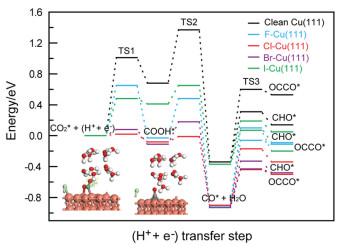
Figure 1.
The MEP diagram for CO2 electroreduction into CO via COOH intermediate and subsequent CHO and OCCO generations via CO further electroreduction.

Electrochemical CO2 conversion powered by sustainable electricity has emerged as a prominent research focus for transforming greenhouse gases into value-enhanced chemical compounds and energy carriers. Among these outputs, C2 products including ethylene and ethanol demonstrate particular industrial significance owing to their exceptional energy storage capacities and superior commercial viability compared to C1 products [1-4]. Cu electrocatalysts demonstrate distinctive capabilities in facilitating the electrochemical reduction of CO2 to diverse hydrocarbons and alcohols, exhibiting notable Faradaic efficiency and operational current density as documented in prior studies [5-8]. However, achieving high selectivity of C2 products is still a primary challenge because of complex CO2 electroreduction pathways and small opportunity of C—C coupling reactions [9-11]. The C—C coupling as a crucial step is sensitive to local environment of Cu electrocatalysts such as Cu/electrolyte interfaces [12-14], thereby leading to more attention.
Previous studies have indicated that halide anions exhibit adsorption capabilities on Cu electrodes, a phenomenon partially attributed to their comparatively small solvation energies [15], thereby influencing both catalytic activity and product selectivity in CO2 electroreduction processes [16-19]. Accordingly, many researchers have extensively investigated how halide anions modulate both catalytic activity and product selectivity, particularly for C2 products at the Cu/electrolyte interfaces. For instance, Varela et al. demonstrated that introducing halide anions (Cl−, Br−, I−) to Cu/electrolyte interfaces markedly enhances CO2 electroreduction activity and product selectivity towards CO and CH4 products due to specific adsorption of halide anions [20]. Parallel investigations by Gao et al. illustrated that adsorbed species like CO and OCCO intermediates can be stabilized by introducing halide anions into Cu/electrolyte interfaces during CO2 electroreduction and a higher Faradaic efficiency for C2 products can be realized on Cl−, Br− and I−-adsorbed Cu electrodes [21], mainly attributed to the interactions between adsorbed halide anions and intermediates [22]. Research findings by Huang et al. revealed that Cu electrode surface modified with I− facilitate enhanced electron transfer to CO intermediate, as evidenced by in situ surface-enhanced Raman spectroscopy analysis, thereby resulting in the enhanced C2 selectivity [23]. Wang et al. further demonstrated that Cu electrode modified with F− achieved exceptional Faradaic efficiency and elevated current density levels for productions of C2 products during CO2 electroreduction [24], in which a hydrogen-assisted C—C coupling reaction through two adsorbed CHO intermediate is suggested. Although the enhanced C2 selectivity can be achieved by halide anion incorporation in electrolytes, precisely determining the influence mechanism of halide anions towards C2 products is still challenging by various spectroscopy techniques due to complexity of Cu/electrolyte interfaces. Moreover, inconsistency of C—C coupling mechanism remains existence.
Density functional theory (DFT) serves as an essential computational approach for elucidating influence mechanism of halide anions on CO2 electroreduction activity and product selectivity, especially C—C coupling mechanism. Using DFT calculations, the study conducted by Janik et al. demonstrated that the halide anions can be adsorbed specifically on the Cu electrocatalysts and revealed that the adsorbed I− can weaken CO adsorption and favor subsequent generations of C—H bond during CO2 electroreduction [25,26]. The conclusion obtained by Janik et al. is opposite to experimental results by Huang et al., in which Cu electrode modified with I− can result in the stronger CO adsorption [23]. A C—C bond generation pathway through two adsorbed CHO dimerization was proposed on Cu electrode surface modified with F− by Wang et al. using DFT thermodynamical methodology without modeling of Cu/electrolyte interfaces [24]. However, the DFT calculations from Calle-Vallejo et al. showed that C—C bond can be generated by dimerization of the adsorbed CO intermediate towards OCCO dimer on unmodified Cu electrodes without solvation model [27], suggesting that the potential alteration of C—C coupling mechanism by halide anions. The theoretical report from Liu et al. indicated that C—C bond generation preferentially takes place on halide anions-modified Cu surface via the coupling reaction of the adsorbed CO and CHO intermediates by DFT thermodynamical calculations without modeling of interfaces [28]. Unfortunately, Cu/aqueous interfaces involving halide anions have been not efficiently simulated in previous theoretical studies. Furthermore, the influence mechanism of halide anions on selectivity towards C2 products remains elusive at present, which may result in diversity of C—C coupling mechanisms. Thus, Cu/electrolyte interfaces including halide anions such as F−, Cl−, Br− and I− anions by DFT calculations are simulated in this study (Fig. S1 in Supporting information), by which the origin of enhanced selectivity towards C2 products during CO2 electroreduction under halide anions is revealed and C—C coupling mechanism is suggested. Subsequently, an alternative CO2 electroreduction mechanism for C2 product generations at Cl−-adsorbed Cu(111)/H2O interface is presented. The present work will offer a theoretical framework for regulation of CO2 electroreduction reactions.
Production activity of CO as a crucial intermediate during CO2 electroreduction is firstly inspected with and without halide anions via COOH species [8,29,30]. As shown in Fig. 1, minimum energy pathway (MEP) analyses reveal substantially decreased barriers for CO production at F, Cl, Br and I atoms-adsorbed Cu(111)/H2O interphases compared to clean Cu(111)/H2O interface. Thus, we conclude that Cu surface modified with halide atoms demonstrate substantially improved electrocatalytic activity for CO production during CO2 electroreduction. This activity enhancement appears correlated with structural modifications in chemisorbed CO2 species, particularly the elongation of C═O bonds from approximately 1.17 Å to 1.30 Å accompanied by angular contraction of O═C═O configurations from nearly linear (180°) to approximately 122 degrees due to the adsorption of halide atoms at Cu(111)/H2O (Table S1 in Supporting information). The recent experimental studies from Cao et al. also showed that the enhanced CO2-to-CO conversion on Fe tetraphenylporphyrin-based electrocatalysts can be ascribed to activation of CO2 molecule and promotion of C—O bond cleavage [31-33]. The Löwdin charge distribution analyses demonstrate electron transfer at Cu(111)/H2O interface, where all halogen atoms exhibit notable electron enrichment as detailed in Table S2 (Supporting information), further suggesting generations of the adsorbed halide anions. Meanwhile, the notable electron transfer can be observed between CO2 and halide anions-adsorbed Cu(111)/H2O interfaces and thus anion radical ·CO2δ− with negative charge (ca. 1.0 e) can be formed (Table S3 in Supporting information). There are almost not electron transfers between CO2 molecule and clean Cu(111)/H2O interface (only ca. 0.04 e), explaining the enhanced CO generation activity because of adsorption of halide anions. Notably, we find that Cl−-adsorbed Cu(111)/H2O interface exhibits the highest CO production activity with minimal activation barrier of ca. 0.05 eV. In fact, the previous experimental work illustrated that the highest activity for CO2 electroreduction into CO on Cl− anions-modified Cu electrodes is also able to be achieved compared with other halide anions [20]. CO further electroreduction can determine C1 or C2 product selectivity. Productions of CHO species by CO surface hydrogenation and OCCO dimer by dimerization via Langmuir-Hinshelwood mechanism are only considered in the present work due to very difficult production of COH species based on our most recent theoretical study [34]. MEP analysis suggest that electrochemical CO reduction to CHO intermediate is more facile to appear than production of OCCO dimer at clean Cu(111)/H2O. The previous experimental observations reported by Koper et al. showed that CO2 electroreduction also appears towards C1 products via CHO intermediate on clean Cu(111) [35]. The computational investigations by Nørskov and Goddard et al. further corroborate the preferential generation of CHO intermediates over OCCO dimer on pure Cu(111) surface [8,36,37]. These agreements among our present study and early experimental and theoretical work may be able to validate the reasonability of our established theoretical models.
As shown in Fig. 1, the required barriers for the adsorbed CO hydrogenation to CHO intermediate at Cu(111)/H2O interfaces modified with F− and Cl− is elevated compared to unmodified Cu(111)/H2O interface, suggesting that CHO production may be unfavorable because of specific adsorption of F and Cl atoms. Meanwhile, we observe that the calculated barriers for CO dimerization to form OCCO dimer are significantly reduced at F− or Cl−-modified Cu(111)/H2O interphases compared to those required for CHO generation, indicating that OCCO production become more energetically favorable during subsequent electrochemical CO reduction because of the modification of F− and Cl−. Moreover, it is also found that F− and Cl−-modified Cu(111)/H2O interfaces exhibit reduced barriers for OCCO dimer generation compared to unmodified Cu(111)/H2O interface, illustrating that decoration of F− or Cl− is in favor of production of OCCO dimer. Although Cu(111)/H2O interfaces modified with Br− and I− show marginally smaller barriers (ca. 0.60 eV) for CO hydrogenation into CHO intermediate relative to unmodified interface, production barriers of OCCO dimer are notably decreased to ca. 0.45 eV, being smaller than production barriers of CHO intermediate. Notably, the present study reveals that Cu(111)/H2O interfaces modified with Br− and I− exhibit markedly reduced OCCO generation barriers (ca. 0.45 eV) compared to the unmodified interface (ca. 0.90 eV). Thus, it can be concluded that incorporating F−, Cl−, Br− and I− into Cu electrodes facilitates CO dimerization to OCCO dimer (Fig. 2), a critical step toward producing C2 products through subsequent reactions. By considering overall MEP pathways of CO2 electroreduction into OCCO dimer, it can be speculated that Cu(111)/H2O interface modified with Cl− has the optimum CO2 electroreduction activity towards OCCO dimer because of the lowest barrier (ca. 0.05 eV) and most negative reaction free energy. The obtained C—C coupling mechanism aligns closely with the early experimental observations by Gao et al. [21], namely, C—C coupling proceeds via CO dimerization of at Cu/electrolyte interfaces modified with halide anions. The further quantitative Löwdin charge distribution analyses of co-adsorbed CO at clean and halide anions-modified Cu(111)/H2O interfaces indicate that net electron gains of the co-adsorbed CO molecules reduce (Table S4 in Supporting information), implying decreased repulsive interactions between the co-adsorbed CO. Therefore, it can be concluded that the decreased repulsive interactions make CO dimerization into OCCO species be more facile to occur, which can result in enhanced C2 product selectivity, as abovementioned more favorable generations of dimer OCCO at Cu(111)/H2O interfaces modified with halide anions.
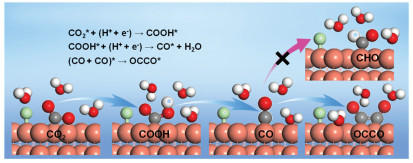
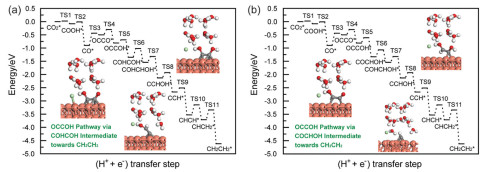
The adsorbed OCCO electroreduction processes are further studied at Cu(111)/H2O interface modified with Cl− in this work due to its optimal electrocatalytic activity towards OCCO production. OCCOH and OCCHO intermediates both could be parallelly produced beginning with OCCO dimer because the very small and surmountable barrier at room temperature (RT) of ca. 0.20 and 0.35 eV is required, respectively (Fig. S2a in Supporting information). OCCOH further electroreduction by surface hydrogenation or hydrogenative dissociation readily produces COHCOH, OCC, COCHOH and COHCHO species with remarkably small barrier of ca. 0.10, 0.05, 0.20 and 0.25 eV respectively required for each pathway (Fig. S2b in Supporting information). COCHOH and COHCHO can be also effortlessly generated by OCCHO surface hydrogenation because the very small barriers of ca. 0.30 and 0.05 eV are required at Cl−-modified Cu(111)/H2O interface. OCCHO further electroreduction also demonstrates facile generation of CHOCHO intermediate with an exceptionally small barrier of ca. 0.10 eV. However, OCCHO hydrogenation into COCH2O species is unfavorable with a substantially higher barrier of ca. 0.45 eV (Fig. S3a in Supporting information). Based on energetics, we can conclude that COCHOH and COHCHO both are readily produced by subsequent electrochemical reduction of OCCOH and OCCHO intermediates due to their respective small and surmountable barriers at RT (Fig. S4 in Supporting information). Subsequently, generations of CHOCHOH and COCH2OH species via hydrogenation of COCHOH are associated with the respective very small barriers of ca. 0.20 and 0.10 eV (Fig. S3b in Supporting information). The required barriers for COHCHOH and CHCO along with H2O molecule generations via respective COCHOH surface hydrogenation and hydrogenative dissociation both are also only ca. 0.25 eV. CHOCHO surface hydrogenation into CHOCH2O is energetically unfavorable because a high barrier is required (ca. 0.50 eV), whereas CHOCHOH generation is facile to take place during CHOCHO surface hydrogenation because of the small and surmountable barrier of ca. 0.35 eV at RT (Fig. S5a in Supporting information). COHCHO further electroreduction into CCHO with release of a H2O molecule, CHOCHOH, COHCH2O and COHCHOH are unfavorable in energetics since a substantial barrier of ca. 0.55, 0.60, 0.75 and 1.15 eV needs to be respectively overcome, especially COHCH2O generation leads to a endergonic reaction free energy profile (ca. 0.35 eV) (Fig. S5b in Supporting information). Thus, we can speculate that COHCHO may be only a spectator because of its facile production and difficult electroreduction. Further electrochemical reduction of COHCOH intermediate demonstrates smaller barrier (ca. 0.30 eV) for producing COHCHOH, whereas the alternative pathway producing CCOH requires overcoming a higher barrier of ca. 0.40 eV (Fig. S6a in Supporting information), suggesting more facile production of COHCHOH. A surmountable barrier of ca. 0.40 eV at RT suggests that CHCO species could be also formed by OCC hydrogenation (Fig. S6b in Supporting information). However, OCC hydrogenation to CCHO and CCOH species are unfavorable in energetics since a notably substantial barrier of ca. 0.85 and 1.15 eV needs to be overcome, respectively. By comprehensively considering MEP, we find that CHOCHOH and COHCHOH are mainly produced by COCHOH hydrogenation since the lowest barriers are required (Fig. S7 in Supporting information). CHCO is inclined to be formed by COCHOH hydrogenative dissociation (Fig. S8a in Supporting information).
CHOCHOH, COHCHOH, CHCO and COCH2OH further electroreduction pathways are considered. CHOCHOH surface hydrogenation to CHOHCHOH species demonstrates superior energetic feasibility, requiring a barrier of merely ca. 0.15 eV (Fig. S9a in Supporting information). The generations of CCHOH and CHCOH species together with a H2O molecule via COHCHOH hydrogenative dissociation are facile to appear with the very small barriers of merely ca. 0.20 and 0.10 eV, whereas COHCHOH hydrogenation to CHOHCHOH and COHCH2OH species are energetically unfavorable because the notably higher barrier of ca. 0.50 and 0.60 eV is respectively needed (Fig. S9b in Supporting information). Subsequent CHCO hydrogenation into CH2CO, CHCHO and CHCOH are energetically unfavorable with the respective unclimbable barriers of ca. 0.60, 0.85 and 0.50 at RT (Fig. S10a in Supporting information). CHOCH2OH species can be produced during COCH2OH surface hydrogenation with a remarkably small barrier of only ca. 0.05 eV, CH2CO generation with the release of a H2O molecule is energetically unfavorable via COCH2OH hydrogenative dissociation, requiring overcoming a larger barrier of ca. 0.95 eV. Although COHCH2OH production is kinetically favorable with a small barrier of only ca. 0.20 eV via COCH2OH surface hydrogenation, it is thermodynamically unfavorable due to an endergonic reaction profile characterized by a positive reaction free energy of ca. 0.15 eV (Fig. S10b in Supporting information). Based on MEP analyses, we can conclude that CHCOH is mainly produced by COHCHOH hydrogenative dissociation (Fig. S8b in Supporting information). CHOCH2OH are predominantly produced by COCH2OH hydrogenation (Fig. S11a in Supporting information). There is a notable propensity for CHOCHOH to undergo hydrogenation to CHOHCHOH (Fig. S11b in Supporting information).
Subsequent to further electroreduction of CHOHCHOH, CHOCH2OH, CCHOH and CHCOH species, hydrogenative dissociation or surface hydrogenation of CHOHCHOH species into CHCHOH and CHOHCH2OH are energetically unfavorable, which stems from requirement to overcome the respective considerably high barrier of ca. 0.70 and 2.30 eV, strongly suggesting that CHOHCHOH species likely remain a non-reactive bystander (Fig. S12a in Supporting information). CH2CHO can be produced by CHOCH2OH hydrogenative dissociation via a non-activated process, whereas generations of CH2OCH2OH and CHOHCH2OH species via CHOCH2OH surface hydrogenation are energetically less favorable because the respective higher barrier of ca. 0.50 and 1.10 eV is required (Fig. S12b in Supporting information). Production of CCH is more facile to take place through subsequent CCHOH hydrogenative dissociation with a substantially smaller barrier of only ca. 0.20 eV, whereas CCHOH surface hydrogenation to CCH2OH and CHCHOH species require overcoming respectively a higher barrier of ca. 0.60 and 0.50 eV overcome for (Fig. S13a in Supporting information). CHCOH hydrogenation into CHCHOH species is more facile to appear since the surmountable barriers of ca. 0.30 eV at RT is required (Fig. S13b in Supporting information). Although the barrier of ca. 0.35 eV for hydrogenative dissociation of CHCOH into CCH is also surmountable at RT, the reaction free energy is positive (ca. 0.10 eV), rendering this pathway endergonic. CHCOH surface hydrogenation into CH2COH is energetically unfavorable with a notably higher barrier of ca. 0.75 eV. By comparing the energetics of each elementary reaction step, we can conclude that CHCHOH is predominantly produced by CHCOH hydrogenation (Fig. S14a in Supporting information). CCH, CHCHOH and CH2CHO further electroreduction are analyzed, surface hydrogenation of CCH to CHCH species is energetically more favorable with a significantly smaller barrier of ca. 0.15 eV (Fig. S15a in Supporting information). CHCHOH hydrogenation or hydrogenative dissociation into CH2CHOH, CHCH2OH and CHCH are kinetically unfavorable with the respective unclimbable barriers of ca. 0.50, 0.60 and 0.65 eV at RT (Fig. S15b in Supporting information). CH2CHO surface hydrogenation into CH3CHO species is facile to appear due to the negligible barrier of ca. 0.15 eV. Conversely, the alternative pathway producing CH2CH2O through CH2CHO surface hydrogenation proves energetically prohibitive, requiring a substantial barrier of ca. 0.80 eV (Fig. S16a in Supporting information). Although CH2CHOH generation barrier is surmountable at RT (ca. 0.35 eV), the reaction free energy is marginally positive (ca. 0.05 eV), being thermodynamically unfavorable. Based on the energetics, we conclude that CHCH generation is notably more favorable via CCH hydrogenation (Fig. S14b in Supporting information). Subsequent CH3CHO and CHCH further electroreduction pathways are scrutinized. Surface hydrogenation CH3CHO into CH3CH2O species is more energetically favorable with a readily surmountable barrier of ca. 0.25 eV at RT (Fig. S16b in Supporting information). CHCH2intermediate readily formed through CHCH surface hydrogenation, requiring a climbable barrier at RT (ca. 0.35 eV). Subsequent CHCH2 and CH3CH2O hydrogenation could potentially yield final C2 products like CH3CH2OH and CH2CH2. Notably, CH3CH2OH production via CH3CH2O hydrogenation faces significant kinetic limitations, requiring a barrier of ca. 0.50 eV that is unclimbable at RT. In contrast, the final CH2CH2production via CHCH2 hydrogenation demonstrates favorable energetics with a climbable barrier of ca. 0.35 eV at RT. Thus, we conclude that Cl−-modified Cu(111)/H2O interface can selectively promote production of C2H4 product.
As described above, Cu(111)/H2O interface modified with Cl− has the optimum CO2 electroreduction activity towards OCCO dimer. Moreover, C—C coupling proceeds via dimerization of two adsorbed CO at Cu/electrolyte interfaces modified with halide anions by Langmuir-Hinshelwood mechanism, which can lead to the final C2H4 product via serial elementary reaction steps. Our present obtained C—C coupling mechanism is well consistent with early investigations from computations and experiments [38-40], which can confirm rationality of our present established theoretical models. Considering parallel generations of OCCOH and OCCHO species starting from OCCO dimer, two pathways are proposed at Cu(111)/H2O interface modified with Cl−, which is respectively defined as OCCOH and OCCHO pathways. On the basis of the above comprehensive analyses of MEP, Cl−-modified Cu(111)/H2O interface can selectively reduce CO2 into C2H4 product. Thus, the optimum production pathways towards C2H4 product can be delineated in Figs. 3 and 4a, in which various intermediates and geometry configurations of some key intermediates are given. In the overall energy diagrams of C2H4 production, the calculated free energy of ca. −4.65 eV for this reaction suggests that C2H4 production through CO dimerization appears readily because of its highly exothermic nature at Cu(111)/H2O interface modified with Cl−. The dimerization of CO into OCCO dimer is identified as rate-determining step because of the highest barrier (ca. 0.50 eV). This finding aligns with the recent investigations conducted by Kastlunger et al., in which CO dimerization to OCCO is also known as rate-limiting step of productions of C2 products during CO2 electroreduction on Cu electrodes based on the constant-potential DFT kinetics and experiments [41]. Although OCCO generation barrier is insurmountable at RT at Cu(111)/H2O interface modified with Cl−, the barrier of only ca. 0.05 eV is required for overall conversion process of CO2 to OCCO dimer, being notably smaller than the value of unmodified Cu(111) surface (ca. 1.38 eV). Our present work on selective promotion of Cl− anion on CO2 electroreduction towards C2H4 product will be able to offer a theoretical guideline for how to selectively regulate reaction pathways and products.
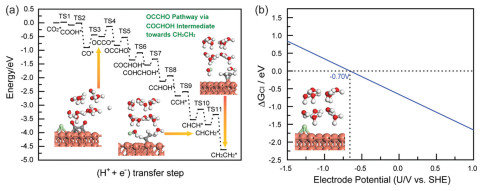
As abovementioned, Cl−-adsorbed Cu(111)/H2O interface significantly improves the selectivity of CO2 electroreduction for C2H4 production. Nevertheless, origin of enhanced CO2 electroreduction product selectivity is still elusive. Consequently, the adsorption free energies of Cl atom varied with electrode potentials are investigated in order to examine whether the adsorbed Cl atom can affect CO2 electroreduction selectivity. The computational details are elaborated in Supporting information. The corresponding relationship between the adsorption free energy and electrode potential is given in Fig. 4b. It can be observed that the calculated equilibrium potentials USHE is ca. −0.70 V (vs. SHE) under standard conditions when the adsorption free energy of Cl atom ΔGads = 0, being in well agreeable with thermodynamically calculated potential of CO2 electroreduction into C2H4 product (ca. −0.76 V vs. SHE) under standard conditions [42-44]. Namely, the adsorption of Cl atom can drive the electrode potentials into potential range of CO2 electroreduction into C2H4 product. Thereby, it can be concluded that the specifically adsorbed Cl− anion enhances CO2 electroreduction selectivity towards C2H4 product. The electrode potential of ca. −0.70 V (vs. SHE) is converted to the RHE scale to compare with experimental values using URHE = USHE + 0.059 pH at pH 6.8 on the Cu electrodes, ca. −0.30 V, namely, CO2 electroreduction proceeds towards C2H4 product via CO dimerization into OCCO dimer at ca. −0.30 V (vs. RHE). The recent reported experiment conducted by Wang et al. employing the operando attenuated total reflectance surface improved infrared absorption spectroscopy also showed that OCCO is a crucial reaction intermediate for C2H4 generation on Cu-based electrocatalyst surfaces and is produced by two adsorbed CO species at −0.40 V (vs. RHE) [45]. The excellent consistencies between our theoretical studies and experimental results confirm both the validity of our employed theoretical modeling and accuracy of calculated results.
Based on the calculated barriers of elementary reaction steps, the onset potential (Uonset vs. RHE) of each elementary reaction step is calculated at present by employing the computational models from Nørskov and Asthagiri et al. in order to further identify the optimum pathways of CO2 electroreduction towards C2H4 product and pinpoint potential-limiting step [8,38,46,47]. The determination of onset potential is on the basis of our present DFT calculated thermodynamical reaction free energies and kinetic barriers. In general, the more positive onset potential means more facile reaction and faster reaction rate. The computational details are given in Supporting information. The determined equilibrium potentials (U0 vs. RHE) and calculated onset potentials (Uonset vs. RHE) of each elementary reaction step including in OCCOH and OCCHO pathways for C2H4 production during electrochemical CO2 reduction at Cl−-adsorbed Cu(111)/H2O interface are summarized in Table S5 (Supporting information). We observe that the calculated onset potentials of each elementary reaction step are all positive, suggesting that electroreduction pathways of CO2 towards C2H4 product are facile to appear with fast kinetics. However, it is also noted that the onset potential for production of OCCOH intermediate (ca. 1.00 V) is more positive than that of OCCHO generation (ca. 0.45 V), indicating that C2H4 product may be mainly produced by OCCOH pathway. The electrochemical CHCH reduction to CHCH2 species could be the potential-limiting step in both OCCOH and OCCHO pathways because of the least positive onset potential of ca. 0.40 V. As demonstrated above, the chemically adsorbed CO2 molecules at Cu(111)/H2O interfaces modified with halide anions can lead to the notably decreased CO generation barriers compared to unmodified Cu(111)/H2O interface, therefore enhancing catalytic activity in CO₂ electroreduction processes targeting CO. The enhanced production of CO activity could be also ascribed to more positive onset potentials at Cu(111)/H2O interface modified with halide anions. The onset potential at Cl−-adsorbed Cu(111)/H2O interface is calculated as ca. 1.05 V through comprehensive analysis of CO production energy pathway, whereas the onset potential is determined to be only a negative value at unmodified Cu(111)/H2O interface (ca. −1.50 V), further explaining enhanced CO production activity. In the mean time, as a key step of CO2 selective electroreduction into C2H5OH product at Cl−-adsorbed Cu(111)/H2O interface, the onset potential of COCH2OH electroreduction to COHCH2OH species is determined to be ca. −0.05 V, indicating that generation of COHCH2OH species is unfavorable although the small barrier of merely ca. 0.20 eV is required. Thus, calculations of onset potential also indicate that only C2H4 product can be selectively produced for CO2 electroreduction due to modification of Cl− anion.
To sum up, the present computational findings demonstrate that the adsorption of halide anions enhance CO generation activity and are beneficial to CO dimerization to OCCO dimer. Significantly, Cl−-adsorbed Cu(111)/H2O interface has the optimum activity and selectivity for generation of OCCO dimer during electrochemical CO2 reduction. Thus, an CO2 electroreduction mechanism towards C2 products that defined as OCCOH and OCCHO pathways is proposed at Cl−-adsorbed Cu(111)/H2O interface. Notably, our current investigations reveal that adsorption of Cl−can selectively reduce CO2 into C2H4 product. The studies on onset potentials and function relationship between adsorption free energy of Cl atom and electrode potential further explain why the Cl−-adsorbed Cu(111)/H2O interface can significantly improve electrochemical CO2 reduction selectivity towards C2H4 product. The excellent conformances between our current computational studies and experimental results confirm the validity of theoretical modeling utilized in this work and accuracy of calculated results.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Lihui Ou: Writing – original draft, Methodology, Formal analysis, Data curation, Conceptualization. Zhancheng Liu: Visualization, Software, Resources. Dai-Huo Liu: Writing – review & editing, Software, Formal analysis. Zhi Zhang: Resources, Investigation, Data curation.
This work was supported by the Natural Science Foundation of Hunan Province (No. 2025JJ50059), Key Program of Hunan University of Arts and Science (No. 23ZZ03), Aid Program for Science and Technology Innovative Research Team in Higher Educational Institutions of Hunan Province and National Natural Science Foundation of China (No. 21303048).
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111920.
T. Yan, X. Chen, L. Kumari, et al., Chem. Rev. 123 (2023) 10530–10583. doi: 10.1021/acs.chemrev.2c00514
Y. Xie, P. Ou, X. Wang, et al., Nat. Catal. 5 (2022) 564–570. doi: 10.1038/s41929-022-00788-1
Y.Y. Birdja, E. Perez-Gallent, M.C. Figueiredo, et al., Nat. Energy 4 (2019) 732–745. doi: 10.1038/s41560-019-0450-y
J.E. Huang, F. Li, A. Ozden, et al., Science 372 (2021) 1074–1078. doi: 10.1126/science.abg6582
Q. Chang, J.H. Lee, Y. Liu, et al., JACS Au 2 (2022) 214–222. doi: 10.1021/jacsau.1c00487
S. Nitopi, E. Bertheussen, S.B. Scott, et al., Chem. Rev. 119 (2019) 7610–7672. doi: 10.1021/acs.chemrev.8b00705
A. Bagger, L. Arnarson, M.H. Hansen, et al., J. Am. Chem. Soc. 141 (2019) 1506–1514. doi: 10.1021/jacs.8b08839
H. Guo, Y. Wang, H. Liu, et al., Carbon Neutral. 3 (2024) 471–487. doi: 10.1002/cnl2.122
H. Li, Y. Jiang, X. Li, et al., J. Am. Chem. Soc. 145 (2023) 14335–14344. doi: 10.1021/jacs.3c03022
M. Zheng, P. Wang, X. Zhi, et al., J. Am. Chem. Soc. 144 (2022) 14936–14944. doi: 10.1021/jacs.2c06820
W. Ge, Y. Chen, Y. Fan, et al., J. Am. Chem. Soc. 144 (2022) 6613–6622. doi: 10.1021/jacs.2c02486
E. Sargeant, P. Rodriguez, Electrochem. Sci. Adv. 3 (2023) e2100178. doi: 10.1002/elsa.202100178
W.C. Ma, S.J. Xie, T.T. Liu, et al., Nat. Catal. 3 (2020) 478–487. doi: 10.1038/s41929-020-0450-0
S.J. Shin, H. Choi, S. Ringe, et al., Nat. Commun. 13 (2022) 5482. doi: 10.1038/s41467-022-33199-8
D.V. Tripkovic, D. Strmcnik, D. van der Vliet, et al., Faraday Discuss. 140 (2008) 25–40.
K. Ogura, J.R. Ferrell, A.V. Cugini, et al., Electrochim. Acta 56 (2010) 381–386. doi: 10.1016/j.electacta.2010.08.065
H. Yano, T. Tanaka, M. Nakayama, et al., J. Electroanal. Chem. 565 (2004) 287–293. doi: 10.1016/j.jelechem.2003.10.021
K. Ogura, M. Salazar-Villalpando, JOM 63 (2011) 35–38. doi: 10.1007/s11837-011-0009-2
N. Vasiljevic, M. Wood, P.J. Heard, et al., J. Electrochem. Soc. 157 (2010) D193–D198. doi: 10.1149/1.3298890
A.S. Varela, W. Ju, T. Reier, P. Strasser, ACS Catal. 6 (2016) 2136–2144. doi: 10.1021/acscatal.5b02550
D.F. Gao, I.T. McCrum, S. Deo, et al., ACS Catal. 8 (2018) 10012–10020. doi: 10.1021/acscatal.8b02587
D.F. Gao, I. Sinev, F. Scholten, et al., Angew. Chem., Int. Ed. 58 (2019) 17047–17053. doi: 10.1002/anie.201910155
Y. Huang, C.W. Ong, B.S. Yeo, ChemSusChem 11 (2018) 3299–3306. doi: 10.1002/cssc.201801078
W.C. Ma, S.J. Xie, T.T. Liu, et al., Nat. Catal. 3 (2020) 478–487. doi: 10.1038/s41929-020-0450-0
I.T. McCrum, S.A. Akhade, M.J. Janik, Electrochim. Acta 173 (2015) 302–309. doi: 10.1016/j.electacta.2015.05.036
S.A. Akhade, I.T. McCrum, M.J. Janik, J. Electrochem. Soc. 163 (2016) F477–F484. doi: 10.1149/2.0581606jes
F. Calle-Vallejo, M.T.M. Koper, Angew. Chem. Int. Ed. 52 (2013) 7282–7285. doi: 10.1002/anie.201301470
X.F. Ma, L. Xing, X.Q. Yao, et al., ChemPhysChem 24 (2023) e202200502. doi: 10.1002/cphc.202200502
Y. Guo, Y. Gao, B. Guo, et al., Carbon Neutral. 3 (2024) 4–31. doi: 10.1007/s43979-023-00077-1
X. Tu, X. Liu, Y. Zhang, J. Zhu, H. Jiang, Green Carbon 2 (2024) 131–148. doi: 10.1016/j.greenca.2024.03.006
Z.Y. Yin, M.C. Zhang, Y.C. Long, et al., Angew. Chem. Int. Ed. 64 (2025) e202500154. doi: 10.1002/anie.202500154
K. Guo, X.L. Li, H.T. Lei, et al., Angew. Chem. Int. Ed. 61 (2022) e202209602. doi: 10.1002/anie.202209602
H.Y. He, Z.Y. Qiu, Z.Y. Yin, et al., Chem. Commun. 60 (2024) 5916–5919. doi: 10.1039/d4cc01630k
L.H. Ou, W.L. You, J.L. Jin, Y.D. Chen, Phys. Chem. Chem. Phys. 25 (2023) 23977–23987. doi: 10.1039/d3cp01900d
K.J.P. Schouten, Y. Kwon, C.J.M. van der Ham, Z. Qin, M.T.M. Koper, Chem. Sci. 2 (2011) 1902–1909. doi: 10.1039/c1sc00277e
W.J. Durand, A.A. Peterson, F. Studt, F. Abild-Pederson, J.K. Nørskov, Surf. Sci. 605 (2011) 1354–1359. doi: 10.1016/j.susc.2011.04.028
H. Xiao, T. Cheng, W.A. Goddard Ⅲ, J. Am. Chem. Soc. 138 (2016) 483–486. doi: 10.1021/jacs.5b11390
K. Jiang, R.B. Sandberg, A.J. Akey, et al., Nat. Catal. 1 (2018) 111–119. doi: 10.1038/s41929-017-0009-x
T.T. Zhuang, Z.Q. Liang, A. Seifitokaldani, et al., Nat. Catal. 1 (2018) 421–428. doi: 10.1038/s41929-018-0084-7
X. Miao, T.W. He, G. Kour, et al., Chem. Sci. 15 (2024) 3330–3338. doi: 10.1039/D3SC06471A
G. Kastlunger, L. Wang, N. Govindarajan, et al., ACS Catal. 12 (2022) 4344–4357. doi: 10.1021/acscatal.1c05520
Y.C. Yao, T. Shi, W.X. Chen, et al., Nat. Commun. 15 (2024) 1257. doi: 10.1038/s41467-024-45704-2
Z.L. Wang, Y.C. Li, X. Zhao, et al., J. Am. Chem. Soc. 145 (2023) 6339–6348. doi: 10.1021/jacs.2c13384
X.F. Qiu, H.L. Zhu, J.R. Huang, P.Q. Liao, X.M. Chen, J. Am. Chem. Soc. 143 (2021) 7242–7246. doi: 10.1021/jacs.1c01466
S.F. Wang, F.H. Li, J. Zhao, et al., Nat. Commun. 15 (2024) 10247. doi: 10.1038/s41467-024-54636-w
J.K. Nørskov, J. Rossmeisl, A. Logadóttir, et al., J. Phys. Chem. B 108 (2004) 17886–17892. doi: 10.1021/jp047349j
W.J. Luo, X.W. Nie, M.J. Janik, A. Asthagiri, ACS Catal. 6 (2016) 219–229. doi: 10.1021/acscatal.5b01967

Figure 1 The MEP diagram for CO2 electroreduction into CO via COOH intermediate and subsequent CHO and OCCO generations via CO further electroreduction.

Figure 3 The overall energy diagrams towards C2H4 product: (a) OCCOH pathway via COHCOH species; (b) OCCOH pathway via COCHOH species at Cl−-modified Cu(111)/H2O interface.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:

