Laboratory of Theoretical and Computational Nanoscience, National Center for Nanoscience and Technology, Chinese Academy of Sciences, Beijing 100190, China
b.
University of Chinese Academy of Sciences, Beijing 100049, China
c.
MOE Key Laboratory of Macromolecular Synthesis and Functionalization, International Research Center for X Polymers, ZJU-YST Joint Research Center for Fundamental Science, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou 310027, China
d.
College of Chemistry, Key Laboratory of Theoretical & Computational Photochemistry of Ministry of Education, Beijing Normal University, Beijing 100875, China
e.
School of Physics and Astronomy, Beijing Normal University, Beijing 100875, China
f.
School of Materials Science and Engineering, Nanyang Technological University, Singapore 639798, Singapore
zhujia@nanoctr.cn (J. Zhu). 1 These authors contributed equally to this work.
Received Date:
14 June 2025 Accepted Date:
30 September 2025 Revised Date:
27 September 2025 Available Online:
15 February 2026
Abstract:
Charge-transfer complexes (CTCs) have emerged as promising n-type organic thermoelectric (TE) materials due to their inherent high electrical conductivity and tunable transport polarities. In this study, we performed a comprehensive first-principles investigation on the TE properties of nine CTCs comprised of 2,7-dialkyl[1]benzothieno[3,2-b][1]benzothiophenes (CnBTBT, n = 4, 8, 12) as donors and fluorinated derivatives of tetracyanoquinodimethane (FmTCNQ, m = 0, 2, 4) as acceptors, aiming to identify high-performance n-type organic TE materials and elucidate the underlying structure–property relationships. Our calculation results, based on the Boltzmann transport equation and deformation potential theory, reveal that the length of the alkyl side chains and the number of fluorine substitutions significantly impact their electronic structures and TE properties. Notably, the CnBTBT–FmTCNQ CTCs with shorter alkyl chains and more fluorine substitution demonstrate superior n-type characteristics, particularly C4BTBT–F4TCNQ, which achieves an excellent power factor of 671 µW cm-1 K-2 at an optimal charge carrier concentration. Our findings not only clarify the critical role of molecular engineering in CTC-based TE materials but also provide valuable guidance for developing high-efficiency organic TE materials with versatile practical applications.
Organic thermoelectric (OTE) materials, which enable the direct conversion between thermal and electrical energy via the Seebeck and Peltier effects, have emerged as ideal candidates for powering wearable electronics and Internet of Things devices [1–9]. They offer compelling advantages over inorganic counterparts, including superior mechanical flexibility for conformal applications [10], solution processability enabling scalable and cost-effective fabrication [5], and intrinsically low thermal conductivity (typically in the range of 0.1–1 W m-1 K-1 [4]) which alleviates the materials design challenge by permitting focused enhancement of the power factor (PF=S2σ, where S is the Seebeck coefficient and σ is the electrical conductivity) to maximize the dimensionless thermoelectric figure of merit (zT=S2σT/κ, where T is the absolute temperature and κ is the thermal conductivity). Despite these advantages, a critical bottleneck persists: while p-type OTE materials have made considerable progress, the development of stable high-performance n-type OTE materials remains challenging, thereby constraining the fabrication and practical application of organic thermoelectric devices [11].
Over the recent decades, organic charge-transfer complexes (CTCs) have garnered increasing attention due to their intriguing photophysical properties [12–15]. Recently, a growing number of investigations have focused on their TE properties of CTCs, driven by their unique advantages [16,17]. On the one hand, the distinctive donor–acceptor composition of CTCs allows high conductivity. For instance, the single crystal of TTF–TCNQ with 1:1 stoichiometry shows metallic behavior with a high conductivity of 200–600 S/cm [17]. Furthermore, the compound of (BEDT-TTF)2Cu(NCS)2 demonstrates superconducting behavior [17]. On the other hand, CTCs offer a promising pathway to achieve n-type TE transport through the modulation of donor and acceptor molecules, ameliorating the scarcity of n-type TE materials [18,19]. Moreover, they exhibit high stability owing to the charge-transfer interaction. Indeed, accumulating evidence has demonstrated that several CTCs have already exhibited promising n-type thermoelectric characteristics [20,21]. To realize this potential, structural modifications of the constituent molecules like side-chain engineering [22] and frontier orbital engineering [23] through functional group substitution can be employed to tune the electronic properties and optimize TE performance. However, the structure–property relationships governing such modification-based molecular engineering strategies remain poorly understood, limiting the rational design of high-performance n-type CTCs. Additionally, the preparation and TE performance characterization of CTC crystals pose significant experimental challenges due to their complex procedures. Consequently, theoretical studies play a crucial role in predicting the TE performance and elucidating structure–property relationships [24], offering valuable guidance for obtaining high-performance n-type CTCs in experiments.
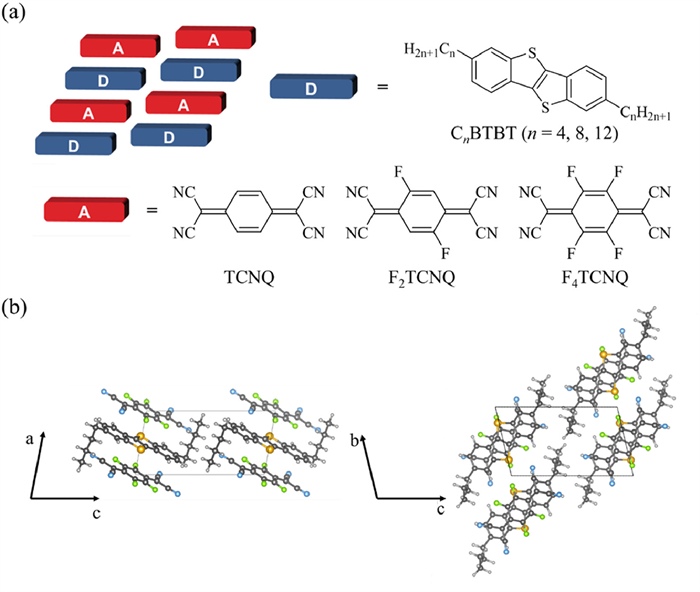
In light of these considerations, we focus on the CnBTBT–FmTCNQ [13] CTC system (denoted as CnFm, as shown in Fig. 1). This system warrants our investigations not only because it represents a systematically tunable series of mixed-stacking CTCs, but also owing to: (ⅰ) Its well-defined structural modifications (alkyl chain length n and fluorination degree m), along with their structural similarity, rendering them particularly amenable for structure–property relationships elucidation; (ⅱ) its high crystalline quality and ordered molecular packing, which facilitate reliable first-principles band structure calculations; and (ⅲ) its stability, promising charge carrier mobilities and, in certain configurations, n-type-dominant transport characteristics [13,25], indicating its potential as an efficient n-type TE material [22]. In this work, we present a comprehensive first-principles investigation of all nine CnFm complexes. Through systematic modulation of n and m, we compute key parameters including crystal and band structures, electronic coupling, and ultimately, the TE properties. Our results reveal that fluorination of the acceptor is a critical factor in enhancing n-type thermoelectric performance, with the C4BTBT–F4TCNQ complex emerging as a particularly promising candidate. This systematic study endeavors to not only provide valuable insights into the thermoelectric performance of the CnFm series but also to establish rational design strategies for the development of high-performance n-type organic thermoelectric materials.
Figure 1
Figure 1.
(a) Schematic diagram of the mix-stacked pattern of CnBTBT–FmTCNQ (denoted as CnFm, the same below) and the involved donors and acceptors in this work. (b) Crystal structure of C4F4 as an example of the CnFm CTC crystal series.
To elucidate the complex relation between molecular structure and thermoelectric properties of CnFm complexes, a theoretical framework based on density functional theory (DFT) and the band-like transport mechanism was utilized. Given that the high crystalline quality and ordered molecular packing observed in such mixed-stacking CTCs are conducive to electronic state delocalization, a band-like transport model was deemed appropriate [23]. In fact, experimental studies on high-purity organic single crystals have progressively accumulated evidence for band-like transport [17,26–28]. The observed thermally activated conduction is frequently attributed to the influence of traps from structural disorder and chemical impurities rather than to an intrinsic hopping mechanism [29,30]. Accordingly, the thermoelectric transport coefficients were evaluated using the Boltzmann transport equation within the constant electron–phonon coupling approximation (CEPCA) via deformation potential (DP) theory [31–33]. This approach has been established as an efficient and robust methodology for predicting the charge transport properties of organic small-molecule crystals [23,32,34–37], making it well-suited for mixed-stacking organic CTCs. It has been successfully implemented to elucidate CTC system structure–property relationships in our previous studies [38,39] as well as by other research groups [20,23]. Computational details are provided in Supporting information.
Our investigations commenced with a comprehensive analysis of the crystal structures of the nine CnFm complexes, all of which adopt a characteristic mixed-stacking arrangement where donor and acceptor molecules alternate along the a-axis (Fig. 1b), a motif might have higher Seebeck coefficient than separated-stacking motif for its semi-conductive behavior [40]. This alternating –D–A–D–A– sequence minimizes direct orbital overlap between D–D or A–A pairs along the stacking direction (calculated to be nearly 0 meV). Such a packing motif inherently favors a super-exchange mechanism for charge transport along this π–π stacking pathway, wherein an intervening molecule of the opposite type functions as a “bridge” to facilitate electronic coupling between adjacent identical molecules. This is consistent with previous studies on similar mixed-stacking CTCs [41,42]. A hallmark of this super-exchange-mediated transport is typically manifested in a quasi-symmetric electronic band structure near the Fermi level, which is indeed observed in our calculations (Fig. 2 and Fig. S3 in Supporting information). This crystal structure and dominant transport mechanism naturally lead to a pronounced anisotropy in the electronic band structure, characterized by significant band dispersion along the a-axis, indicative of potentially efficient charge transport, while the bands along the inter-stack directions (b- and c-axes) are comparatively flat (Fig. 2 and Fig. S3). Such anisotropic band structure is known to be beneficial for thermoelectric applications, where the flat bands in the inter-stack directions contribute to a sharp density of states near the Fermi level, while the dispersive bands along the stacking direction ensure adequate electrical conductivity [43]. Moreover, the calculated effective transfer integrals (Veff) for electron transport via super-exchange along the a-axis are consistently and significantly larger than those for hole transport across all nine CnFm compounds (Table 1). This disparity suggests that electrons are more effectively delocalized and transported through the super-exchange “bridges” than holes in these systems, partly due to the deep HOMO/HOMO–1 energy levels of FmTCNQ which are less effective as bridges for holes [44]. Furthermore, across all nine CnFm complexes, the electronic structure consistently exhibits features conducive to n-type transport, such as conduction bandwidths (CBW) that are generally larger than valence bandwidths (VBW), and electron effective masses (me*) along the a-axis that are typically smaller than hole effective masses (mh*) (Tables S2 and S3 in Supporting information). The results of the DP theory show that the differences in the strength of the electron–phonon coupling among CnFm are not significant (Table S5 in Supporting information), which indicates that the TE performance differences among them may mainly be reflected in the electronic structure. Based on these findings, our subsequent analysis of transport properties will focus on n-type transport along the a-axis.
Figure 2
Figure 2.
The calculated band structure and density of states of (a) C4F0, (b) C4F2, and (c) C4F4, respectively. The fermi level is set to 0 eV.
Table 1.
The calculated effective transfer integrals of electrons (Veffe) and holes (Veffh) for the CnFm CTC crystals (m = 0, 2 and 4; n = 4, 8 and 12).
An examination of the frontier molecular orbitals (FMOs) for the molecules provides basic insight into the expected roles of fluorine atoms and alkyl side chain. Our calculations consistently show that the FMOs are predominantly localized on their respective π-conjugated backbones. Significantly, these key orbitals exhibit virtually no electron density on the alkyl side chains of the CnBTBT molecules (representative FMOs are depicted in Fig. S5 in Supporting information). This primary observation suggests that alterations directly targeting the π-conjugated systems, such as the fluorination of the TCNQ acceptor, are likely to exert a more direct and substantial influence on electronic properties. Conversely, modifications to the alkyl side chains are anticipated to primarily impact the material’s characteristics through steric effects and modulation of the molecular packing [13]. This hypothesis regarding the distinct roles of π-core and side-chain modifications will guide the following discussion on the effects of fluorination and alkyl chain length.
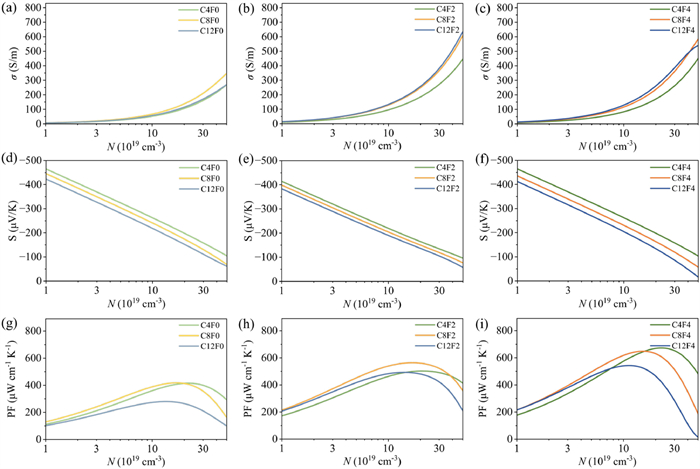
The incorporation of fluorine atoms into the TCNQ acceptor moiety is more effective than alkyl chain modification for enhancing the n-type thermoelectric performance of these CnFm complexes. This “electronic tuning” via fluorination profoundly impacts several key factors. Firstly, increasing “m” systematically enhances the electron affinity of the acceptor molecule, thereby promoting a more significant degree of charge transfer (DCT, Fig. S2 in Supporting information) and strengthening the intermolecular electronic coupling [45,46]. This is directly reflected in a general trend of decreasing π–π stacking distances (dπ–π, Fig. S1 in Supporting information) and, more importantly, an increase in the effective electronic transfer integrals (Table 1) along the a-axis that is particularly evident when comparing CnF0 with CnF2 and CnF4. Secondly, Boltzmann-transport calculations reveal a substantial rise in σ upon fluorination: The σ of CnF2 is typically twice as high as that of CnF0, whereas CnF2 and CnF4 are comparable (Figs. 3a–c). The mobility of C8Fm is consistent with the experimental results in trend, that is, C8F2 has the largest mobility, which is comparable to that of C8F4, while C8F0 is much lower [13]. Finally, a consistent and significant reduction in the bandgap is observed with increasing fluorine substitution across all alkyl chain lengths, with the CnF4 exhibiting the narrowest bandgaps (Fig. S1). Such narrow bandgaps are beneficial for achieving higher intrinsic carrier concentrations or requiring lower doping levels to reach optimal thermoelectric performance.
Figure 3
Figure 3.
Thermoelectric properties of CnF0 (a, d, g), CnF2 (b, e, h), and CnF4 (c, f, i) as a function of carrier concentration at 300 K. The plots show: (a–c) n-type electrical conductivity, (d–f) n-type Seebeck coefficient, and (g–i) power factor. All values are for the π–π stacking direction of the CTC crystals.
The specific m also subtly influences the density of states (DOS) near the CBM, which directly impacts the Seebeck coefficient according to the Mott relation [47,48]. Compared with CnF0 and CnF4, CnF2 shows a less steep rise DOS near CBM (Fig. 2 and Fig. S3) stemming from the lower molecular symmetry of the F2TCNQ acceptor [44], which may cause lower n-type Seebeck coefficient magnitudes (|Sn|). This reduced symmetry (Fig. S4 in Supporting information) enhanced electronic coupling of F2TCNQ along b-axis (Table S4 in Supporting information) and greater band dispersion, thereby affecting the overall DOS profile near the CBM. Consequently, the |Sn| of the CnF2 are consistently lower than others (Figs. 3d–f). Because fluorination raises the electrical conductivity to a similar extent in the CnF2 and CnF4 series, the smaller |Sn| for CnF2 leads to the overall PF trend: CnF0 < CnF2 < CnF4. For instance, within the C4Fm, the optimal PF systematically increases from approximately 414 µW cm-1 K-2 for C4F0 to 502 µW cm-1 K-2 for C4F2 and culminates at a remarkable 671 µW cm-1 K-2 for C4F4 (Fig. 3 and Table 2), underscoring fluorination as a pivotal design element for high-performance n-type CTCs.
Table 2
Table 2.
n-Type thermoelectric properties of CnFm, including optimal doping carrier concentration and power factor at optimal doping concentration.
Compared to direct electronic structure modulation by acceptor fluorination, the alkyl side chains on the CnBTBT donor primarily function as structural modulators. This is reflected in that the side chain length has little influence on electronic structure parameters such as the Veff (Table 1), DCT (Fig. S2), band gap (Fig. S1), bandwidth (Table S2) and m* (Table S1 in Supporting information). In contrast, their most notable effect is the expansion of the unit cell volume with increasing chain length (Table S1). For a fixed “m”, a larger unit cell at the same volumetric carrier concentration (N) means that each π–π pillars accommodates more charge carriers, which lowers the |S|. This interpretation might be corroborated by the nearly parallel and equally spaced |Sn|–N curves observed for both p-type (Figs. S6d–f in Supporting information) and n-type transport (Figs. 3d–f) when “n” is varied at a given “m”. The influence of alkyl chain length on electrical conductivity is more complex (Figs. 3a–c). It has been reported that slight changes in molecular packing of CnFm caused by steric hindrance of longer side chains, like the π–π stacking distance and stacking angle, may lead to higher mobility [13,17]. Thus, longer side chains show higher σ in our calculation. Notably, σ is observed to increase substantially from C4Fm to C8Fm, but shows little further improvement from C8Fm to C12Fm. With increasing side chain length, the rate of mobility enhancement tends to saturate or may even decline [49]. At a given carrier concentration, chain elongation therefore decreases the |S| but increases the σ. These opposing tendencies could cancel each other out in the PF. As a result, C4Fm and C8Fm complexes exhibiting comparable PF at the same m value, while C12Fm complexes consistently display the lowest PF. This non-monotonic behavior shows that although the alkyl chains are not primary electronic contributors, they can also modulate charge transport pathways indirectly by steric hindrance. However, despite these complex dependencies, the overall effect of alkyl chain length on PF is still lower than that of acceptor fluorination (Fig. 3).
In conclusion, this comprehensive first-principles investigation systematically elucidates the effects of the number of fluorine atoms and the length of alkyl side chain on the n-type thermoelectric properties of CnFm. The packing motif of CnFm CTCs indicate that charge transport along the π–π stacking direction is dominant and proceeds via the super-exchange mechanism. Fluorination of the TCNQ acceptor emerges as the more impactful strategy, serving primarily to tune the electronic properties. Increasing the number of “m” directly tunes the electronic structure by enhancing electron affinity, promoting charge transfer, strengthening super-exchange coupling, and reducing the bandgap, which results in a substantial boost in n-type electrical conductivity and, consequently, the PF. Although the alkyl side chains on the CnBTBT donor are not directly involved in the coupling, they play an indirect role by slightly regulating the crystal packing and changing the unit cell volume. Thus, the C4BTBT–F4TCNQ (C4F4) was identified as the most promising candidate. It exhibits the highest predicted n-type optimum power factor, further benefiting from a relatively small intrinsic band gap, which may help achieve high carrier concentrations with moderate doping. However, the flexible alkyl chains are likely to scatter heat-carrying phonons and thus depress the lattice thermal conductivity, a contribution to ZT that our approach does not capture. In addition, some electron-rich side chains may be involved in coupling, which may lead to higher conductivity and is worthy of further study. Our calculations, which model ideal single crystals, do not explicitly account for defect states or Coulomb scattering from dopant ions, as we focus on field-effect modulation which minimizes structural disruption. The real samples will inevitably feature dopants, crystal defects, dynamic disorder and grain boundaries that may reshape both mobility and optimal doping levels. Nevertheless, this study still deepens our understanding of the thermoelectric structure–property relationships of mixed-stacking CTCs like CnFm and provides rational design principles for the development of future high-performance n-type organic thermoelectric materials.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal reslationships that could have appeared to influence the work reported in this paper.
We thank Dr. Yishan Wang for helpful discussions on carrying out the calculations. We are grateful to Ms. Ping Lu for her valuable work on the initial investigations. We extend our appreciation to Prof. Zhigang Shuai for generously sharing his homemade codes, which significantly facilitated the transport calculations using BoltzTraP. Additionally, we thank Prof. Hua Geng and Prof. Yuanping Yi for their valuable assistance in performing the super-exchange calculations. This work was supported by the Strategic Priority Research Program of the Chinese Academy of Science (No. XDB0520000), the National Natural Science Foundation of China (Nos. 52273170 and 52394271), and the National Key R&D Program of China (No. 2022YFA1203200).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111933.
K. Oki, S. Horike, M. Yamaguchi, et al., Mol. Syst. Des. Eng. 5 (2020) 809–814. doi: 10.1039/d0me00017e
[49]
A. Mishra, S. Mahalik, A. Mishra, Chem. Commun. 61 (2025) 3649–3668. doi: 10.1039/d4cc06017b
Figure 1
(a) Schematic diagram of the mix-stacked pattern of CnBTBT–FmTCNQ (denoted as CnFm, the same below) and the involved donors and acceptors in this work. (b) Crystal structure of C4F4 as an example of the CnFm CTC crystal series.
Figure 3
Thermoelectric properties of CnF0 (a, d, g), CnF2 (b, e, h), and CnF4 (c, f, i) as a function of carrier concentration at 300 K. The plots show: (a–c) n-type electrical conductivity, (d–f) n-type Seebeck coefficient, and (g–i) power factor. All values are for the π–π stacking direction of the CTC crystals.
Table 1.
The calculated effective transfer integrals of electrons (Veffe) and holes (Veffh) for the CnFm CTC crystals (m = 0, 2 and 4; n = 4, 8 and 12).

 DownLoad:
DownLoad:


 下载:
下载:




