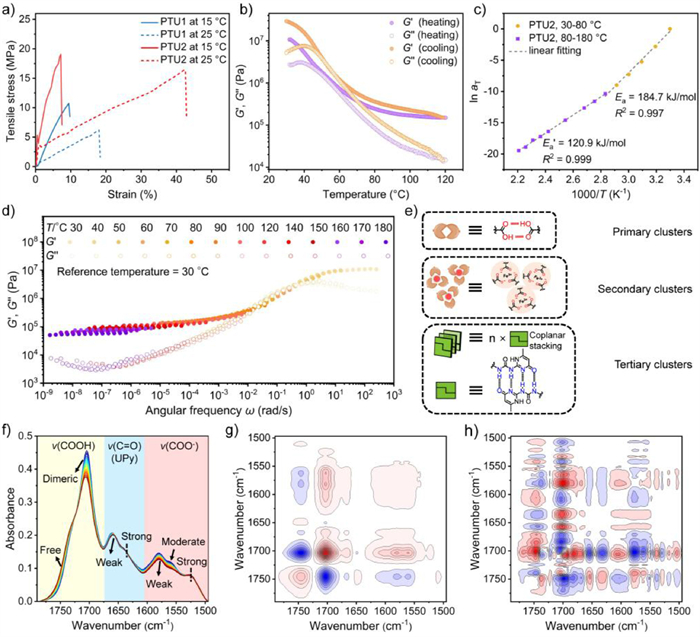
Given that PTU2 exhibits substantial mechanical reinforcement compared to PTU1, we identified characteristic elastic deformation regions and yielding phenomena in the stress-strain curves (Fig. 3a and Fig. S16 in Supporting information), indicating its prospective utility in engineering plastic applications. We then monitored the response and dissipation capacity using a series of frequency-dependent and temperature-variable rheological analyses (Figs. 3b-d and Fig. S17 in Supporting information) to understand the extent of dissipation of the multilayer interactions within PTU2 in response to thermal heating. The analysis of the temperature dependence of the rheological results during the alternative heating and cooling processes from 30 ℃ to 120 ℃ (Fig. 3b) revealed that the storage modulus of PTU2 was consistently higher than the loss modulus, exhibiting a pronounced solid-like characteristic and dominant elastic behavior. Even above the Tg, the polymer can maintain a relatively strong recovery and a certain degree of elasticity rather than flowing, ascribed to the internal multiple hydrogen bonding and electrostatic interactions that impart the material with high thermal stimulus tolerance. To quantitatively get the temperature-dependent exchange kinetics of the dynamic polymeric network, the apparent activation energy (Ea) and the master curves of the PTU2 network were evaluated and plotted by the time-temperature superposition (TTS) principle (Figs. 3c and d). The PTU2 showed a temperature-dependent linear characteristic with a notable slope transition at 80 ℃: The Ea below 80 ℃ was 184.7 kJ/mol, while Ea′ decreased to 120.9 kJ/mol above 80 ℃. Interpret Ea as the dynamic dissociation energy of disulfide bonds [46,47], which is relatively lower than the literature values due to the presence of protons within the polymer network that can act as catalysts, thereby reducing its activation energy [48]. The Ea′ indicates that the thermal activation of hydrogen bond networks and ionic clusters manifests at temperatures exceeding 80 ℃. The robustness of the hydrogen bond network enables its reconfiguration and migration at higher temperatures, while the combination of multiple interactions equips the PTU2 with a broad range of responsive capabilities (Fig. 3d and Fig. S17 in Supporting information).
Figure 3
Figure 3.
Mechanism analysis for the multiple energy dissipation clusters. (a) Representative tensile stress-strain curves of PTU1 (blue) and PTU2 (red) at 15 ℃ (solid line) and 25 ℃ (dotted line). (b) The temperature-dependent storage and loss moduli of PTU2 were recorded using a rheometer at a constant frequency of 1 Hz. (c) Linear fitting of temperature-dependent aT for PTU2 to the Arrhenius equation. (d) Time-temperature superposition (TTS) for the frequency sweep plot of G′ (storage modulus) and G′′ (loss modulus) for PTU2 at a reference temperature of 30 ℃. (e) Schematic diagram of multiple energy dissipation clusters. (f) Temperature-variable transition IR spectra of PTU2 upon heating from 24 ℃ to 80 ℃ (interval: 2 ℃). (g) 2DCOS synchronous and (h) asynchronous spectra generated from (f) In 2DCOS spectra, red colors represent positive intensities, while blue colors represent negative intensities.
To gain a deeper understanding of the stiffness and dynamics of hydrogen bonding and multiple interactions within the polymeric network for energy dissipation (Fig. 3e) [49–53], temperature variable transmission infrared spectra of PTU2 were recorded from 25 ℃ to 80 ℃ (Fig. 3f). The thermal sensitivity varies among different clusters. The carboxylic acid pairs (serve as primary clusters) undergo the most significant reduction in spectral intensities with negligible wavenumber shifts, while the decrease for carboxylate groups (ionic clusters, serve as secondary clusters) was the next most, and UPy motifs (microphase, serve as tertiary clusters) showed the least reduction in spectral intensities. Two-dimensional correlation spectra (2DCOS) display the thermal-responsive sequence of multiple clusters (Figs. 3g and h). Based on Noda’s judging rule [54], synchronous and asynchronous spectra are recorded synchronously [9,41]. The responsive sequence of multiple clusters to thermal treatment is as follows (→ denotes prior to or earlier than, Table S2 in Supporting information): 1684 → 1693 → 1705 → 1525 → 1749 → 1562 → 1579 → 1658 → 1635 cm-1, corresponding to v(COOH) (including 1684 → 1693 cm-1, oligomer or cluster, primary clusters) → v(COOH) (dimer, primary clusters) → v(COO−) (strong, secondary clusters) → v(COOH) (free, primary clusters) → v(COO−) (moderate, secondary clusters) → v(COO−) (weak, secondary clusters) → v(C=O) (UPy, weak, tertiary clusters) → v(C=O) (UPy, strong, tertiary clusters). If ignoring the changes related to the same species and quantity transformation, the sequence of clusters can be simplified as carboxylic acid pairs → ionic clusters → UPy motifs-induced microphase, consistent with expected sequences of multiple energy dissipation. The earliest response of carboxylic acid pairs to thermal stimuli suggests that the dynamics and flow of the polymer chains are dominated by primary clusters, as indicated by the Tg. However, the microphase domains within the networks exhibit a high tolerance to thermal heating, which is not easily affected by changes in chain mobility behavior, while simultaneously restricting the extensive movement of chains, ultimately leading to low strain and high strength. Overall, the formation of microphase domains induced by UPy motifs and the separation optimized by ionic clusters enables polymeric toughening of the polymeric material, expanding its application in industrial plastics and enhancing its energy dissipation against external mechanical forces and thermal heating.
The broad-range mechanical and thermal stimuli responsiveness and tolerance endow the polymeric material with promising structural reconfigurability and programmability. Based on rheological characterization and 2DCOS analysis, the primary activation process of the hydrogen bond networks and ionic clusters occurs above 80 ℃. Therefore, we selected 100 ℃ as the shape memory programming temperature and 60 ℃ as the recovery temperature. This temperature selection enables the effective maintenance of the stabilizing effects of both the microphase separation induced by hydrogen bond networks and the ionic clusters on the network structure while simultaneously preserving the dynamic recovery properties of disulfide bonds. As shown in Figs. 4a-f, the temporary shapes of PTU2 thin films could be recovered to their original shapes in different directions when immersed in a 60 ℃ water bath, showing rapid one-way shape memory under mild trigger conditions. Specifically, we fabricated several original shapes to illustrate the deformation processes in different directions, like one pair of axisymmetric directions in butterfly wings (Fig. 4a) and two pairs of axisymmetric but opposite directions in the wings and fuselage of the airplane (Fig. 4b), and four different and non-axisymmetric directions in the windmill (Fig. 4c). All three fabrication processes can be recovered to original shapes upon mild thermal treatment in seconds, exhibiting the ability to fully recover large-angle deformations (Figs. 4d-f and Video S1 in Supporting information). The use of a water bath enables rapid and complete heat transfer, whereas the shape recovery on a hot plate may take slightly longer due to uneven heating (Fig. S18 and Video S2 in Supporting information). Avoiding the use of high temperatures while enabling rapid and large-strain response and recovery allows the SPMs to have a wide range of application scenarios [55–58].
Figure 4
Figure 4.
Photos and characterization of the PTU2 shape memory behaviors. (a-c) Digital photos of PTU2 thin films with different initial shapes being thermally programmed into corresponding temporary shapes (butterfly, airplane, windmill, respectively). Scale bar: 1 cm. (d-f) Digital photos of the temporary shapes recovering to their original shapes in different directions when immersed in a 60 ℃ water bath. Scale bar: 1 cm. (g) Storage modulus, loss modulus, and tan delta curves of the PTU2 sheet obtained by DMA under a constant frequency of 1 Hz. (h) Repeated and reversible actuation of PTU2 programmed by uniaxial stretching.
To further explore the thermo-mechanical properties of PTU2, we performed dynamic mechanical analysis (DMA) to investigate its one-way shape memory behavior (Figs. 4g and h, and Fig. S19 in Supporting information). The tan delta curve displays a peak around 30 ℃, corresponding to the glass transition, which is higher than room temperature. A second peak, observed in the range of 60~65 ℃, is attributed to the thermal dissociation of micro-crystalline domains, consistent with the material’s heat deflection temperature (HDT) [56]. Despite the crossover point of the storage modulus and loss modulus curves being located at ~42 ℃, within the temperature range of 30–50 ℃, the storage modulus and loss modulus are close, suggesting that the material exhibits a substantial energy equilibrium and pronounced viscoelastic behavior over this temperature interval. One shape memory cycle demonstrates a 30% strain at 60 ℃, which is fixed upon cooling to get the temporary shape and then rapidly recovers its original shape when reheated to 60 ℃ with 96.7% shape fixity (Rf) and 83.2% shape recovery (Rr) (Fig. S20 in Supporting information). Notably, the shape recovery temperature is only 60 ℃, indicating that simple heating can enable the rapid recovery of the polymer shape, thereby endowing it with considerable practical applicability. Furthermore, in the multi-cycle repeated shape memory testing, PTU2 demonstrated good cyclic behavior. Due to the influence of the carboxylic acid pairs on the densely cross-linked network, full recovery after repeated cycling was challenging, resulting in a minor deficiency in the anti-fatigue characteristics. In brief, incorporating the electrostatic interactions of ionic clusters and their facilitation of UPy-induced microphase separation provides a novel approach for designing industrially applicable shape-memory dynamic SPMs.
In conclusion, the mutually reinforcing quadruple H-bonding UPy-induced microphase separation and iron(Ⅲ)-to-carboxylate ionic clusters present a new strategy and methodology. Leveraging dynamic interactions through the deep cross-integration of conventional plastics and supramolecular chemistry enables the creation of novel SPMs with superior mechanical qualities and outstanding energy dissipation capacity. The systematic optimization of polythioctic acid networks through the strategic manipulation of stoichiometric approaches and multiphase engineering, rather than conventional mechanical doping approaches, has enabled the achievement of engineering plastic-grade performance via intrinsic property modulation. This breakthrough represents a significant milestone in bridging the gap between laboratory research and industrial-scale manufacturing protocols. The strategic integration and directional functionalization of hydrogen bonding, coupled with the exploitation of low-strain electrostatic interactions, have synergistically enhanced the strength and toughness of the materials. Furthermore, this approach has also imparted polymeric materials with a broad range of tolerance to external mechanical and thermal stimuli. Exploring facile triggering and favorable performance in shape memory applications has catalyzed the industrial-scale implementation and utilization of SPMs. Through the precise modulation of the hard and soft segments within the polymeric network and the optimization of the network chain rigidity or crosslinking density, the prospect of a bright future for SPMs is envisioned.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the National Natural Science Foundation of China (No. 22375063), Science and Technology Commission of Shanghai Municipality (No. 23JC1401700), and the Fundamental Research Funds for the Central Universities. We thank Research Center of Analysis and Test of East China University of Science and Technology for the help on the characterization. Thanks to Professor Shengtong Sun (Donghua University) for important technical guidance and support in the analysis of the two-dimensional correlation spectra (2DCOS).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111946.
Figure 1
Synthesis and the mechanical properties of polymeric materials. (a) Cartoon diagram of the preparation of polydisulfide-based SPMs: PTU1 and PTU2. (b) The SAXS curve of monomer TU with the proposed interstack interaction between UPy motifs. (c) Comparison of Young’s modulus, toughness, and strain rates of PTU1 and PTU2. Error bars represent standard deviations. (d) The DSC spectrum of PTU1 and PTU2 with their Tg.
Figure 2
The toughening mechanism of PTU2 and the microstructural characterization. (a) Cartoon diagram of the mechanism of ionic cluster-optimized microphase separation. (b) The Raman spectra of monomer TA (grey curve), monomer TU (green curve), polymer PTU1 (blue curve, the same as follows), and polymer PTU2 (red curve, the same as follows). (c) The experimental SAXS curve of PTU1 and PTU2. (d) The first derivative of SAXS intensity. Inset: 2D image of PTU2. (e-g) SEM cross-section images of pristine, liquid-nitrogen-fractured, and tensile-fractured PTU2 samples, respectively.
Figure 3
Mechanism analysis for the multiple energy dissipation clusters. (a) Representative tensile stress-strain curves of PTU1 (blue) and PTU2 (red) at 15 ℃ (solid line) and 25 ℃ (dotted line). (b) The temperature-dependent storage and loss moduli of PTU2 were recorded using a rheometer at a constant frequency of 1 Hz. (c) Linear fitting of temperature-dependent aT for PTU2 to the Arrhenius equation. (d) Time-temperature superposition (TTS) for the frequency sweep plot of G′ (storage modulus) and G′′ (loss modulus) for PTU2 at a reference temperature of 30 ℃. (e) Schematic diagram of multiple energy dissipation clusters. (f) Temperature-variable transition IR spectra of PTU2 upon heating from 24 ℃ to 80 ℃ (interval: 2 ℃). (g) 2DCOS synchronous and (h) asynchronous spectra generated from (f) In 2DCOS spectra, red colors represent positive intensities, while blue colors represent negative intensities.
Figure 4
Photos and characterization of the PTU2 shape memory behaviors. (a-c) Digital photos of PTU2 thin films with different initial shapes being thermally programmed into corresponding temporary shapes (butterfly, airplane, windmill, respectively). Scale bar: 1 cm. (d-f) Digital photos of the temporary shapes recovering to their original shapes in different directions when immersed in a 60 ℃ water bath. Scale bar: 1 cm. (g) Storage modulus, loss modulus, and tan delta curves of the PTU2 sheet obtained by DMA under a constant frequency of 1 Hz. (h) Repeated and reversible actuation of PTU2 programmed by uniaxial stretching.

 DownLoad:
DownLoad:


 下载:
下载:





