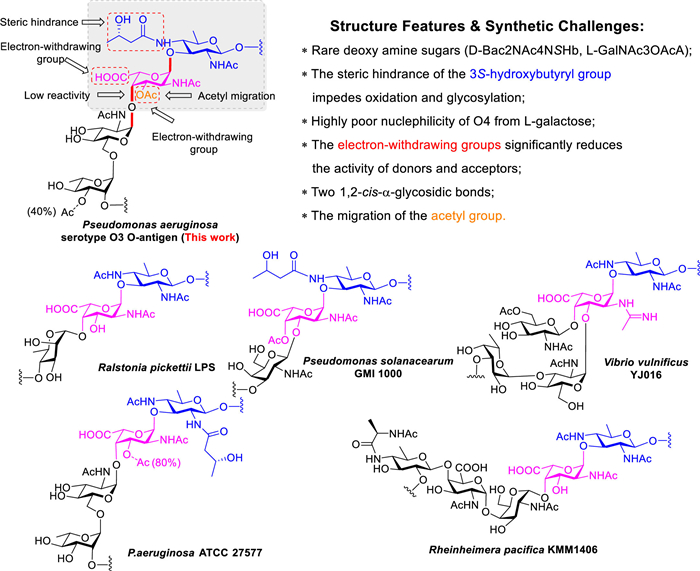
Figure 1.
Representative structures of bacterial glycans containing α-L-GalNAcA-(1→3)-β-D-Bac disaccharide or similar motifs.
Chemical synthesis of the highly functionalized O-antigen repeating unit from Pseudomonas aeruginosa serotype O3 for glycoconjugate vaccine development
Guochao Lv , Guangzong Tian , Guodong Chen , Shengyong Zhu , Jialong Bao , Chunjun Qin , Xiaopeng Zou , Jing Hu , Peter H. Seeberger , Jian Yin

Pseudomonas aeruginosa, a Gram-negative opportunistic pathogen known for its remarkable environmental adaptability [1], is a leading cause of nosocomial infections [2]. Immunocompromised individuals are particularly vulnerable to its severe manifestations, which include antibiotic-resistant wound infections, ventilator-associated pneumonia (VAP), and chronic pulmonary colonization in patients with cystic fibrosis (CF) [3,4]. The reported overall mortality rates associated with P. aeruginosa exceed 70%, with the directly attributable fatality rate reaching as high as 40% [5]. Although P. aeruginosa infections are primarily treated with antibiotics, their effectiveness is increasingly compromised by both intrinsic and acquired resistance mechanisms [6,7]. Conventional antibiotic therapies often fail to completely eradicate the bacteria and may inadvertently contribute to the emergence of resistance [8]. Consequently, there is an urgent need for alternative strategies to treat and prevent P. aeruginosa infections. Vaccines targeting this pathogen present a promising strategy to disrupt the infection cycle and mitigate the spread of antibiotic resistance, particularly among high-risk populations [9]. However, currently, there are no commercially available vaccines against P. aeruginosa [10].
The lipopolysaccharide (LPS) is recognized as a major virulence factor of P. aeruginosa. Strains expressing LPS O-antigens are classified into 20 serotypes under the International Antigenic Typing System (IATS), based on the chemical composition of their O-antigens [11]. Extensive research has demonstrated that O-antigen based vaccines can elicit the production of specific antibodies that exhibit protective efficacy in preclinical animal studies [12]. Research on carbohydrate-based vaccines, particularly those targeting O-antigens, has persisted for decades. In 1971, Fisher et al. developed the first vaccine targeting P. aeruginosa by extracting O-antigens from seven different serotypes (O1, O2, O3, O5, O6, O10, and O11). Clinical trials indicated that the vaccine induced high-titer antibodies in most participants; however, significant adverse effects hindered its clinical adoption [13]. In 1989, the Swiss Serum and Vaccine Institute developed a conjugate vaccine, Aerugen, which combined the O-antigens of eight serotypes (O1, O2, O3, O4, O5, O6, O11, and O12) with exotoxin A [14]. Although promising, this vaccine failed to demonstrate sufficient efficacy in Phase Ⅲ trials for preventing P. aeruginosa infections in cystic fibrosis patients, leading to its discontinuation [15]. Despite these setbacks, O-antigens continue to show promise in the development of glycoconjugate vaccines. Chemical synthesis is well-known for facilitating the production of uncontaminated O-antigens and assisting in the identification of key epitopes, thereby making them central to carbohydrate vaccine research [16–18]. In recent years, several O-antigen-related glycans of P. aeruginosa have been synthesized. For instance, Li et al. synthesized the O-antigen trisaccharide of the P. aeruginosa serotype O7 in 2017 [19]. Kulkarni et al. and our team prepared the trisaccharide O-antigen of serotype O11 [20,21]. Additionally, the O10 and O19 trisaccharide repeating units were synthesized by Gao et al. and our team [22,23]. More recently, we also synthesized the O-antigen repeating units of serotypes O4, O5, and O17 [13,24,25]. According to Lu and colleagues, serotype O3 accounts for 4.1% of all clinical infections caused by P. aeruginosa [5].
The O3 O-antigen structure of P. aeruginosa has been shown to consist of a rare tetrasaccharide repeating unit characterized as follows: [→2)-α-L-Rha-(1→6)-α-D-GlcNAc-(1→4)-α-L-GalNAc3OAcA-(1→3)-β-D-Bac2NAc4NSHb-(1→] (Fig. 1) [26]. Approximately 40% of these repeating tetrasaccharides feature an acetyl group at the 3-O position of L-rhamnose (L-Rha). Noteworthily, nonstoichiometric O-acetylation is frequently observed within the LPS of P. aeruginosa [26]. This tetrasaccharide, which contains three rare amino sugars, possesses various functional groups at specific positions [27,28]. A 3S-hydroxybutyryl (Hb) amide linker is attached to the C4 position of the D-bacillosamine (D-Bac) unit, while both the L-galactose (L-Gal) and D-glucose (D-Glc) units carry an acetylamino group at the C2 position. The unique composition of rare sugars, combined with an unusual side chain, is seldom found in host cellular structures, rendering this tetrasaccharide O-antigen a promising candidate for vaccine development [29,30]. The identification of the D-Bac unit has revealed correlations with pathogenicity in specific contexts, establishing this tetrasaccharide as a compelling focus for pathogenicity studies [31]. Although the α-L-GalNAcA-(1→3)-β-D-Bac disaccharide motif and its derivatives are prevalent in many bacterial polysaccharides [22,32–35], there have been no reports of the selective oxidation of the primary alcohol on the L-Gal unit to galacturonic acid in the presence of the 3S-Hb group on the D-Bac unit (Fig. 1). Current research on the use of the L-Gal unit as an acceptor in disaccharide assembly is notably limited, especially when the linkage site is at the C4 hydroxyl group and both acetyl and carboxyl electron-withdrawing groups are simultaneously present. Recently, the Gao group reported the synthesis of the LPS O-antigen of P. aeruginosa ATCC 2757 using an artificially tuned sequence, positioning the L-Gal unit at the non-reducing end to avoid the construction of the (1→4)-L-Gal glycosidic bond [22]. Our team also reported the synthesis of the LPS O-antigens of P. aeruginosa serotypes O10 and O19, where the (1→4)-L-Gal glycosidic linkage was constructed; however, their synthetic sequences lacked an acetyl group on the L-Gal unit [23], thereby increasing the synthetic challenge of this work. To date, the chemical synthesis of the O3 O-antigen tetrasaccharide repeating unit of P. aeruginosa has not been reported, presenting significant synthetic challenges. First, the monosaccharides corresponding to this LPS, including D-GlcNAc, L-GalNAc3OAcA, and D-Bac2NAc4NSHb, are rare sugars that are not commercially available. Second, when an acetyl group is located in the ortho position relative to the glycosylation site of L-galactose, it may induce undesirable acetyl migration. Third, glycosylation at the C4 hydroxyl group of the L-Gal unit is particularly challenging due to its low reactivity in the presence of electron-withdrawing groups at adjacent positions. Fourth, the target structure contains various functional groups, including acetyl, carboxyl, acetamido, and a 3S-hydroxybutyryl (Hb) chain, with the 3S-Hb group contributing significant steric hindrance. Therefore, adopting a well-considered orthogonal protection strategy and a carefully designed reaction sequence is essential to ensure the feasibility and efficiency of the synthesis. Herein, we report the first chemical synthesis of the P. aeruginosa serotype O3 O-antigen tetrasaccharide. A linker featuring a free amino group was incorporated at the reducing end of the tetrasaccharide, allowing for subsequent modifications or conjugation with other molecules [36,37]. This advancement will significantly enhance biological and immunological investigations of this O-antigen.
The synthesis of the target tetrasaccharide 1 was envisioned to be accomplished through a [1+(1+(1 + 1))] strategy, initiated from the reducing end (Fig. 2). Retrosynthetic analysis revealed that tetrasaccharide 1 could be derived from protected glycan 2 through functional group interconversions (FGI) followed by global deprotection. This linear glycosylation approach employed building blocks 4, 6, 7, and 8. An orthogonal amine linker containing a β-O-glycosidic linkage was incorporated into the synthetic oligosaccharides to enable subsequent conjugation or immobilization. Inspired by the work of the Kulkarni group, the 3S-Hb group was introduced at the C4 amino position of building block 7 during the monosaccharide stage [38]. The levulinoyl (Lev) group on the C6 hydroxyl of 6 acted as a remote participating group to promote the stereoselective assembly of the α-D-GlcNAc linkage and served as a temporary protecting group for (1→6) branching. Moreover, the neighboring Lev group of 4 was strategically utilized to efficiently construct the 1,2-trans-L-Rha linkage. An electron-donating 2-naphthylmethyl ether (Nap) was used to protect the C3 hydroxyl of 8, thereby enhancing the reactivity of the adjacent C4 hydroxyl group. Compounds 6 and 8 featured a non-participating azido group at their C2 positions, which served as a masked amino group to facilitate 1,2-cis-glycosylation. Furthermore, the azido group exhibited stability under various reaction conditions, including oxidative steps commonly used in carbohydrate synthesis, and could be easily converted into the desired acetamido group through selective reduction followed by N-acetylation. Conversely, a trichloroacetamido (TCA) group was selected to protect the amino group on 7 to facilitate the stereoselective formation of the β-glycosidic bond. The amino group in the linker at the reducing end of the glycan was protected using benzyl (Bn) and benzyloxycarbonyl (Cbz) groups, allowing for clear differentiation from other amino functionalities. Given that galacturonic acid typically exhibits low reactivity in glycosylation reactions [39], we devised a strategy to replace L-galacturonic acid with 4,6-O-benzylidene-protected L-galactose during the synthesis process. This intermediate could be converted into L-galacturonic acid through post-glycosylation oxidation, ensuring efficient assembly while overcoming the reactivity limitations associated with the direct use of L-galacturonic acid in glycosylation.
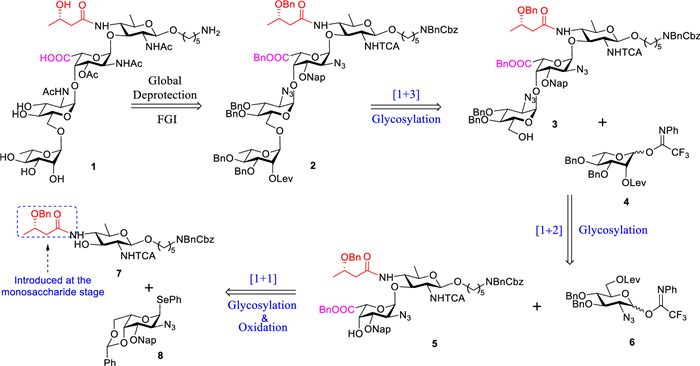
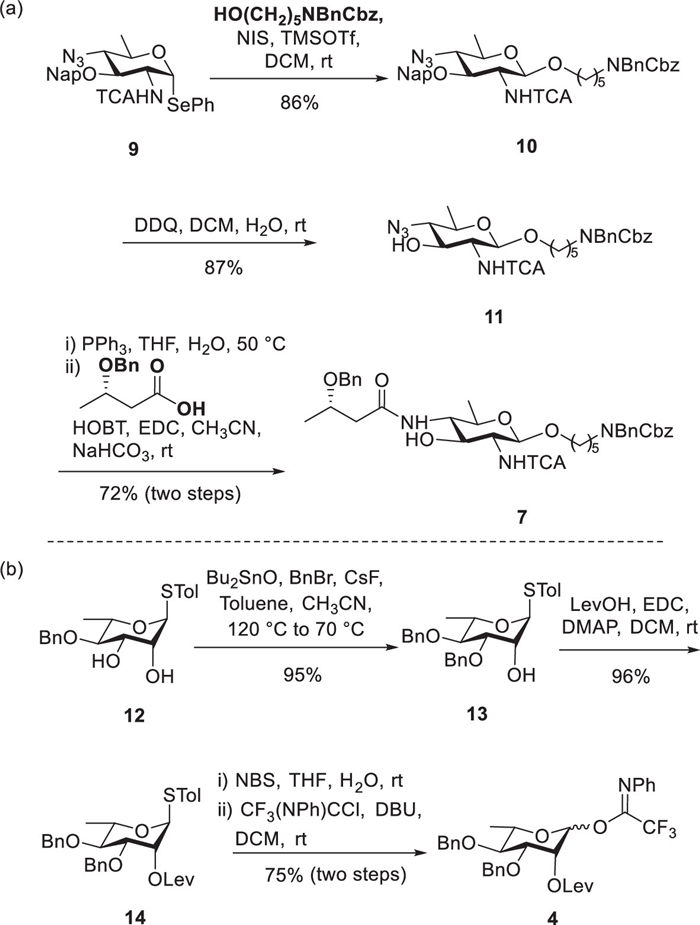
We initiated the synthesis by preparing orthogonally protected rare sugar building blocks (Scheme 1). The synthesis of building blocks 6 and 8 was conducted using modified procedures based on established literature methods [40,41]. The synthesis of acceptor 7 commenced from known D-Bac derivatives [42]. As depicted in Scheme 1a, glycosylation of donor 9 with a protected amino-pentanol linker (N-Bn-N-Cbz-5-aminopentane-1-ol), promoted by trimethylsilyl trifluoromethanesulfonate (TMSOTf) and N-iodosuccinimide (NIS) as activators, afforded product 10 in 86% yield with exclusive β-stereoselectivity (anomeric proton of the donor residue: 4.85 ppm, 3JH1/H2 = 8.4 Hz). The Nap protecting group was removed through 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ)-mediated oxidative cleavage. Subsequently, the azide functionality underwent Staudinger reduction to produce the corresponding amine, which was then coupled with 3S-hydroxybutyric acid under 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), hydroxybenzotriazole (HOBT), and sodium bicarbonate (NaHCO3) conditions, resulting in the C4-amide acceptor 7 with an overall yield of 72% across both steps. The synthesis of building block 4 from the known L-rhamnose diol 12 is shown in Scheme 1b [43]. A tin-mediated regioselective protection of the C3 hydroxyl group with dibutyltin oxide (Bu2SnO) and benzyl bromide (BnBr) produced 13 in 95% yield. Treatment with levulinic acid (LevOH) and EDC efficiently converted 13 into the corresponding ester 14 in 96% yield. Following the levulinylation of the C2 hydroxyl group, the thioglycoside 16 was further transformed into the trifluoroacetimidate donor 4, achieving an overall yield of 75%.
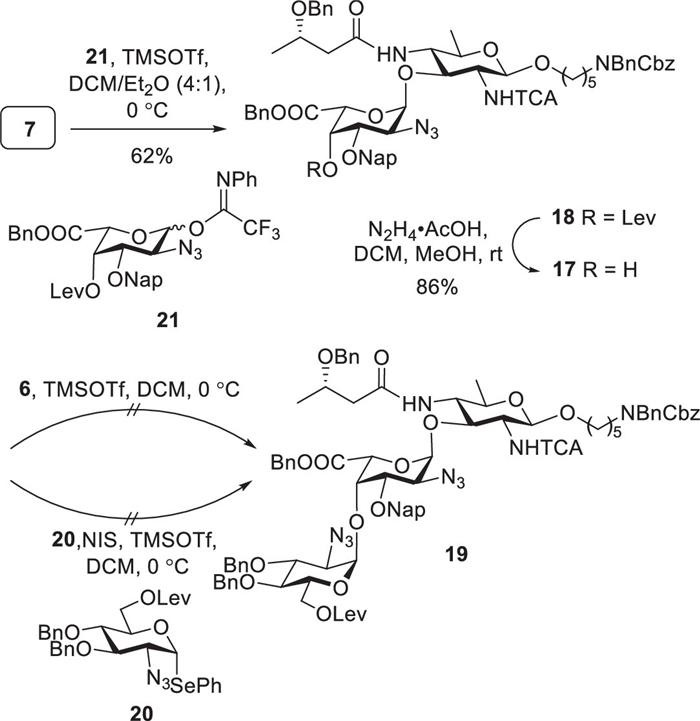
With all building blocks in hand, we proceeded to synthesize disaccharide 17 (Scheme 2). The construction of the galactosidic linkage commenced with the glycosylation of donor 8 with acceptor 7, affording disaccharide 15 in 79% yield with complete α-stereoselectivity (anomeric proton of the donor residue: 4.94 ppm, 3JH1/H2 = 3.4 Hz) [44]. Disaccharide 15 was subsequently treated with trifluoroacetic acid (TFA) in the presence of water, yielding diol 16 in 72%. Attempts to selectively oxidize the C6 hydroxyl group to the corresponding carboxylic acid using 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) and (diacetoxyiodo)benzene (BAIB) were unsuccessful. To obtain the target compound 17, we evaluated a series of oxidation methods under varying experimental conditions (Table S1 in Supporting information) [13,45–49], but none proved effective. Drawing upon foundational studies conducted by our group, we undertook preliminary confirmatory experiments to investigate this outcome. We observed that the selective oxidation of primary alcohols proceeded efficiently on compound S3 (see Supporting information), but not on compound 16. The primary structural difference between the two disaccharides is the presence of the 3S-Hb group in 16. It is plausible that the steric hindrance introduced by this substituent interferes with the selective abstraction of the α-hydrogen from the primary alcohol by the TEMPO intermediate, thereby impeding the oxidation process [50,51].

Subsequently, we adopted an alternative synthetic route that involved the selective oxidation of the C6 hydroxyl group to produce L-galacturonic acid 21 (see Supporting information). The TMSOTf-catalyzed glycosylation of acceptor 7 with donor 21 afforded the desired α-glycoside 18 (anomeric proton of the donor residue: 5.08 ppm, 3JH1/H2 = 3.7 Hz) with a yield of 62% (Scheme 3). The subsequent removal of the levulinyl group transformed 18 into disaccharide acceptor 17. Unfortunately, several attempts to synthesize trisaccharide 19 through the glycosylation of disaccharide acceptor 17 using selenoglycoside 20 and the corresponding trifluoroacetimidate 6 were unsuccessful. The failure of this coupling reaction is likely attributed to the inherently poor nucleophilicity of the axial C4 hydroxyl group of L-galactose in compound 17, which limits its reactivity toward glycosyl donors. Furthermore, the steric hindrance imposed by the 3S-Hb group may further contribute to the failure of the glycosylation reaction.
Given the challenges posed by the rare L-galacturonic acid unit in glycosylation reactions and the limited historical precedent concerning its behavior as a glycosyl donor or acceptor, we prioritized the construction of the L-galactoside bond within our synthetic strategy. To mitigate the steric hindrance imposed by the 3S-Hb group, we tentatively protected the amine using a 2,2,2-trichlorethoxycarbonyl (Troc) group. After thorough consideration, we designed and implemented a [1+(2 + 1)] synthetic strategy for the preparation of the target tetrasaccharide (Fig. 3).
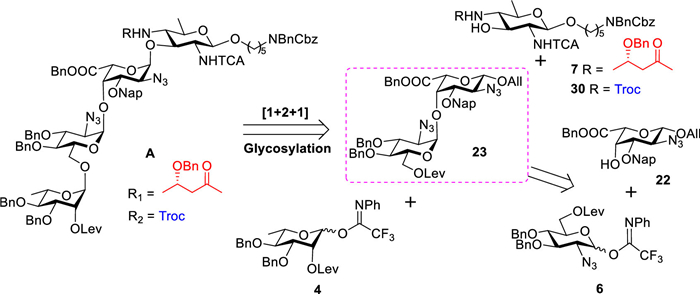
The union of acceptor 22 with trifluoroacetimidate donor 6, in the presence of TMSOTf at 0 ℃, afforded disaccharide 23 with a yield of 62% and complete α-selectivity (anomeric proton of the donor residue: 5.50 ppm, 3JH1/H2 = 3.6 Hz) (Scheme 4) [41]. The removal of the allyl group was accomplished by using [Ir(COD)(CH3Ph2P)2]PF6, resulting in the complete conversion of 23 into the corresponding vinyl ether. Following this, hydrolysis with iodine (I2) and water in tetrahydrofuran (THF) yielded the desired alcohol quantitatively. The introduction of the trifluoroacetimidate then furnished donor 24 with an overall yield of 87%. Unfortunately, the glycosylation of disaccharide donor 24 with acceptor 7 was unsuccessful. Analysis revealed the presence of decomposed donor byproducts and unreacted acceptor, suggesting that the steric hindrance from the 3S-Hb group in 7 may impede the glycosylation reaction. Furthermore, we hypothesize that disaccharides inherently exhibit greater steric hindrance than monosaccharides when acting as donors, which further limits the nucleophilic attack of the acceptor. To address these challenges, disaccharide 24 was utilized to glycosylate acceptor 30 (see Supporting information) in the presence of TMSOTf, a condition that proved effective for trisaccharide formation. We successfully obtained trisaccharide 25 (α:β = 8:1, α: anomeric proton of the donor residue: 5.13 ppm, 3JH1/H2 = 3.4 Hz) in 59% yield. Subsequent cleavage of the levulinoyl ester using hydrazine acetate yielded trisaccharide acceptor 26, which was further glycosylated with building block 6 to produce tetrasaccharide 27 (anomeric proton of the donor residue: 4.66 ppm, 1JC1/H1 = 171.4 Hz) in 61% yield. The conversion of the azide to the corresponding acetamido was accomplished by using thioacetic acid (AcSH) in pyridine, resulting in the acetamido-sugar 28 in 77% yield. However, oxidative cleavage of the Nap ether with DDQ resulted in a low yield (Table S2 in Supporting information, entry 1). Increasing the equivalents of DDQ or extending the reaction time led to significant degradation of the starting materials (Table S2, entries 2 and 3) [52]. We attempted to substitute 1,2-dichloroethane (DCE) for dichloromethane or to add the additive β-pinene to facilitate the removal of Nap ether (Table S2, entries 4 and 5) [53,54]; however, only trace amounts of products were detectable by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF). Ceric ammonium nitrate (CAN) and HF·pyridine were also evaluated as alternative methods, but did not yield the expected amount of the product (Table S2, entries 6 and 7) [55,56]. Lowary and co-workers reported that selectively cleaving Nap ethers in the presence of abundant benzyl ethers (seven in this case) is particularly challenging due to competitive debenzylation [52].
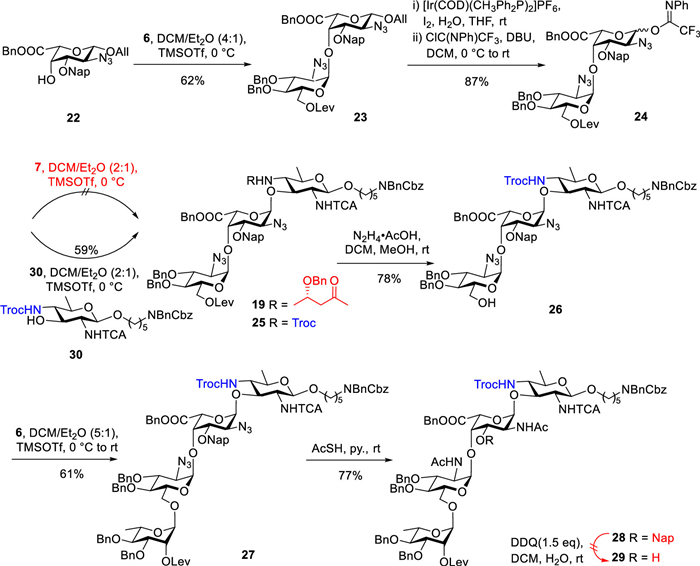
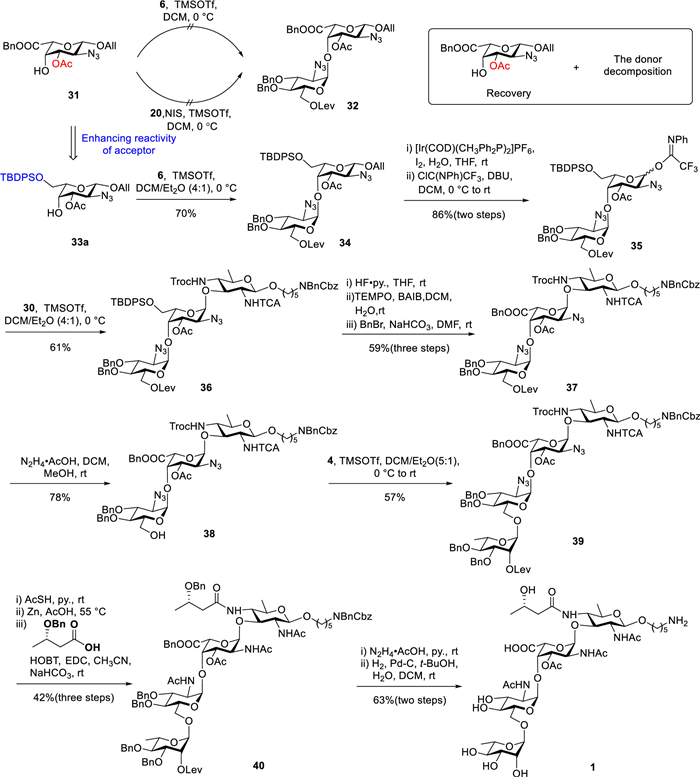
Disappointed by these results, we revised our synthetic route by replacing the Nap ether with an acetyl ester (Ac). As depicted in Scheme 5, we synthesized acceptor 31 (for details, see Supporting information). Unfortunately, glycosylation of acceptor 31 with donors 6 and 20 was unsuccessful; instead, the acceptor was quantitatively recovered unchanged after workup. Achieving glycosylation at the C4 position of L-galactose is particularly challenging when both an acetyl group and a carboxyl group are present, as these electron-withdrawing groups significantly diminish the reactivity of the acceptor. To address this limitation, we selectively protected the C6 hydroxyl group using a tert–butyldiphenylsilyl (TBDPS) ether to enhance the reactivity of the C4 hydroxyl group derivative 33a for subsequent glycosylation [57,58]. However, we observed the migration of the acetyl group to the adjacent C4 hydroxyl group of the L-Gal unit under alkaline conditions. Notably, acetyl migration rarely occurs during the reaction itself and is typically significant only after the concentration of the reaction solution. Encouragingly, the prompt separation and purification of the crude product enabled us to maintain the acetyl migration within an acceptable threshold (< 7%). Fortunately, monosaccharide allyl derivative 33a was glycosylated with donor 6, activated by TMSOTf, to generate disaccharide 34 (anomeric proton of the donor residue: 5.12 ppm, 3JH1/H2 = 3.7 Hz) with a yield of 70% and high α-stereoselectivity. Disaccharide 34 was subsequently transformed into disaccharide donor 35 by removing the allyl group and installing an N-pentyltrifluoroacetimidate on the anomeric hydroxyl, resulting in 86% yield over two steps. Donor 35 was then glycosylated with acceptor 30, leading to a mixture of α and β anomers of trisaccharide 36 (α:β = 10:1, α: anomeric proton of the donor residue: 5.07 ppm, 1JC1/H1 = 172.6 Hz) in 61% yield. After successfully obtaining the protected trisaccharide, the C6-OTBDPS group in compound 36 was cleaved using HF·pyridine. Subsequent oxidation mediated by TEMPO and BAIB, followed by benzyl ester formation using BnBr and NaHCO3 in N,N-dimethylformamide (DMF), afforded trisaccharide 37 with a yield of 59% over three steps. Cleavage of the levulinoyl ester using hydrazine acetate then furnished the C6-OH trisaccharide acceptor 38 in 78% yield. Monosaccharide donor 4 was coupled with trisaccharide acceptor 38 to generate the desired α-glycoside tetrasaccharide 39 (anomeric proton of the donor residue: 4.53 ppm, 1JC1/H1 = 170.3 Hz) in 57% yield. Reduction of the two azide groups using AcSH in pyridine was followed by treatment of the tetrasaccharide with excess zinc powder in acetic acid at 55 ℃, effectively removing the Troc group and reducing the N-trichloroacetamide to the corresponding N-acetamide. Subsequent coupling of the resulting C4 amino group with the 3S-hydroxybutyryl acid derivative afforded the desired tetrasaccharide 40 in 42% yield over three steps. Finally, removal of the levulinoyl group followed by hydrogenation delivered the target tetrasaccharide 1 with a yield of 63% over two steps.
In summary, the target tetrasaccharide of the P. aeruginosa serotype O3 O-antigen was synthesized for the first time through the careful design and implementation of an elaborate synthetic strategy. We observed that significant steric hindrance from the 3S-Hb not only impeded the oxidation of primary alcohols but also negatively impacted the efficiency of glycosylation reactions. The inability to selectively cleave the Nap ether necessitated a revision of the synthetic route. Notably, the selective removal of a Nap ether during the late stages of synthesis, particularly in the presence of multiple benzyl groups, remains challenging to predict. During disaccharide assembly, we found that the reactivity of the C4 hydroxyl group in the acceptor was significantly influenced by substituent effects. Replacing the C6 carboxylate ester with a TBDPS group in the L-Gal acceptor proved to be an effective strategy for enhancing the efficiency of glycosylation assembly. Additionally, we observed acetyl migration under alkaline conditions, emphasizing the necessity for rapid workup to minimize migration during purification. The complex synthesis of tetrasaccharide 1 provided meaningful insights into the assembly of other aminoglycosides with functionalized structures, particularly those incorporating the 3S-hydroxybutylamino group. Furthermore, the installation of a linker containing a free amino group at the glycan reducing end enables subsequent conjugation or immobilization, facilitating the immunological evaluation of the O3 O-antigen and the development of an anti-P. aeruginosa synthetic glycoconjugate vaccine.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Guochao Lv: Writing – original draft, Methodology, Investigation, Formal analysis, Data curation. Guangzong Tian: Writing – review & editing, Supervision, Methodology, Investigation, Formal analysis. Guodong Chen: Investigation. Shengyong Zhu: Investigation. Jialong Bao: Investigation. Chunjun Qin: Investigation, Funding acquisition. Xiaopeng Zou: Investigation. Jing Hu: Investigation, Funding acquisition. Peter H. Seeberger: Supervision. Jian Yin: Writing – review & editing, Supervision, Resources, Project administration, Funding acquisition, Formal analysis, Conceptualization.
The authors are grateful to the National Key R & D Program of China (No. 2023YFC2308000), the National Natural Science Foundation of China (Nos. 22478153, 22477046, 22177041), the Max Planck Society International Partner Group Program, the China Scholarship Council (CSC), and the Fundamental Research Funds for the Central Universities for funding. P.H.S. thanks the Max-Planck Society for generous financial support.
Supplementary material associated with this article can be found, in the online version, at doi:
M.W. Azam, A.U. Khan, Drug Discov. Today 24 (2019) 350–359. doi: 10.1016/j.drudis.2018.07.003
P. Laborda, F. Sanz-García, S. Hernando-Amado, et al., Curr. Opin. Microbiol. 64 (2021) 125–132. doi: 10.1016/j.mib.2021.09.010
I. Bianconi, B. Alcalá-Franco, M. Scarselli, et al., Front. Immunol. 9 (2019) 3021. doi: 10.3389/fimmu.2018.03021
G.P. Priebe, J.B. Goldberg, Expert. Rev. Vacc. 13 (2014) 507–519. doi: 10.1586/14760584.2014.890053
Q. Lu, P. Eggimann, C.E. Luyt, et al., Crit. Care 18 (2014) R17. doi: 10.1186/cc13697
J.C. Vázquez-Ucha, D. Rodríguez, C. Lasarte-Monterrubio, et al., J. Med. Chem. 64 (2021) 6310–6328. doi: 10.1021/acs.jmedchem.1c00369
J.A. Driscoll, S.L. Brody, M.H. Kollef, Drugs 67 (2007) 351–368. doi: 10.2165/00003495-200767030-00003
G.M. Rossolini, E. Mantengoli, Clin. Microbiol. Infect. 11 (2005) 17–32. doi: 10.1111/j.1469-0691.2005.01161.x
Z.J. Hou, J.J. Wang, X.X. Zhang, et al., Chin. Chem. Lett. 34 (2023) 107804. doi: 10.1016/j.cclet.2022.107804
M. Killough, A. Rodgers, R. Ingram, Vaccines 10 (2022) 1100. doi: 10.3390/vaccines10071100
Y.A. Knirel, O.V. Bystrova, N.A. Kocharova, et al., J. Endotoxin Res. 12 (2006) 324–336.
M.W. Fisher, H.B. Devlin, F. Gnabski, J. Bacteriol. 98 (1969) 835–836. doi: 10.1128/jb.98.2.835-836.1969
G.Z. Tian, J. Hu, C.J. Qin, et al., J. Am. Chem. Soc. 146 (2024) 18427–18439. doi: 10.1021/jacs.4c03814
S.J. Cryz, J.C. Sadoff, E. Fürer, Microb. Pathog. 6 (1989) 75–80. doi: 10.1016/0882-4010(89)90010-7
A. Sharma, A. Krause, S. Worgall, Hum. Vaccines 7 (2011) 999–1011. doi: 10.4161/hv.7.10.16369
C. Anish, B. Schumann, C.L. Pereira, et al., Chem. Biol. 21 (2014) 38–50. doi: 10.1016/j.chembiol.2014.01.002
L.X. Li, J. Hu, C.J. Qin, et al., Chin. Chem. Lett. 36 (2025) 110797. doi: 10.1016/j.cclet.2024.110797
W.Q. Song, J.T. Cai, X.P. Zou, et al., Chin. Chem. Lett. 29 (2018) 27–34. doi: 10.1016/j.cclet.2017.09.061
H. Liu, Y.F. Zhang, R.H. Wei, et al., J. Am. Chem. Soc. 139 (2017) 13420–13428. doi: 10.1021/jacs.7b06055
A. Behera, D. Rai, S.S. Kulkarni, J. Am. Chem. Soc. 142 (2019) 456–467.
C.J. Qin, Z.H. Liu, M.R. Ding, et al., J. Carbohydr. Chem. 39 (2020) 374–397. doi: 10.1080/07328303.2020.1839479
X.Y. Yang, H. Zhang, Q.P. Zhao, et al., JACS Au 4 (2024) 2351–2362. doi: 10.1021/jacsau.4c00321
X.P. Zou, C.J. Qin, G.Z. Tian, et al., Org. Lett. 26 (2024) 9198–9202. doi: 10.1021/acs.orglett.4c03167
G.Z. Tian, J.L. Bao, G.D. Chen, et al., Chin. J. Chem. 43 (2024) 743–749.
X.P. Zou, J.X. Zhang, Y.L. Lu, et al., Chin. J. Chem. 43 (2025) 2174–2180. doi: 10.1002/cjoc.70098
Y.A. Knirel, Rev. Microbiol. 17 (1990) 273–304. doi: 10.3109/10408419009105729
G.Z. Tian, C.J. Qin, J. Hu, et al., Molecules 28 (2023) 7112. doi: 10.3390/molecules28207112
G.Z. Tian, X.L. Wang, J. Hu, et al., Chin. Chem. Lett. 26 (2015) 922–930. doi: 10.1016/j.cclet.2015.04.026
A. Adibekian, P. Stallforth, M.L. Hecht, et al., Chem. Sci. 2 (2011) 337–344. doi: 10.1039/C0SC00322K
D.B. Werz, R. Ranzinger, S. Herget, et al., ACS Chem. Biol. 2 (2007) 685–691. doi: 10.1021/cb700178s
M.J. Morrison, B. Imperiali, Biochemistry 53 (2014) 624–638. doi: 10.1021/bi401546r
E. Vinogradov, L. Nossova, A. Swierzko, et al., Carbohydr. Res. 339 (2004) 2045–2047. doi: 10.1016/j.carres.2004.05.014
G. Orgambide, H. Montrozier, P. Servin, et al., J. Biol. Chem. 266 (1991) 8312–8321.
N.A. Komandrova, M.S. Kokoulin, A.I. Kalinovsky, et al., Carbohydr. Res. 394 (2014) 1–6.
S.N. Senchenkova, A.S. Shashkov, Y.A. Knirel, et al., Carbohydr. Res. 344 (2009) 1009–1013. doi: 10.1016/j.carres.2009.03.021
P. Shen, H. Lin, Y.K. Bao, et al., Chin. Chem. Lett. 34 (2023) 107679. doi: 10.1016/j.cclet.2022.07.022
H. Lin, Y.C. Li, K. Zhou, et al., Chin. Chem. Lett. 35 (2024) 108670. doi: 10.1016/j.cclet.2023.108670
S. Biswas, B.K. Ghotekar, S.S. Kulkarni, Org. Lett. 23 (2021) 6137–6142. doi: 10.1021/acs.orglett.1c02239
J. Sianturi, P. Priegue, J. Hu, et al., Angew. Chem. Int. Ed. 61 (2022) e202209556. doi: 10.1002/anie.202209556
K.X. Shou, S.S. Liu, Y.Q. Zhang, et al., Chin. J. Chem. 42 (2024) 1593–1598. doi: 10.1002/cjoc.202400121
C.J. Qin, H.L. Hou, M.R. Ding, et al., Chin. J. Nat. Med. 20 (2022) 387–392.
J. van Mechelen, J. Voorneveld, H.S. Overkleeft, et al., Org. Biomol. Chem. 18 (2020) 2834–2837. doi: 10.1039/d0ob00256a
H.R.H. Elsaidi, T.L. Lowary, Chem. Sci. 6 (2015) 3161–3172. doi: 10.1039/C4SC04004J
S.Y. Li, W.W. Zhuge, X. Sun, et al., Chin. Chem. Lett. 35 (2024) 109089. doi: 10.1016/j.cclet.2023.109089
H. Zhang, X.H. Wang, Y.H. Meng, et al., JACS Au 2 (2021) 97–108.
B. Hagen, J.H.M. van Dijk, Q.J. Zhang, et al., Org. Lett. 19 (2017) 2514–2517. doi: 10.1021/acs.orglett.7b00747
R.E.J.N. Litjens, R. den Heeten, M.S.M. Timmer, et al., Chem. Eur. J. 11 (2005) 1010–1016. doi: 10.1002/chem.200400862
M. Vetro, B. Costa, G. Donvito, et al., Org. Biomol. Chem. 13 (2015) 1091–1099. doi: 10.1039/C4OB01602E
M. Yadav, C.L. Liotta, R. Krishnamurthy, Bioorg. Med. Chem. Lett. 28 (2018) 2759–2765. doi: 10.1016/j.bmcl.2018.01.066
M. Emmadi, S.S. Kulkarni, J. Org. Chem. 83 (2018) 14323–14337. doi: 10.1021/acs.joc.8b02037
S. Zhang, P.H. Seeberger, Chem. Eur. J. 27 (2021) 17444–17451. doi: 10.1002/chem.202103234
K. Sahloul, T.L. Lowary, J. Org. Chem. 80 (2015) 11417–11434. doi: 10.1021/acs.joc.5b02083
P. Wang, J.L. Wang, W.J. Yin, et al., Org. Lett. 24 (2022) 971–976. doi: 10.1021/acs.orglett.1c04363
D. Lloyd, M. Bylsma, D.K. Bright, et al., J. Org. Chem. 82 (2017) 3926–3934. doi: 10.1021/acs.joc.7b00065
Y. Li, B. Roy, X.Y. Liu, Chem. Commun. 47 (2011) 8952–8954. doi: 10.1039/c1cc13264d
V. Cattaneo, D. Oldrini, A. Corrado, et al., Org. Chem. Front. 3 (2016) 753–758. doi: 10.1039/C6QO00144K
C.J. Qin, B. Schumann, X.P. Zou, et al., J. Am. Chem. Soc. 140 (2018) 3120–3127. doi: 10.1021/jacs.8b00148
C.J. Qin, L.X. Li, G.Z. Tian, et al., J. Am. Chem. Soc. 144 (2022) 21068–21079. doi: 10.1021/jacs.2c05953

Figure 1 Representative structures of bacterial glycans containing α-L-GalNAcA-(1→3)-β-D-Bac disaccharide or similar motifs.

Figure 2 Retrosynthetic analysis of the P. aeruginosa serotype O3 O-antigen tetrasaccharide 1.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:





