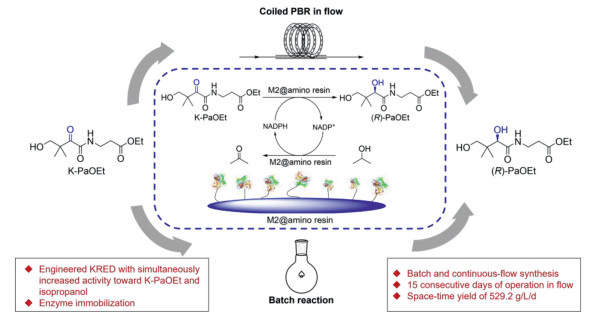
Figure 1.
Immobilized ketoreductase mutant-catalyzed stereoselective reduction of K-PaOEt to (R)-PaOEt in batch and continuous-flow.
Batch and continuous-flow asymmetric synthesis of D-pantothenic acid precursor enabled by immobilized ketoreductase mutant
Pan Hu , Xiaofan Wu , Yi An , Xianjing Zheng , Liang Gao , Yuan Tao , Yajiao Zhang , Zedu Huang , Fener Chen

Capitalizing on the revolutionary advancement of modern biotechnologies such as genome mining, gene synthesis, and protein engineering, enzyme catalysis has undergone rapid development since 1980′s, and established as the third pillar of catalysis, joined by transition-metal catalysis and organocatalysis [1-3]. Enzyme catalysis often features excellent catalytic efficiency, unparalleled chemo-, regio-, and stereoselectivity, as well as mild reaction conditions. Nowadays, enzymes can be utilized in different forms, for instance, isolated enzyme (either purified or cell-free extracts (CFE)), whole cell expressing the target enzyme, and immobilized enzyme. In particular, immobilized enzyme has gained increasing popularity especially in industry, because it possesses additional appealing properties over free enzyme, including superior stability under industrially relevant conditions (e.g., high temperature, extreme pH, and presence of organic solvent), simplified downstream process, as well as the enhanced recyclability and reusability [4-6]. Moreover, immobilized enzyme is well suited for the integration to flow chemistry, another green and enabling technology receiving much attention in recent years, thereby harnessing both the benefits of enzyme catalysis and that of flow chemistry, for example, enhanced mass and/or heat transfer, mitigation of product inhibition, and improved productivity [7-26].
With enormous development in past several decades, the NAD(P)H dependent ketoreductase (KRED)-catalyzed reduction of prochiral ketones has become a mature and often preferred means for the synthesis of valuable chiral secondary alcohols both in academic and industrial communities [27-32]. In this context, we recently evolved a versatile variant M3 (F97L/S173I/P243L) of the ketoreductase SSCR through the structure-guided semi-rational protein engineering, constructed a recombinant Escherichia coli strain co-expressing M3 and glucose dehydrogenase (GDH), and applied the whole cells of this strain to catalyze the reduction of ethyl 2′-ketopantothenate (K-PaOEt, Fig. 1) to ethyl (R)-pantothenate ((R)-PaOEt, Fig. 1) in a space-time yield (STY) of 520 g L-1 d-1 with an excellent enantioselectivity (> 99% ee) [33]. The subsequent hydrolysis of (R)-PaOEt gave a nearly quantitative conversion to D-pantothenic acid (D-PaA), an essential vitamin with the annual production capacity of its commercial form (calcium pantothenate) being over 23,000 tons [34,35]. Although effective, using whole cells for catalysis complicated the downstream product isolation process and it was also difficult to recycle and reuse those biocatalysts. In order to address these issues and strength the practical applicability of our synthetic method further, herein, we report the protein engineering enabled development of a SSCR mutant with simultaneously improved activity toward K-PaOEt and isopropanol, the immobilization of this variant to an amino resin, as well as the application of the obtained immobilized enzyme to the batch and continuous-flow synthesis of (R)-PaOEt (Fig. 1).
In our previous biocatalytic synthesis of (R)-PaOEt, we invoked a coupled enzyme approach, specifically using glucose dehydrogenase and glucose to regenerate the cofactor NADPH [33]. Under those conditions, a saturated aqueous solution of Na2CO3 had to be titrated to maintain the reaction pH, particularly under high-substrate loadings, because significant amount of gluconic acid resulted from the oxidation of glucose followed by hydrolysis, would dramatically lower the pH of the reaction system. Thus, other cofactor regeneration approaches were also pursued in order to avoid this kind of inconvenient operation. To this end, a very recent study from the Zhu and Wu group suggested the incorporation of the M242F mutation into SSCR could boost this enzyme’s activity toward the NADP+-dependent oxidation of isopropanol [36]. Prompted by this exciting finding, we surmised that it might be possible to develop an efficient SSCR variant capable of reducing K-PaOEt by using isopropanol as the sacrificial co-substrate to regenerate NADPH. To test this hypothesis, through using site-directed mutagenesis, the M242F mutation was firstly incorporated into six SSCR variants previously showing improved activity toward K-PaOEt relative to the wild-type (WT) SSCR, including single mutants F97L, F97M, and S173I, and double mutants F97L/S173I and F97M/S173I, as well as the triple mutant F97L/S173I/P243L (M3). In addition, single mutant M242F was also constructed using the same way, with WT as the parental enzyme. Next, the activity of thus prepared mutants was examined toward K-PaOEt and isopropanol, in the form of CFE. As depicted in Fig. 2A, for K-PaOEt, all the seven newly constructed variants except F97M/M242F exhibited higher activity (113%−179%) than WT, but lower activity than M3, the best-performing variant identified previously [33]. On the other hand, compared with WT, variants carrying the M242F mutation all displayed a significantly increased activity in the oxidation of isopropanol (2.2–9.5-fold), whereas M3 showed inferior activity relative to WT (Fig. 2A). Consistent with the literature observation [36], our results indicated the M242F mutation was crucial for acquiring high activity toward isopropanol.
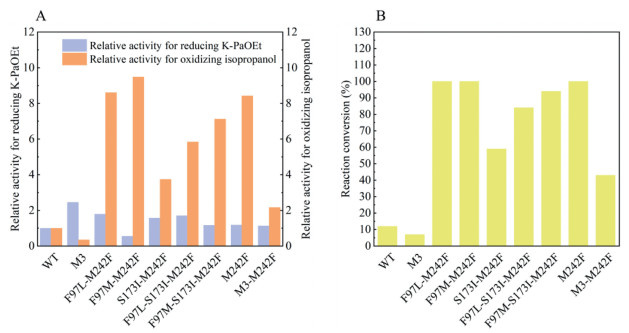
Moreover, we evaluated these mutant enzymes along with WT and M3 for reducing 50 mmol/L of K-PaOEt, by using isopropanol as the hydrogen donor to regenerate the required cofactor NADPH. As shown in Fig. 2B, to our delight, all the M242F mutation-containing variants gave obviously enhanced conversions over WT and M3 (43%−100% versus 12% and 7%). In particular, full conversion was achieved for mutants F97L/M242F, F97M/M242F, and M242F under the reaction conditions employed. It is worth pointing out that mutant F97M/M242F possessed only 55% and 22% activity of WT and M3 (Fig. 2A), respectively, toward K-PaOEt, nevertheless, outcompeting the latter enzymes for the reduction of K-PaOEt. All of these results underscored that sufficient activity of oxidizing isopropanol was one determining factor to achieve good conversion in this substrate-coupled cofactor regeneration system. This improved catalytic performance might be attributed to the possibly narrowed substrate entrance tunnel induced by the M242F mutation, thereby reducing the free release of isopropanol from the substrate-binding site, according to the elegant related study from the Zhu and Wu group [36]. To differentiate the performance of variants F97L/M242F, F97M/M242F, and M242F, we increased the substrate concentration to 100 mmol/L and meanwhile reduced the amount of enzyme. Under such more demanding conditions, F97L/M242F and F97M/M242F outperformed M242F, giving 36%, 38%, and 25% conversions, respectively. Finally, higher conversion was obtained with F97L/M242F than that with F97M/M242F (65% and 56%, respectively), when prolonging the reaction time. Importantly, the optical purities of (R)-PaOEt afforded in these two enzyme-catalyzed reduction reactions were determined both as > 99% ee. Taken together, the double mutant F97L/M242F, hereafter referred to as M2, was identified as the best enzyme variant for the current reaction system, and applied in the remaining studies. Furthermore, we determined the specific activity and kinetic parameters of the engineered enzyme M2 and WT toward K-PaOEt and isopropanol using purified proteins as well. According to the results presented in Table S1 (Supporting information), M2 exhibited 1.7-fold and 1.5-fold enhancement in specific activity and catalytic efficiency (kcat/KM), respectively, toward K-PaOEt compared with WT. On the other hand, a notable improvement on the oxidation of isopropanol was seen for M2 over WT, as evidenced by the greater than 10-fold improved specific activity (0.34 versus 0.03 U/mg) and catalytic efficiency (1.40 versus 0.12 L mol-1 s-1).
Six commercially available resins were screened as potential immobilization carriers, including an epoxy resin (LXTE-600), an amino resin (LXTE-700), an amino-epoxy resin (LXTE-706), a carboxyl resin (LXTE-800), an ion exchange resin (LXTE-902), and a macroporous resin (LXTE-1000). For the initial tests, we attempted the immobilization of M2 onto these resins using the glutaraldehyde (GLA) activation method, and evaluated the performance of these carriers using immobilization yield (IY) and activity recovery (AR). Specifically, the immobilization yield is defined as the ratio of the mass of enzyme immobilized over the mass of free enzyme used for the immobilization, while the activity recovery is defined as the ratio of the activity of the immobilized enzyme over the activity of free enzyme used for the immobilization. Notably, purified protein of M2 was utilized for immobilization, as it would be more conducive to quantification. As summarized in Table S2 (Supporting information), the use of the carboxyl resin and the ion exchange resin apparently failed to immobilize the enzyme, whereas immobilization yields ranging from 49.3% to 94.7% were achieved for the other four resins. Intriguingly, conversion of K-PaOEt to (R)-PaOEt was only observed when the immobilized enzyme M2@amino resin was employed as the biocatalyst, resulting in an activity recovery of 54.8%. Hence, the amino resin LXTE-700 was identified as the suitable carrier and used for the remaining process development.
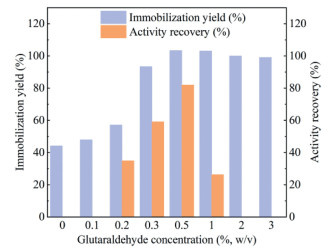
Firstly, the effect of glutaraldehyde concentration on the immobilization was exploited, by examining a concentration range between 0 and 3% (w/v). On the one hand, the immobilization yields rose with the increase of glutaraldehyde used from 0 to 0.5% (Fig. 3). Noteworthy, an immobilization yield of 44.2% was attained without the use of glutaraldehyde, which was attributed to the non-specific adsorption of proteins to the resins. When 0.5% or higher concentrations of glutaraldehyde were employed, immobilization yields of close to 100% were realized, implying apparently all M2 proteins (0.5 mg/mL, 1 mL) were bound to the resins. On the other hand, activity recovery was only detected when the concentration of glutaraldehyde was between 0.2% and 1%, with the peak value of 82.0% being achieved in the presence of 0.5% of glutaraldehyde (Fig. 3). Excessive use of glutaraldehyde (> 0.5%) gave rise to the decreased activity recovery or even complete deactivation of the enzymes, possibly due to the unwanted reactions occurred between some catalytically important Lys residues and glutaraldehyde. Altogether, 0.5% of glutaraldehyde was disclosed as the optimal concentration for the present enzyme immobilization.
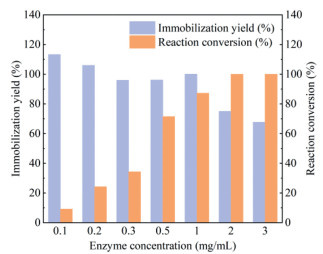
Next, a variety of enzyme concentrations ranging from 0.1 mg/mL to 3 mg/mL were utilized to investigate their effects on the immobilization yield and conversion rate. When the free enzyme concentrations for immobilization were between 0.1 mg/mL and 1 mg/mL, the apparent immobilization yields were determined and calculated as 96%−113%, suggesting that all proteins were immobilized onto the carrier (Fig. 4). Of note, in the case of using low enzyme concentration for immobilization (such as 0.1 mg/mL), as complete immobilization was achieved, no enzyme remained in the solution. Under such conditions, an apparent immobilization yield exceeding 100% sometimes would be encountered, which could be mainly attributed to experimental detection limit (please also see Supporting information for detailed discussion). In comparison, lower immobilization yields of 75% and 68%, respectively, were achieved with 2 mg/mL and 3 mg/mL of free enzyme, meaning the free enzyme was in excess relative to the resin and some of them were wasted. In terms of reaction conversion, poor conversion rates (9.1%−34.2%) were accomplished when using M2@amino resin prepared with no > 0.3 mg/mL of free enzyme as the biocatalyst (Fig. 4). We speculated that 0.3 mg/mL was below the threshold of free enzyme required to saturate the immobilization carrier, thereby the resulting immobilized enzyme was insufficient to achieve high conversion. In line with this assumption, the use of M2@amino resin constructed from 0.5 mg/mL and 1 mg/mL of free enzyme provided significantly improved conversions of 71.5% and 87.2%, respectively (Fig. 4). Taking both the activity of the immobilized enzyme (related to reaction conversion) and the cost of enzyme preparation into consideration, we chose 0.5 mg/mL of free enzyme for the subsequent studies.
The immobilized enzyme M2@amino resin, which was prepared under the optimal conditions identified above, was then characterized using various techniques. We initially wanted to use Fourier transform infrared (FTIR) to qualitatively verify the enzyme immobilization, by taking advantage of the characteristic absorption of amide I of proteins which is typically in the range from 1630 cm-1 to 1690 cm-1. Notably, an absorption around 1640 cm-1 was seen in all the three spectra acquired for the starting amino resin, amino resin-GLA, and M2@amino resin (Fig. S4A in Supporting information). Nonetheless, an additional weak absorption approximately at 1660 cm-1 appeared only in the spectrum of M2@amino resin (Fig. S4A), thus to some extent supporting the successful immobilization of M2. Another line of evidence was provided via the thermal gravimetric analysis (TGA). In the range of 200–540 ℃ at these TGA curves, a much more significant weight loss was observed for amino resin-GLA and M2@amino resin relative to the unmodified amino resin (82.5% and 84.9% versus 44.6%, Fig. S4B in Supporting information), which was ascribed to the calcination of the incorporated organic material including protein and glutaraldehyde. Furthermore, this slightly higher weight loss of M2@amino resin over that of amino resin-GLA, combined with the fact that M2@amino resin experienced an additional weight loss between 480 ℃ and 520 ℃, both supported that M2 was indeed immobilized onto the resin. In order to better understand the distribution of enzymes on the resin, a fluorescent immobilized M2 (FITC-M2@amino resin), assembled through labelling the protein with fluorescein isothiocyanate (FITC) followed by immobilization, was studied using confocal laser scanning microscopy (CLSM). The obtained image indicated the enzyme was distributed uniformly on the surface of the resin (Fig. S4C in Supporting information).
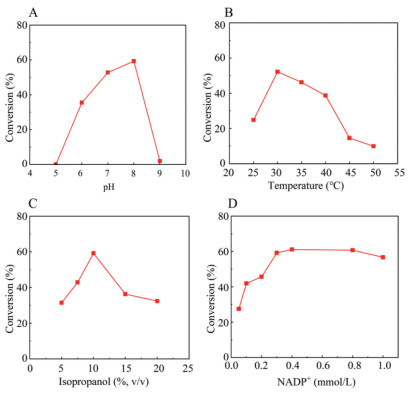
In the course of studying the pH dependence (5.0–9.0) and temperature effect (25–50 ℃) of M2@amino resin-catalyzed reduction of K-PaOEt, we found that pH 8.0 and 30 ℃ were the optimal reaction pH and temperature, respectively (Figs. 5A and B). Then, the effect of isopropanol loading was also investigated. As shown in Fig. 5C, highest conversion was obtained with the use of 10% (v/v) of isopropanol, while using higher or lower amounts of isopropanol both resulted in inferior conversions. Similarly, the influence of cofactor dosage was examined. A gradual increase of conversion (27.5%−59.2%) was observed when the loading of NADP+ was steadily enhanced from 0.05 mmol/L to 0.3 mmol/L, whereas no significant difference in the conversion rate was seen upon further increasing the amount of NADP+ even to 1 mmol/L (Fig. 5D). Considering both the reaction efficiency and economic aspect, a NADP+ concentration of 0.3 mmol/L was selected.
The storage stability of M2@amino resin at 4 ℃ was exploited. As depicted in Fig. S5 (Supporting information), no obvious change of activity was seen after 60 days of storage, as evidenced by the complete conversion of K-PaOEt. Furthermore, as high as 95.9% of conversion was still achieved, even after storage for 80 days. In contrast, the free enzyme experienced a clear drop of activity after 40 days of storage, and a moderate conversion of 67.4% was encountered when applying the free enzyme stored after 80 days as the biocatalyst. These results highlighted the superior long-term storage stability of M2@amino resin over free enzyme.
Next, we tested the recyclability and reusability of M2@amino resin in the consecutive batch reduction of K-PaOEt to (R)-PaOEt, with the immobilized enzymes being recovered after each cycle and reused in the next reaction under identical conditions. To our delight, full conversion was maintained after 12 cycles of reuse (Fig. S6 in Supporting information). The excellent recyclability and reusability of M2@amino resin were further exemplified by the conversion rates of 91.2%, 79.1%, and 65.3% obtained after 17, 24, and 30 cycles of reuse, respectively (Fig. S6). Under these reaction conditions, a space-time yield (STY) of 84 g L-1 d-1 was accomplished.
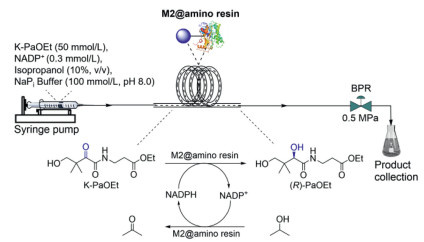
Finally, we wanted to test the feasibility of M2@amino resin-catalyzed reduction of K-PaOEt in a continuous-flow mode. According to the characteristics of the immobilized enzyme M2@amino resin and our previous experience with flow biocatalysis [16,37], we decided to utilize a coiled packed-bed reactor (PBR) for the current study. As presented in Fig. 6, a mixture of K-PaOEt (50 mmol/L), NADP+ (0.3 mmol/L), isopropanol (10%, v/v) in NaPi buffer was continuously fed into the reactor containing M2@amino resin using a syringe pump, and a back-pressure regulator (BPR) was included to render the solution move evenly through the system. The product (R)-PaOEt-containing out-stream was continuously collected, and if necessary, could be extracted with organic solvent for the purpose of monitoring the reaction progress using NMR. With this setup, three coiled PBRs, assembled by packing 0.9, 1.8, and 3.6 g of M2@amino resin into 1/8 tubes (1.6 * 3.175 mm), respectively, were evaluated and compared in terms of STY and conversions under various flow rates (0.01–0.05 mL/min).
On the one hand, as the flow rate decreased, the conversion of K-PaOEt to (R)-PaOEt was enhanced, regardless of which reactor used (Table 1). On the other hand, the residence time required to achieve complete conversion was found to be largely dependent on the amount of immobilized enzyme employed. For example, for the reactor filled with 0.9 g of M2@amino resin, full conversion of the substrate was only observed with the flow rate of 0.01 mL/min (entry 5, Table 1), corresponding to a residence time of 60 min. In comparison, these numbers were revealed to be 0.03 mL/min and 36.6 min (entry 8, Table 1), when 1.8 g of immobilized enzyme was applied. We further tried the reactor containing 3.6 g of M2@amino resin. The reaction went to completion again under the flow rate of 0.03 mL/min (corresponding to a residence time of 66.6 min, entry 13, Table 1), but to our disappointment, incomplete consumption of substrate was found when the flow rate was lifted to 0.04 mL/min (entry 12, Table 1). On account of conversion and residence time, the optimal conditions of this in-flow biocatalytic reaction were identified as follows: a PBR filled with 1.8 g of M2@amino resin, a flow rate of 0.03 mL/min (corresponding to a residence time of 36.6 min), 30 ℃, and 5.0 bar backpressure. Under such conditions, K-PaOEt was completely converted with a STY of 529.2 g L-1 d-1, which was 6.3 times higher than that with the batch synthesis.
 DownLoad:
CSV
DownLoad:
CSV
| Entry | Flow rate (mL/min) | Volume (mL) | tR (min) | Conversion (%) |
| 1 | 0.05 | 0.6 | 12 | 28 |
| 2 | 0.04 | 15 | 41 | |
| 3 | 0.03 | 20 | 56 | |
| 4 | 0.02 | 30 | 98 | |
| 5 | 0.01 | 60 | 100 | |
| 6 | 0.05 | 1.1 | 22 | 51 |
| 7 | 0.04 | 27.5 | 72 | |
| 8 | 0.03 | 36.6 | 100 | |
| 9 | 0.02 | 55 | 100 | |
| 10 | 0.01 | 110 | 100 | |
| 11 | 0.05 | 2 | 40 | 78 |
| 12 | 0.04 | 50 | 95 | |
| 13 | 0.03 | 66.6 | 100 | |
| 14 | 0.02 | 100 | 100 | |
| 15 | 0.01 | 200 | 100 |
The operational stability of our flow biocatalytic system was then examined under the optimal reaction conditions. Remarkably, the conversion rate remained 100% and only slightly declined to 96.6% after 15 and 17 consecutive days of operation, respectively. Meantime, a certain amount of effluent was collected during this operation. Upon extraction, drying and removing solvent, (R)-PaOEt of > 99% ee was furnished in 90% isolated yield with a chemical purity of > 95%.
To summarize, a ketoreductase mutant designated as M2, carrying two point mutations of F97L and M242F relative to the wild-type SSCR, was constructed by site-directed mutagenesis, exhibited simultaneously improved activity toward K-PaOEt and isopropanol, and could effectively catalyze the stereoselective reduction of K-PaOEt to (R)-PaOEt by using isopropanol as the sacrificial co-substrate to regenerate NADPH. Through screening six commercially available carriers, an amino resin LXTE-700 was identified as the best solid support for the immobilization of M2 via the glutaraldehyde activation method. Upon optimization of the immobilization process and reaction conditions, the fabricated immobilized enzyme M2@amino resin demonstrated excellent recyclability and reusability, maintaining full conversion of K-PaOEt to (R)-PaOEt, the direct precursor of D-pantothenic acid, after 12 cycles of reuse. Finally, this immobilized enzyme-catalyzed synthesis of optically pure (R)-PaOEt was implemented in continuous-flow, accomplishing a 6.3 times higher space-time yield than that with the batch synthesis (529.2 versus 84 g L-1 d-1). The 100% conversion rate achieved after 15 consecutive days of operation showcased the outstanding operational stability of our developed flow biocatalysis system.
The authors declare no competing financial interest.
Pan Hu: Writing – review & editing, Writing – original draft, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Xiaofan Wu: Writing – review & editing, Writing – original draft, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Yi An: Methodology, Investigation, Data curation. Xianjing Zheng: Methodology, Investigation, Data curation. Liang Gao: Methodology, Investigation, Data curation. Yuan Tao: Methodology, Investigation, Data curation. Yajiao Zhang: Writing – review & editing, Writing – original draft, Supervision, Project administration, Conceptualization. Zedu Huang: Writing – review & editing, Writing – original draft, Supervision, Resources, Project administration, Funding acquisition, Conceptualization. Fener Chen: Writing – review & editing, Writing – original draft, Supervision, Resources, Project administration, Conceptualization.
We thank the Science and Technology R & D Major Project of Jiangxi Province (No. 20244AFI92001), the National Natural Science Foundation of China (Nos. 22071033 and 21801047) for the financial supports.
Supplementary material associated with this article can be found, in the online version, at doi:
S. Wu, R. Snajdrova, J.C. Moore, K. Baldenius, U.T. Bornscheuer, Angew. Chem. Int. Ed. 60 (2021) 88–119. doi: 10.1002/anie.202006648
R. Buller, S. Lutz, R.J. Kazlauskas, et al., Science 382 (2023) eadh8615. doi: 10.1126/science.adh8615
S. Simić, E. Zukić, L. Schmermund, et al., Chem. Rev. 122 (2022) 1052–1126. doi: 10.1021/acs.chemrev.1c00574
J.M. Bolivar, J.M. Woodley, R. Fernandez-Lafuente, Chem. Soc. Rev. 51 (2022) 6251–6290. doi: 10.1039/d2cs00083k
R.A. Sheldon, S. van Pelt, Chem. Soc. Rev. 42 (2013) 6223–6235. doi: 10.1039/C3CS60075K
R.C. Rodrigues, Á. Berenguer-Murcia, D. Carballares, R. Morellon-Sterling, R. Fernandez-Lafuente, Biotechnol. Adv. 52 (2021) 107821. doi: 10.1016/j.biotechadv.2021.107821
J. Britton, S. Majumdar, G.A. Weiss, Chem. Soc. Rev. 47 (2018) 5891–5918. doi: 10.1039/c7cs00906b
Z. Tang, Y. Oku, T. Matsuda, Org. Process Res. Dev. 28 (2024) 1308–1326. doi: 10.1021/acs.oprd.3c00405
M.P. Thompson, I. Peñafiel, S.C. Cosgrove, N.J. Turner, Org. Process Res. Dev. 23 (2019) 9–18. doi: 10.1021/acs.oprd.8b00305
P. De Santis, L.E. Meyer, S. Kara, React. Chem. Eng. 5 (2020) 2155–2184. doi: 10.1039/d0re00335b
A. Naramittanakul, S. Buttranon, A. Petchsuk, P. Chaiyen, N. Weeranoppanant, React. Chem. Eng. 6 (2021) 1771–1790. doi: 10.1039/d1re00189b
A.I. Benítez-Mateos, F. Paradisi, J. Flow Chem. 14 (2024) 211–218. doi: 10.1007/s41981-023-00283-z
L.E. Meyer, M. Hobisch, S. Kara, Curr. Opin. Biotechnol. 78 (2022) 102835. doi: 10.1016/j.copbio.2022.102835
Y.J. Hu, J. Chen, Y.Q. Wang, et al., Chem. Eng. J. 437 (2022) 135400. doi: 10.1016/j.cej.2022.135400
P. Hu, X. Wu, Z. Huang, F. Chen, Chin. J. Pharm. 56 (2025) 421–434.
C. Hu, Z. Huang, M. Jiang, et al., ACS Sustainable Chem. Eng. 9 (2021) 8990–9000. doi: 10.1021/acssuschemeng.1c01419
D. Andrés-Sanz, A. Maiz-Iginitz, J.M. Bolivar, et al., Green Chem. 26 (2024) 4563–4573. doi: 10.1039/d4gc00369a
Q. Chen, Y. An, M. Feng, et al., Green Chem. 24 (2022) 9508–9518. doi: 10.1039/d2gc03082a
G. García-Marquina, J. Langer, M. Sánchez-Costa, et al., ACS Sustainable Chem. Eng. 10 (2022) 9899–9910. doi: 10.1021/acssuschemeng.2c02279
B. Shen, R. Ding, J. Dai, et al., Green Synth. Catal. 2 (2021) 367–373.
R. Greifenstein, T. Ballweg, T. Hashem, et al., Angew. Chem. Int. Ed. 61 (2022) e202117144. doi: 10.1002/anie.202117144
V. Williams, Y. Cui, J. Zhao, et al., Org. Process Res. Dev. 26 (2022) 1984–1995. doi: 10.1021/acs.oprd.1c00383
W. Zhang, S.F. Li, J.Q. Zhu, et al., Biochem. Eng. J. 197 (2023) 108964. doi: 10.1016/j.bej.2023.108964
P. Xie, J. Lan, J. Zhou, et al., Bioresour. Bioprocess. 11 (2024) 70. doi: 10.1186/s40643-024-00786-0
L. Chu, X. Zhang, J. Li, et al., Chin. Chem. Lett. 35 (2024) 108896. doi: 10.1016/j.cclet.2023.108896
X. Deng, M. Fan, M. Wu, et al., Chin. Chem. Lett. 35 (2024) 108684. doi: 10.1016/j.cclet.2023.108684
Z. Li, H. Yang, J. Liu, Z. Huang, F. Chen, Chem. Rec. 21 (2021) 1611–1630. doi: 10.1002/tcr.202100062
W. Zhang, Z.Q. Shao, Z.X. Wang, et al., Int. J. Biol. Macromol. 274 (2024) 133264. doi: 10.1016/j.ijbiomac.2024.133264
L. Qiao, Z. Luo, H. Chen, et al., Chem. Commun. 59 (2023) 7518–7533. doi: 10.1039/d3cc01474f
B. Su, L. Xu, X. Xu, et al., Green Synth. Catal. 1 (2020) 150–159.
X. Yue, Y. Li, D. Sang, et al., Chin. Chem. Lett. 34 (2023) 108178. doi: 10.1016/j.cclet.2023.108178
Z. Guo, Z. Wu, X. Wu, et al., Green Chem. 26 (2024) 7818–7824. doi: 10.1039/d4gc01478b
P. Hu, X. Wu, Y. Zhang, et al., Green Chem. 26 (2024) 2124–2134. doi: 10.1039/d3gc04344d
M. Eggersdorfer, D. Laudert, U. Létinois, et al., Angew. Chem. Int. Ed. 51 (2012) 12960–12990. doi: 10.1002/anie.201205886
P. Cheng, J. Wang, Y. Wu, et al., Enzyme Microb. Tech. 126 (2019) 77–85. doi: 10.1016/j.enzmictec.2019.04.001
H. Zhang, X. Chen, T. Lv, et al., ACS Catal. 13 (2023) 9960–9968. doi: 10.1021/acscatal.3c01569
Y. Zhang, M. Liu, Z. Yang, et al., Green Chem. 25 (2023) 3223–3235. doi: 10.1039/d2gc04894a

Figure 1 Immobilized ketoreductase mutant-catalyzed stereoselective reduction of K-PaOEt to (R)-PaOEt in batch and continuous-flow.

Figure 2 (A) Relative activity of the cell-free extracts of the wild-type (WT) SSCR and its variants. The activity of the WT was defined as 1.0. (B) WT- or its variant-catalyzed reduction of K-PaOEt to (R)-PaOEt. Reaction conditions (1 mL): K-PaOEt (50 mmol/L), NADP+ (1 mmol/L), isopropanol (10%, v/v), and 40 g/L cell-free extracts (CFE) (wet cell weight) of KREDs in NaPi buffer (100 mmol/L, pH 7.0). Reaction mixtures in the Eppendorf tubes were shaken in a temperature-controlled orbital shaker at 30 ℃ and 200 rpm for 3 h.

Figure 3 The immobilization yield and activity recovery of M2@amino resin prepared under different concentrations of glutaraldehyde (0–3%, w/v).

Figure 4 The immobilization yield and reaction conversion achieved with M2@amino resin prepared using different concentrations of free enzyme (0.1–3 mg/mL).

Figure 5 Optimization of reaction conditions on pH (A), temperature (B), concentration of isopropanol (C), and NADP+ dosage (D).

Figure 6 Schematic view of the continuous-flow synthesis of (R)-PaOEt catalyzed by M2@amino resin.
Table 1. Reaction optimization for M2@amino resin-catalyzed reduction of K-PaOEt to (R)-PaOEt in a continuous-flow mode.
| Entry | Flow rate (mL/min) | Volume (mL) | tR (min) | Conversion (%) |
| 1 | 0.05 | 0.6 | 12 | 28 |
| 2 | 0.04 | 15 | 41 | |
| 3 | 0.03 | 20 | 56 | |
| 4 | 0.02 | 30 | 98 | |
| 5 | 0.01 | 60 | 100 | |
| 6 | 0.05 | 1.1 | 22 | 51 |
| 7 | 0.04 | 27.5 | 72 | |
| 8 | 0.03 | 36.6 | 100 | |
| 9 | 0.02 | 55 | 100 | |
| 10 | 0.01 | 110 | 100 | |
| 11 | 0.05 | 2 | 40 | 78 |
| 12 | 0.04 | 50 | 95 | |
| 13 | 0.03 | 66.6 | 100 | |
| 14 | 0.02 | 100 | 100 | |
| 15 | 0.01 | 200 | 100 |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们