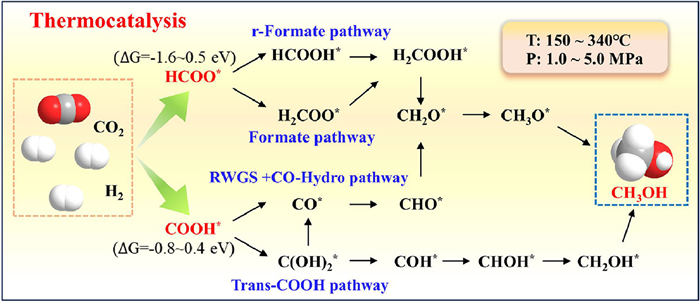
Figure 1.
Pathways of thermal catalytic CO2-to-methanol conversion with SACs.
Single-atom catalysts for CO2-to-methanol conversion: A critical review
Jingying Wang , Jianhui Zhao , Shaopo Wang , Jingjie Yu , Ning Li

The global energy landscape has undergone a profound transformation in recent decades, with fossil fuel combustion driving atmospheric CO2 concentration to unprecedented levels (exceeding 410 ppm) [1–3]. The exponential rise of greenhouse gas has caused environmental challenges including accelerated global warming and existential threats to ecological stability and sustainable development [1,2]. The Intergovernmental Panel on Climate Change (IPCC) warned that potential atmospheric CO2 concentrations could reach 590 ppm by the end of the century under business-as-usual scenarios [4]. Catalytic transformation of CO2 captured from ambient air or industrial exhaust gases into value-added chemical feedstocks serves as a critical enabler for carbon neutrality.
The inherent thermodynamic stability of CO2 molecules necessitates substantial energy inputs for activation and conversion. While thermo-catalytic hydrogenation remains the most mature approach for CO2 utilization, emerging strategies such as photocatalysis and electrocatalysis have demonstrated remarkable potential for sustainable carbon cycling. These processes yield diverse products ranging from C1 compounds (CO [5,6], CH4 [7,8], CH3OH [9,10], HCOOH [11,12]) to C2+ hydrocarbons and oxygenates (C2H4 [13,14], C2H5OH [15,16], CH3COOH [17,18]). Among them, methanol emerges as a particularly valuable target due to its dual role as a clean energy source and versatile chemical feedstock [19–22]. In energy applications, it is utilized in methanol gasoline to reduce exhaust emissions and fuel cell technology for new energy vehicles [19]. Within the chemical industry, the derivative formaldehyde serves as a critical component in manufacturing resins, plastics and adhesives [20]. Additionally, methanol functions as a solvent for pharmaceutical synthesis, raw material for pesticides and reaction medium in biodiesel production [21].
The efficient conversion of CO2 to methanol remains hindered by multifaceted scientific and technological barriers. The main challenges include catalyst design and performance optimization, reaction condition regulation, system integration, scalability and economic viability barriers [19–22]. Particularly, catalyst innovation continues to be of paramount importance. Recent advances in SACs have revolutionized CO2 conversion paradigms through maximizing atomic efficiency and tailored electronic configurations [23–26]. Distinct from conventional nanoparticle catalysts, SACs feature isolated metal centers anchored on support matrices, enhancing coordination unsaturation [27–29]. The unique architecture enables exceptional catalytic performance in CO2 activation, with precise control over reaction pathways and product selectivity. Although existing reviews have systematically addressed SACs synthesis, characterization and support engineering [23–32], critical knowledge gaps persist in three key areas: Comparative analysis of SACs performance across thermal, photocatalytic and electrocatalytic systems; mechanistic elucidation of CO2-to-methanol conversion under different energy inputs; systematic evaluation of reaction condition impacts on catalytic metrics. Previous works predominantly focus on specific catalytic modalities [33–35] or general CO2 conversion chemistries [36–38], lacking systematic summarization and comparison of the mechanisms and affecting factors with SACs for CO2-to-methanol conversion via different technologies.
To bridge this knowledge gap, this review first summarizes the preparation methods of SACs, unique advantages in CO2 reduction and the single-atoms/support materials for CO2-to-methanol conversion in Supporting information. We then dissect reaction mechanisms governing SACs-mediated CO2-to-methanol conversion through thermal hydrogenation, photocatalytic and electrocatalytic technologies. A comparative analysis of recent breakthroughs across these domains is presented, with particular emphasis on microenvironmental influences and affecting factors on methanol production. Through systematic benchmarking of activity-selectivity-application triads, we delineate technology-specific advantages and operational constraints. Ultimately, the challenges in CO2-to-methanol conversion faced by SACs are addressed, providing clear optimization targets and improvement directions for future research. This review indicates directions for bridging the gap between laboratory-scale demonstrations and practical implementation in CO2-to-methanol conversion field.
Thermocatalytic CO2 hydrogenation to methanol represents a thermodynamically governed process that converts CO2 into methanol through H2-mediated reduction under elevated temperatures (150-340 ℃) and pressures (1.0-5.0 MPa). The technology is progressing toward industrial implementation through laboratory studies and pilot-scale demonstrations [39]. Complemented by density functional theory (DFT) calculations mapping reaction energetics, the conversion process is identified as three critical steps: CO2 chemisorption, H2 dissociation and selective intermediate hydrogenation [39]. Notably, the transformation of adsorbed CO2 into active intermediates on SACs surfaces constitutes the rate-determining step [40].
SACs enhance catalytic kinetics through three synergistic mechanisms: High-density isolated sites promote CO2 activation via strong chemical adsorption; facile heterolytic H2 dissociation generates reactive H*; precise control over intermediate hydrogenation steers selectivity toward methanol. For instance, in the Rh/In2O3 system, Rh atoms strengthened CO2 adsorption while enabling H* spillover [40]. The dual function facilitated sequential hydrogenation of activated CO2 through the HCOO* intermediate, achieving efficient methanol synthesis.
Macroscopically, CO2 hydrogenation proceeds via two distinct pathways. (1) Direct pathway: Concerted hydrogenation of CO2 and H2 on SACs yields methanol (Eq. 1). (2) Indirect pathway: a two-stage process involving: (a) Reverse water-gas shift (RWGS) to CO (Eq. 2); (b) Subsequent CO hydrogenation (Eq. 3).
|
|
(1) |
|
|
(2) |
|
|
(3) |
Reaction pathways in SACs-mediated systems for CO2-to-methanol conversion diverge depending on intermediate species [41,42]. As illustrated in Fig. 1, four distinct mechanisms dominate: r-Formate [43,44], formate [45,46], RWGS+CO-Hydro [47] and trans-COOH [48–50] pathways. The pathways are differentiated by two key intermediates: HCOO* (ΔG = -1.6~0.5 eV) and COOH* (ΔG = -0.8~0.4 eV). The HCOO*-dominated pathway with thermodynamic advantage prevails in most systems. For example, Pt/Mo-NC catalyst followed the r-Formate pathway [51], where Mo single atoms adsorbed CO2 and Pt nanoparticles facilitated H* generation. Similarly, in the Cu/GaZrOx system, methanol formation proceeded through the r-Formate pathway, where single-atom Cu sites (Cu1-O3) activated CO2 and Ga sites mediated H2 dissociation [52], highlighting the critical role of metal-support synergy. Compared to Cu clusters or nanoparticles, atomic Cu dispersion stabilizes intermediates, suppressing RWGS-derived CO formation to improve methanol selectivity.
Rational manipulation of reaction pathways in CO2 hydrogenation to methanol can be achieved through precise engineering of SACs local microenvironments to modulate energy barriers of key intermediates. Notably, when anchoring Cu-SACs with distinct coordination on C3N4, the atomic configuration determined product selectivity. The Cu-N4 sites enabled selective methanol production via the r-Formate pathway, while Cu-N3 configurations favored CO generation through RWGS [53]. The coordination-dependent catalytic behavior is further exemplified in Pt/MoS2 system [54]. The spatially isolated Pt single atoms directly mediated methanol synthesis through trans-COOH pathway, whereas clustered Pt atoms initiated a hydrogenation process from COOH* to formic acid before methanol formation. Remarkably, atomic-scale spatial engineering achieved synergistic catalysis, reducing the overall energy barrier and boosting catalytic activity.
Defect engineering emerges as a potent strategy to fine-tune CO2 adsorption and intermediate stabilization through controlled creation of oxygen vacancies (Ov) and sulfur vacancies (Sv). As demonstrated in Ov-rich In2O3 system, the strategic incorporation of Ni single atoms synergistically enhanced Ov concentration, facilitating adsorption and activation of CO2 and H* species at vacancy sites [55]. Interface engineering between Pd single atoms and Cu/ZrO2 carriers established Cuδ+ and Ov active centers, optimizing CO2 activation and directing HCOO* intermediate formation for methanol synthesis [56]. Intriguingly, spatially differentiated vacancy effects were observed in Cu/MoS2@SiO2 catalysts: edge-positioned Sv sites driven CH4 generation, while basal-plane Sv oriented for methanol production with a dual-path mechanism combining RWGS and CO hydrogenation [57].
Synergistic catalytic approaches enable dynamic pathway modulation, which is exemplified by cobalt-modified Pt@UiO-66-NH2(Co) hybrid system. Under thermal conditions, CO production dominated via COOH*-mediated pathway, whereas photo-thermal co-activation switched the dominant intermediate to HCOO* for methanol synthesis [58]. The photonic energy input lowered the optimal reaction temperature and suppressed side-reactions, achieving high selectivity toward methanol while reducing energy consumption versus conventional thermal catalysis. Such coupled systems embody the green chemistry principles through energy-efficient operation and waste minimization.
Table 1 systematically summarizes the catalytic performance of SACs in thermally driven CO2 hydrogenation to methanol. The superiority of SACs over conventional catalytic system has been revealed. A representative case involved Pd-Cu/ZrO2 catalyst, where atomic dispersion of Pd elevated methanol production rates from 0.22 mmol gcat-1 h-1 to 0.51 mmol gcat -1 h-1 under mild operational conditions (260 ℃, 1.5 MPa). The 132% enhancement was attributed to the creation of interfacial active centers facilitated by isolated Pd sites [56]. The catalytic synergy in SACs was further exemplified by Pd1/In2O3 system, where In-O-Pd Lewis acid-base pairs demonstrated bifunctional activation of CO2 and H2. The unique configuration achieved an exceptional performance of 33.0% CO2 conversion with 99.4% methanol selectivity at 300 ℃ and 3.0 MPa, outperforming conventional Pd nanoparticle counterparts [59]. Advanced support engineering through MZrOx (M = Zn, Cd, Ga) facilitated the SACs development via interfacial synergy. The optimized Pd-ZnZrOx catalyst exemplified the strategy, delivering a methanol yield of 21.53 mmol gcat-1 h-1 while maintaining high activity through 100 h testing. A 1.8-fold improvement was presented over bare ZnZrOx support, underscoring the critical role of metal-oxide/metal interface engineering in stabilizing active sites and promoting hydrogen dissociation [60].
 DownLoad:
CSV
DownLoad:
CSV
| SACs | Reaction conditions | Conversion rate (%) | Yield (mmol gcat-1 h-1) | Selectivity (%) | Ref. | |||
| CO2/H2 | GHSV (L h-1 gcat-1) | Pressure (MPa) | Temperature (℃) | |||||
| 2% Pt/Mo-NC | 1/3 | – | 3 | 180 | – | 0.27 | 88.0 | [51] |
| 12.1 wt% Cu/C3N4 | 1/3 N2% =4% | – | 3.2 | 150 | – | 4.20 | 95.5 | [53] |
| 0.2 wt% Pt/MoS2 | 1/3 | – | 3.2 | 150 | – | ≈0.41a | 95.4 | [54] |
| 1/3 | – | 3.2 | 210 | – | ≈37.42a | 93.0 | ||
| 7.5 wt% Pt/MoS2 | 1/3 | – | 3.2 | 150 | – | ≈5.25a | 81.3 | |
| 1/3 | – | 3.2 | 210 | – | ≈46.61a | 76.0 | ||
| Cu/MoS2@SiO2 | 1/4 | 24 | 5 | 260 | 12.7 | ≈13.04a | 72.5 | [57] |
| 0.96 wt% Rh/In2O3 | 1/4 Ar% ≈ 17% | 60 | 5 | 300 | 9.3 | 31.21 | 75.0 | [40] |
| 1/4 Ar% ≈ 17% | 30 | 5 | 270 | 3.7 | 5.67 | 71.0 | ||
| 1/4 Ar% ≈ 17% | 45 | 5 | 270 | 4.2 | 12.17 | 87.0 | ||
| 9.7 wt% Ni/In2O3 | 1/3 N2% = 4% | 21 | 5 | 300 | 18.5 | 17.17 | 54.0 | [55] |
| 1/3 N2% = 4% | 21 | 5 | <225 | – | – | 100.0 | ||
| Pd1/In2O3 | 1/5 | – | 3.3 | 200 | 33.0 | ≈63.62a | 99.4 | [59] |
| 0.58 wt% Pt/In2O3 | 1/3 Ar% = 4% | 54 | 2 | 220 | <1.0 | – | 91.1 | [61] |
| 1/3 Ar% = 4% | 54 | 4 | 300 | >9.0 | 23.72 | 65.0 | ||
| 0.75 wt% Pd/In2O3 | 1/4 | 48 | 5 | 280 | 9.7 | 29.96 | 78.0 | [62] |
| 6 wt% Ni/In2O3 | 1/3 N2% = 20% | 48 | 3 | 250 | – | 7.80 | – | [63] |
| 1 wt% Re/In2O3 | 1/4 | 21 | 5 | 300 | – | 16.85 | 72.1 | [64] |
| Ir1Pd1-In2O3 | 1/3 | – | 3 | 250 | 10.5 | – | 97.0 | [69] |
| 1.0 wt% Mo/TiO2 anatase | 1/3 N2% = 20% | 7.5 | 3 | 275 | 5.2 | 0.35 | 10.2 | [65] |
| 2.9 wt% Mo/TiO2 rutile | 1/3 N2% = 20% | 7.5 | 3 | 275 | 6.8 | 1.09 | 24.2 | |
| 0.83 wt% Cu1/ZnO | 1/3 H2O% = 0.11% | 6 | 3 | 170 | 4.9 | – | 99.1 | [72] |
| 1/3 | 6 | 3 | 170 | 1 | – | 93.5 | ||
| 2% Cu/GaZrOx | 1/3 | 6 | 3 | 300 | – | – | 87.0 | [52] |
| Pd-Cu/ZrO2 | 1/3 | 6 | 1.5 | 260 | ≈5 | 0.51 | 38.0 | [56] |
| Pd-ZnZrOx | 1/4 Ar% = 5% | 24 | 5 | 320 | – | 21.54 | 85.0 | [60] |
| 10% Co/5% In-ZrO2 | 1/4 | 24 | 3 | 300 | – | 4.68 | 86.0 | [70] |
| CuZnZrOx | 1/3 | – | 4.5 | 290 | 9.5 | – | 76.0 | [73] |
| 5 mol% Zn-ZrOx | 1/4 | 24 | 5 | 320-340 | – | 14.36 | 80.0 | [74] |
| Cu@FAU | 1/3 | 12 | 3 | 240 | 11.5 | 12.80 | 89.5 | [66] |
| MOF-808-NaCu | 1/3 | – | 3.5 | 275 | – | 9.55 | 93.0 | [67] |
| 1/3 | – | 3.5 | 250 | 4.1 | – | 93.0 | ||
| NU-1000-NH2/PrS-Cu | 1/3 | 12 | 1 | 280 | – | 3.12 | ≈100 | [68] |
| Pt1@MIL101 | 1/3 | – | 3.2 | 150 | – | 1.08 | 90.3 | [75] |
| Pt@UiO-66-NH2(Co)b | 1/3 | – | 1.5 | 240 | – | 1.91 | 74.8a | [58] |
| a The symbol “≈” in the table indicates methanol yields calculated based on data from the references. b The results from reference are obtained under experimental conditions of a 300 W Xenon lamp with a light intensity of 1.0 W/cm2. |
||||||||
The catalytic performance of SACs is governed by three critical factors: The electronic configuration of metal centers, the physicochemical properties of support materials and the precise control of metal loading density. As summarized in Table 1, widely investigated supports such as In2O3, MoS2, TiO2, C3N4, zeolites, Metal-organic frameworks (MOFs) and MZrOx solid solutions exhibited distinct structure-activity relationships in CO2 hydrogenation. Remarkably, In2O3-supported SACs demonstrated exceptional efficiency for methanol synthesis. Isolated transition metal sites (e.g., Rh [40], Pt [61], Pd [59,62], Ni [55,63], Re [64]) synergistically activated H2 dissociation and CO2 chemisorption at the metal-support interface, achieving dramatic enhancements in methanol productivity. A paradigmatic example was Pd1/In2O3, which delivered an unprecedented methanol yield of 63.62 mmol gcat-1 h-1 with 99.4% selectivity [59]. Notably, the metal loading amount exhibits a volcano-type relationship with catalytic performance. For instance, Pt/In2O3 with optimal loadings (0.13-0.58 wt%) maintained high CO2 conversion, whereas excessive Pt loading (2.50 wt%) triggered RWGS dominance, as evidenced by escalated CO selectivity and compromised stability [61].
The architectural engineering of catalyst support critically dictates both activity and product distribution. Crystalline phase modulation and coordination environment optimization emerge as powerful strategies to tailor reaction pathways. Comparative studies revealed that rutile-phase TiO2 nanorods supporting 2.9 wt% Mo (Mo/TiO2-rutile) enabled a methanol production rate of 1.09 mmol gcat-1 h-1 [65], outperforming the anatase counterparts by 3.1-fold. In contrast, C3N4-supported Cu-N4 moieties achieved methanol production rate of 4.20 mmol gcat-1 h-1 and selectivity of 95.5% at 150 ℃ [53]. Spatial confinement strategies further enhance performance. Zeolite-encapsulated Cu@FAU attained methanol production rate of 12.8 mmol gcat-1 h-1 via reactant enrichment in micropores [66]. Besides, the MoS2@SiO2 composite selectively exposed basal Sv, suppressing methanation side reactions and achieving 72.5% methanol selectivity [57]. Notably, MOF-808-NaCu demonstrated dual functionality: Na+ ions simultaneously stabilized Cu sites and modulated local electron density, achieving concurrent enhancements in activity (9.55 mmol gcat-1 h-1) and selectivity (93.0%) [67]. Moreover, the MOF-derived NU-1000-NH2/PrS-Cu, engineered through incorporation of amino (-NH2) and propylthiol (-PrSH) functional groups, acquired methanol production rate of 3.12 mmol gcat-1 h-1 with 100% selectivity at 280 ℃ [68].
Dual SACs transcend the limitations of conventional SACs by enabling cooperative catalysts. The strategic pairing of adjacent metal sites not only preserves single-atom characteristics but also facilitates the adsorption of intermediates. In a groundbreaking design, photo-induced deposition of Ir-Pd dimers on In2O3 boosted CO2 conversion (10.5%) and methanol selectivity (97.0%) [69]. Similarly, for Co-In-ZrO2 catalyst, the Co sites dominated CO2 adsorption and HCOO* stabilization while neighboring In atoms facilitated the hydrogenation of intermediates, elevating methanol selectivity to 86.0% [70].
Photo-assisted thermal catalytic systems harness synergistic effects to achieve concurrent enhancement of reaction selectivity and kinetic performance under mild conditions. SACs frequently exhibit remarkable activity enhancements when subjected to photon irradiation, as exemplified by the Pt@UiO-66-NH2(Co) system. Under conventional thermal catalysis conditions (240 ℃, 1.5 MPa), the system predominantly facilitated CO2 reduction to CO via the COOH* intermediate pathway. Remarkably, the implementation of photo-thermal synergy improved the methanol production rate to 1.91 mmol gcat-1 h-1, corresponding to a 7.8-fold enhancement over thermal catalytic performance [58]. The mechanistic divergence originated from light-induced modifications in both the CO2 activation pathway and the transformation dynamics of key intermediates, demonstrating the critical role of photonic energy in steering reaction selectivity.
The methanol synthesis efficiency exhibits multivariate dependence on reaction parameters encompassing temperature, pressure, gas hourly space velocity (GHSV), CO2/H2 ratios and reactor design. Temperature emerges as the pivotal kinetic determinant, governing product distribution through dual modulation mechanisms: thermodynamic equilibrium constraints and competitive reaction pathway selection. The system manifests fundamental competition between exothermic CO2 hydrogenation (ΔH < 0; Eq. 1) and endothermic RWGS processes (ΔH > 0; Eq. 2). Lower temperature regimes (<220 ℃) thermodynamically favor methanol synthesis yet suffer from CO2 activation barrier. Conversely, elevated temperatures (>280 ℃) enhance reaction kinetics at the expense of thermodynamic selectivity, promoting CO formation through RWGS dominance. Catalytic performance studies have revealed the temperature-dependent regularity. A 0.58 wt% Pt/In2O3 system achieved 91.1% methanol selectivity with <1% CO2 conversion at 220 ℃, while thermal intensification to 300 ℃ elevated conversion rate to >9% but reduced the selectivity to 65% [61]. Analogous behavior in 9.7 wt% Ni-In2O3 system demonstrated complete methanol selectivity below 225 ℃, contrasting with 46% CO selectivity at 300 ℃ [55]. These findings underscore the critical need for quantitative temperature-activity-selectivity correlations to enable synergistic optimization of conversion efficiency and product selectivity.
Pressure modulation provides thermodynamic leverage, shifting equilibrium toward methanol synthesis via increasing the pressure [71]. Optimization of GHSV can regulate reactant-catalyst interaction duration, consequently dictating the products distribution. Elevated GHSV (45 vs. 30 L h-1 gcat-1) in Rh/In2O3 (0.96 wt%) system reduced over-hydrogenation side reactions, enhancing methanol selectivity from 71% to 87% at 270 ℃ [40]. Feedstock composition studies identify CO2/H2 = 1:3-1:5 as the optimal stoichiometric scope, balancing H2-mediated kinetic enhancement against competing side reactions. Notably, introducing 0.11 vol% H2O vapor in Cu1/ZnO mediated system achieved dual enhancement of CO2 conversion and methanol selectivity (99.1%), which was attributed to water-mediated pathway modulation [72].
Reactor selection imposes critical process constraints. Fixed-bed systems offer structural simplicity and catalyst recyclability but suffer from inherent heat transfer limitation. Batch reactors enable enhanced mass/heat transfer and gradient-free operation yet present catalyst recovery challenges and limited operational continuity [27]. Establishing a process intensification framework of thermodynamic modeling, kinetic optimization and reactor engineering is beneficial for methanol synthesis [73–75].
Photocatalytic reduction of CO2 to methanol represents a solar-driven carbon-neutral technology with transformative potential for sustainable energy storage [76–78]. The process faces fundamental thermodynamic challenges due to the exceptional stability of CO2 molecules. The C=O bonds in CO2 molecule exhibits dissociation energies of 750 kJ/mol, which is significantly higher than C-C (336 kJ/mol) and C-H (430 kJ/mol) bonds [79]. The conversion of CO2 to methanol entails the transfer of multiple electrons and an equivalent number of protons (CO2 + 6H+ + 6e− → CH3OH + H2O) [80]. Although the proton-assisted multi-electron reduction process operates at a lower potential (-0.38 V), the efficient generation of methanol still necessitates a suitable catalyst.
The performance of photocatalytic systems is fundamentally determined by the band structure and active sites of catalytic materials. Conventional photocatalysts frequently suffer from suboptimal charge carrier dynamics and insufficient density of active sites, limiting their practical applicability. SACs emerge as a transformative paradigm, combining maximized atom utilization efficiency with atomic-level precision in active site engineering. The unique attributes enable SACs to orchestrate directional electron transfer from semiconductor supports to isolated metal centers. The electron-enriched catalytic sites dramatically enhance CO2-to-methanol conversion, which is a critical advancement overcoming traditional catalyst limitations [81–85].
Mechanistic studies reveal that metal single atoms induce band structure modulation, extending photo-absorption spectra while prolonging charge carrier lifetimes through quantum confinement effects. The functionality synergistically enhances the photon harvesting and redox capability for CO2 conversion. The reduction mechanism was illustrated in Fig. 2. Upon the energy of incident light exceeding the bandgap energy, SACs mediated electrons transfer from valence band (VB) to conduction band (CB). The separated charge carriers subsequently migrate to distinct catalytic sites and drive CO2 reduction. While holes oxidize H2O/sacrificial agents, generating essential protons for methanol formation. The compartmentalized charge utilization suppresses recombination losses, completing the photocatalytic reduction through proton-coupled electron transfer (PCET) processes [86–89].
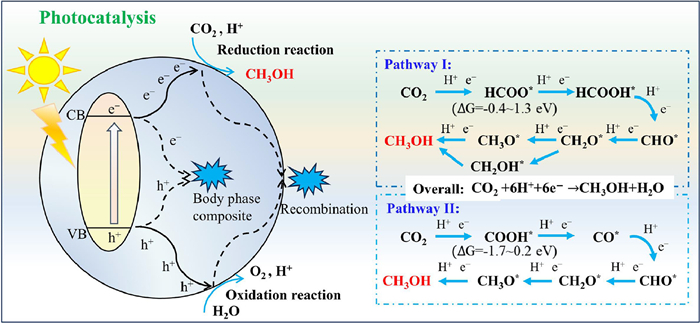
The reaction initiates with thermodynamically challenging CO2 activation, requiring electron transfer to the lowest unoccupied molecular orbital (LUMO) of CO2 to form the CO2•− radical anion (-1.90 V vs. NHE). SACs overcome the rate-determining step through orbital hybridization with adsorbed CO2, forming surface-stabilized *CO2δ− intermediates [90]. The interaction induces molecular bending (O-C-O angle reduction) that lowers the LUMO energy through rehybridization from sp to sp2, thereby reducing the activation barrier. Coordination chemistry dictates subsequent pathways, with oxygen-/carbon-/mixed-coordination modes of *CO2δ− on SACs steering intermediate evolution [91,92].
As shown in Fig. 2, the photocatalytic CO2-to-methanol pathway is dictated by the binding configurations between SACs and activated *CO2δ−, yielding two distinct mechanistic routes: HCOO* (ΔG= -0.4~1.3 eV) and COOH* (ΔG = -1.7~0.2 eV) mediated pathways [89,93–95]. Although the COOH* exhibits a lower free energy, the HCOO* pathway demonstrates broader prevalence across catalytic conditions. For instance, Pd1/PCN (Pd single atom anchored on polymeric carbon nitride (PCN)) demonstrated enhanced charge separation by channeling photogenerated electrons from the PCN conduction band to Pd sites under visible light. The electron redistribution facilitated CO2 activation at Pd centers, initiating sequential proton-coupled hydrogenation steps that culminated in methanol desorption [96]. In contrast, CO2 activation on CuSAs/UiO-66-NH2 followed the COOH* pathway. First-principles calculations revealed that Cu sites reduced energy barriers of rate-limiting steps, particularly during COOH* and CHO* formation [97]. Furthermore, Cu sites functioned as electron-trapping centers, prolonging charge carrier lifetime and enhancing methanol production.
The photocatalytic CO2-to-methanol conversion performance of various SACs was systematically summarized in Table 2. The monoatomic metals applied for photocatalytic reduction are predominantly Cu, followed by Ru, Pd, Co and Pr [98]. Introducing metal single atoms is beneficial for photocatalytic performance improvement. For instance, when Cu single atoms were introduced into UiO-66-NH2, the bandgap was narrowed, promoting electron enrichment and conversion efficiency [97]. The methanol production rate catalyzed by Cu-SAs/UiO-66-NH2 was 5.33 µmol gcat-1 h-1, which was much higher than that of Cu-NPs/UiO-66-NH2 and pristine UiO-66-NH2. Similarly, the introduction of Ru into C3N4 lowered the band gap of the catalyst (RuSA-mC3N4), enhanced light absorption in the visible region, promoted photogenerated electron production and achieved a high methanol yield (250.0 µmol gcat-1 h-1) [99]. In addition, the methanol yield was several times higher than that of the prepared CNF(ZnO) by placing dispersed Ru single atoms on stabilized CNF(ZnO) nanocages, reaching 62.25 µmol gcat-1 h-1 [100].
DownLoad:
CSV
| SACs | Light source | Reaction medium | AQY (%) | Selectivity (%) | Productivity (µmol gcat-1 h-1) | Ref. |
| Pd1/PCN | 300 W xenon lamp (λ > 420 nm) | KHCO3/Na2SO3 | – | 85.1 | 742.4 | [96] |
| Pr1-N4O2/CN | 300 W xenon lamp | ACN/H2O | – | 92.4 | 511.1 | [98] |
| RuSA-mC3N4 | 34 W blue LED light (400-500 nm light density: 22 mW/cm2) | DMF/H2O | – | – | 250.0 | [99] |
| Er1Nd3/CN | 300 W xenon lamp (λ > 420 nm) | TEOA/H2O | – | – | 987.7 | [104] |
| Co/g-C3N4 | 300 W xenon lamp (light density: 1500 mW/cm2) | H2O | – | 96.2 | 235.5 | [105] |
| CuSAs/UiO-66-NH2 | 300 W xenon lamp (λ > 400 nm) | TEOA/H2O | – | – | 5.3 | [97] |
| Cu1@BiOBr | 300 W xenon lamp | H2O | 12.2 | 90 | 627.6 | [103] |
| CuSAs@PCN@UiO-66-NH2 | 300 W xenon lamp (420-800 nm) | TEA/ACN/H2O | – | – | 4150.0 | [106] |
| Ru/CNF(ZnO) | 400 W xenon lamp (400-440 nm) | IPA/ACN/H2O | 1.4 | 85 | 62.3 | [100] |
| Apparent quantum yield (AQY) is the ratio of the number of electrons participating in the reaction to the total number of incident photons at a specific wavelength. | ||||||
Cordination microenvironment of single-atom catalytic centers exerts a profound influence on the electronic density, which directly governs photocatalytic performance [101,102]. Modulation of electron density at active sites through ligand engineering has emerged as an effective strategy for activity enhancement. For instance, oxygen-coordinated Pr (Pr1-N4O2/CN) demonstrated superior electron accumulation capability compared to the nitrogen-coordinated counterparts (Pr1-N6/CN), achieving methanol production rate of 511.1 µmol gcat-1 h-1. The performance surpassed that of pristine carbon nitride (CN) by 28.9-fold and Pr1-N6/CN by 1.9-fold [98]. Similarly, Cu single atoms coordinated with four oxygen atoms in BiOBr nanosheets (Cu1@BiOBr) functioned as efficient electron traps, delivering a methanol yield of 627.6 µmol gcat-1 h-1 [103]. Moreover, the Er-Nd dual SACs fabricated through atomic confinement and coordination strategies, achieved an exceptional methanol production rate of 987.7 µmol gcat-1 h-1 under visible-light irradiation [104].
Precise control the loading quantity of single atoms on support is critical for optimizing atomic dispersion and activity. Either insufficient or excessive loading of Co on the g-C3N4 substrate resulted in reduced active atomic sites. Intriguingly, the optimized Co/g-C3N4-0.2 with cobalt loading of 24.6 wt% presented exceptional methanol production (235.5 µmol gcat-1 h-1) [105]. Furthermore, a “chemical scissors” strategy was developed to achieve site-specific anchoring of Pd atoms on PCN. Through sequential metal loading and selective etching, the approach eliminated secondary metal clusters while preserving isolated Pd atoms, leading to methanol production rate of 742.4 µmol gcat-1 h-1 [96]. The strategy demonstrated exceptional precision in balancing metal loading and atomic utilization efficiency.
Notably, the integration of single-atom catalysis with heterojunction engineering has unlocked unprecedented catalytic synergies. A breakthrough was achieved by the skillfully-designed CuSAs@PCN@UiO-66-NH2, a ternary composite combining Cu single atoms, PCN and MOFs. The configuration capitalized on both heterojunction-induced charge separation and atomic-level active sites, achieving a record methanol yield of 4150 µmol gcat-1 h-1 under optimized conditions [106]. To our knowledge, it represents the highest efficiency reported for photocatalytic CO2-to-methanol conversion to date.
Beyond optimization of the SACs, the implementation of sacrificial electron donor (SED) has emerged as an approach to suppress charge recombination and amplify electron utilization. SED as hole scavenger prolong electron-hole separation lifetime and elevate the quantum yield [107]. Commonly employed SED include triethanolamine (TEOA), triethylamine (TEA), Na2SO3, ethylene-diaminetetraacetic acid (EDTA), isopropanol (IPA) and ascorbic acid. Concomitantly, the incorporation of polar organic solvents (e.g., acetonitrile (ACN), N,N-dimethylformamide (DMF), ethyl acetate (EA)) facilitates homogeneous dispersion of photocatalysts within reaction systems. The solvation effect enhances interfacial charge transfer while mitigating particle aggregation, contributing to catalytic performance improvement. Systematic investigations revealed solvent-dependent methanol yield variations in CO2 photoreduction. Controlled experiments demonstrated that ACN/H2O co-solvent system achieved a higher methanol yield compared to DMF/H2O, attributed to ACN’s superior dielectric properties and coordination capabilities [100]. Besides, comparative evaluation of sacrificial agents identified IPA as the most effective electron donor, delivering a greater methanol production rate than TEA and TEOA [100]. The phenomenon may originate from IPA’s optimal redox potential alignment with the catalyst’s valence band edge, enabling efficient hole scavenging while minimizing side reactions.
The methanol production rate exhibits significant dependence on the aqueous solubility of CO2, which remains intrinsically limited under conventional solid-liquid phase conditions. Alkaline additives (e.g., NaOH, Na2CO3, KHCO3) are employed to promote carbonate speciation through proton abstraction, enhancing CO2 dissolution. However, the intricate multistep ionization equilibria of dissolved CO2, H2CO3, HCO3− and CO32− introduce substantial complexity in quantifying initial CO2 concentrations [108]. Thermodynamic modulation via elevated pressure or temperature adjustment presents an alternative pathway to enhance CO2 adsorption on catalyst surface [109–111]. Despite the critical role of CO2 concentration gradients in methanol synthesis, systematic investigation in SACs mediated system remain absent in literature.
Under ambient condition, electrocatalytic CO2 reduction enables directional methanol synthesis via potential-driven redox reactions with electrodes. The marginal potential difference (+20 mV) between CO2 reduction and water decomposition enables predominant hydrogen evolution reaction (HER), resulting in extra electrical energy consumption [112]. High faraday efficiency (FE) achievement necessitates HER-suppressing electrode materials that prioritize electron allocation toward CO2 reduction. The intrinsic property establishes SACs-based electrocatalytic system as superior candidate for efficient CO2-to-methanol conversion. Furthermore, the technology demonstrates inherent compatibility with renewable sources (e.g., solar/wind), holding significance for carbon neutrality and sustainable development [113–116].
The electrocatalytic CO2 reduction proceed through sequential stages: (1) CO2(g) diffuses through the bulk electrolyte to the gas-liquid interface, followed by dissolution and migration across the electrolyte boundary layer to the catalyst surface, where it undergoes chemisorption at active sites; (2) The adsorbed CO2* accepts electrons to form primary intermediates, subsequently undergoing proton-coupled electron transfer (H+/e−) to generate diverse intermediates and further turn into methanol [117]; (3) Product desorption occurs through diffusive transport into the bulk electrolyte or gas phase, completing the catalytic cycle. In the methanol synthesis system, the cathodic half-reaction follows Eq. 4, while the anodic oxygen evolution reaction (OER) adheres to Eq. 5, yielding the overall reaction described by Eq. 6.
|
|
(4) |
|
|
(5) |
|
|
(6) |
SACs with maximized atomic utilization can optimize CO2 adsorption energy and accelerate charge transfer. The distinctive metal-ligand coordination architecture and tailored electronic states in SACs favor reaction pathway optimization, thereby enhancing CO2 conversion efficiency. As exemplified by Cu anchored on porous carbon nanofibers (CuSAs/TCNFs), the microporous nanofibers with atomic-scale Cu sites contributed to CO2 chemisorption. The mononuclear Cu centers exhibited near-optimal CO* binding energy, enabling methanol-selective catalysis [118]. Similarly, atomic Cu sites of copper cyanamide crystals (Cu2NCN) enhanced electron delocalization, which modulated the CO2 reduction pathways toward methanol/methane [119]. Furthermore, SACs with defective support are favorable for the interface reaction process, As demonstrated by Sn-atom-decorated CuOx catalyst (Sn1/Ov CuO-90), atomic Sn sites coupled with oxygen vacancy achieved higher double-layer capacitance, greater CO2 adsorption capacity and lower interfacial charge transfer resistance, enhancing the CO2 reduction efficiency [120].
During CO2 electroreduction, the binding strength of active sites toward reaction intermediates governs mechanistic pathways and determines product selectivity. The catalytic mechanism of SACs for CO2-to-methanol conversion (Fig. 3) reveals two distinct favorable routes: COOH* (ΔG = 0.1-0.5 eV) and HCOO* (ΔG = -0.1~0.7 eV) pathways. In the COOH* route, CO* adsorption behavior dictates methanol selectivity. The Cu-N4 configuration in CuSAs/TCNFs exhibited moderate CO* binding with ΔG(CO*) = +0.12 eV, suppressing both gaseous CO desorption and C-C coupling side reactions while simultaneously blocking HCOO* formation to prevent formic acid generation [118]. It ensured precise reaction through the hydrogenation sequence as pathway Ⅰ in Fig. 3. Conversely, SA-Cu-MXene preferentially activated the HCOO* pathway through stronger stabilization of HCOO* (ΔG(HCOO*) = -0.01 eV vs. ΔG(COOH*) = +0.24 eV), driving the transformation sequence as pathway Ⅱ in Fig. 3 [121]. The HCOOH*-to-CHO* conversion emerged as the rate-determining step, where the unsaturated Cu sites enabled activation barrier decrease. It demonstrated that SACs composition and coordination exerted pathway regulation through intermediate stabilization, providing fundamental principles for targeted catalyst engineering toward optimized product distributions.
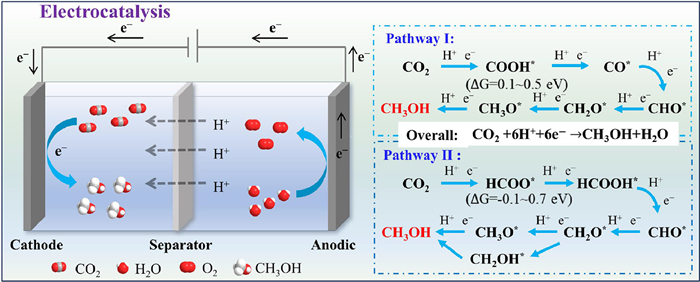
Notably, Dutta et al. demonstrated through theoretical calculations that transition metal SACs (Co, Ni) anchored on C6N6 nanosheets electrocatalytically CO2-to-methanol conversion via pathway Ⅱ [122]. These catalysts exhibited strong visible-light absorption, suggesting the potential applicability in photocatalytic CO2 reduction. It highlighted a promising strategy for achieving efficient CO2 reduction through photo-electrocatalytic synergy.
Recent advancements in SACs for CO2 electroreduction have focused on optimizing activity, selectivity and stability. Cu- and Sn-based SACs demonstrated exceptional methanol synthesis performance [118–121,123]. As summarized in Table 3, the SA-Cu-MXene catalyst achieved a methanol FE of 59.1% at -1.4 V vs. RHE, while maintaining operational stability with current density exceeding Cu particle-MXene and pristine MXene [121]. The porous carbon structure and abundant Cu active sites of CuSAs/TCNFs enhanced CO2 adsorption capacity, yielding a methanol production rate of 68.4 µmol m-2 s-1 at FE of 44.0% [118]. Further improvements in electron transfer efficiency of Cu2NCN reached methanol FE of 70.0% [119]. The Sn1/Ov CuO-90 system demonstrated synergistic enhancement from Sn atomic sites, oxygen vacancy and CuO support, achieving methanol FE of 88.0% through optimized CO2 adsorption and reduced charge transfer resistance [120]. The dual SACs CuIn@ZIF-8, incorporating Cu and In into ZIF-8 frameworks, achieved methanol FE of 55.6% at -0.62 V vs. RHE. The introduction of In atoms not only suppressed the HER but also established electron-enriched Cu sites and electron-deficient In sites through interfacial charge transfer, thereby optimizing the catalytic performance [124].
DownLoad:
CSV
| SACs | Electrolyte | Applied potential (V vs. RHE) | Current density (mA/cm2) | FE (%) | Productivity (µmol m-2 s-1) | Ref. |
| CuSAs/TCNFs | 0.1 mol/L KHCO3 | −0.9 | -93.0 | 44.0 | 68.4 | [118] |
| Cu2NCN | 0.5 mol/L KHCO3 | −0.9 | -92.3 | 70.0 | 1600 | [119] |
| Sn1/Ov CuO-90 | [Bmim]BF4 (25 mol%) aqueous solution | −2.0 | 67.0 | 88.6 | – | [120] |
| SA-Cu-MXene | 0.1 mol/L KHCO3 | −1.4 | 21.3 | 59.1 | – | [121] |
| Cu3(HHTQ)2 | 0.1 mol/L KHCO3 | −0.4 | 45.0 | 53.6 | – | [123] |
| CuIn@ZIF-8 | 0.5 mol/L KHCO3 | −0.6 | – | 55.6 | – | [125] |
| Note: Faradaic efficiency (FE) is the ratio of charge utilized for target product formation to the total charge passed. | ||||||
In addition to the SACs involved in the reaction, the electrolyte solution has a critical impact on the reaction process. High-conductivity electrolytes effectively reduce polarization resistance and elevate current density, improving methanol yield and selectivity [125,126]. Based on existing research, the electrolytes are mainly divided into three primary categories: Aqueous, ionic liquid and organic solvent electrolytes. The widely used aqueous electrolytes benefit from inherent proton-donating capability of water but suffer from intrinsic CO2 solubility limitations (~34 mmol/L at 25 ℃, 1 atm), resulting in constrained mass transfer [127]. To mitigate it, aqueous systems are commonly modified through the addition of pH-regulating agents (KHCO3, NaHCO3, KOH) that simultaneously serve as proton donors. While offering advantages of eco-compatibility, cost-effectiveness and high current density tolerance, the aqueous electrolytes typically exhibit suboptimal product selectivity and current stability.
In comparison, ionic liquid electrolytes exhibit superior performance through enhanced CO2 solvation with solubility of 4-5 times higher than that in aqueous solution and optimized charge transfer characteristics. When employing Sn1/Ov-CuO as catalyst, the [Bmim]BF4/H2O system achieved 88.6% methanol Faradaic efficiency at 67.0 mA/cm2, significantly outperforming 0.5 mol/L KHCO3 aqueous solution system that yielded only 16.4 mA/cm2 with undetectable methanol production [120]. The performance enhancement resulted from three synergistic mechanisms: Improved CO2 adsorption capacity, hydrogen evolution reaction suppression and facilitation of multi-electron transfer pathways.
The reactor mainly includes H-type cells and membrane electrode assembly (MEA) based electrolyzers. The H-type cells offer structural simplicity and convenient control, which are ideal for fundamental studies and catalyst evaluation. Conversely, MEA electrolyzers are demonstrated superior technological application with higher current densities and enhanced product selectivity while effectively suppressing hydrogen evolution [119]. Additional parameters including electrolyte concentration, ion speciation, pH and thermal conditions may further influence CO2 reduction efficiency. However, systematic investigations of these factors in SACs-based electrocatalytic systems remain absent from current literatures, highlighting a critical research gap requiring urgent attention.
This review provides a comprehensive analysis of SACs in CO2-to-methanol conversion systems, with emphasis on mechanistic insights and technical determinants across thermal, photocatalytic and electrocatalytic technologies. SACs maximize atomic efficiency through precisely coordinated active sites and quantum confinement effects. The tunable electronic configuration at metal-support interfaces optimizes adsorption-desorption equilibria for critical intermediates, enabling ΔG values approaching favorable levels for methanol production. Thermal catalysis achieves industrial-scale methanol production under harsh conditions, yet suffers from thermodynamic limitations and catalyst sintering. Photocatalysis exhibits remarkable selectivity through bandgap-engineered charge separation in ambient-condition, but remains constrained by lower quantum yields and inefficient light harvesting. Electrocatalysis demonstrates electrolytes-tunable product distributions (ionic liquid vs. KHCO3 solution), while competitive HER necessitates advanced strategies for CO2 activation and proton transfer regulation.
Based on the current research progress, future development should focus on the precise design of active sites based on single atoms and supports, the synergy of multiple technological pathways including photonic, thermal and electrical methods, the development of intelligent catalytic systems and the in-situ conversion systems for low-concentration CO2 in the atmosphere or industrial waste gases. These efforts will propel the application of SACs in CO2 utilization, providing theoretical guidance and technical references for the development of efficient, low-carbon catalytic systems. We have outlined the possible challenges and directions for future research.
(1) It is necessary to establish a theoretical model for the structure-activity relationship of SACs, with a focus on breakthroughs in metal center screening, coordination microenvironment modulation and carrier engineering strategies. For instance, employing carriers with high specific surface areas and abundant basic sites can enhance the adsorption capacity for CO2, while interface modification and defect engineering can promote the anchoring of single atoms. Designing efficient precursors, introducing in-situ synthesis and plasma-assisted techniques can achieve uniform distribution and utilizing in-situ characterization technologies can enable real-time monitoring and regulation of the site formation process.
(2) Photo-assisted thermal catalysis and electrocatalysis can enhance reaction efficiency, offering strong controllability and improving methanol production rates. These methods also enable more efficient energy utilization, reduce environmental impact and align with the principles of sustainable development. The synergistic effects provide a new direction for optimizing energy utilization efficiency and enhancing reaction performance. However, the complex kinetics and interfacial behaviors of the reaction process require systematic investigation. It is necessary to establish kinetic models for multi-technology coupling to clarify the synergistic mechanisms of interfacial charge transfer and mass transport processes. In the future, in-situ characterization techniques can be employed to quantitatively analyze the dynamic adsorption behaviors of key intermediates under the synergy of multi-technology.
(3) In the future, artificial intelligence (AI) could be employed to enhance the efficiency of SACs in converting CO2 to methanol. Machine learning (ML) as a pivotal branch of artificial intelligence, enables computational systems to autonomously improve performance through experiential learning by extracting patterns from data and optimizing models. As a specialized ML paradigm, reinforcement learning (RL) empowers agents to develop optimal strategies through iterative environmental interactions. Notably, graph neural networks (GNNs) have emerged as transformative deep learning architectures for processing graph-structured data, demonstrating exceptional capability in deciphering complex structural features of catalysts, thereby gaining significant traction in chemical and materials science research. The application of these techniques promises transformative advances in SACs design, particularly in: high-throughput screening of high-performance SACs, precise prediction of adsorption energies and stability metrics for metal atoms at distinct anchoring sites, optimized tailoring of metal-support coordination environments, atomic-scale simulation of catalytic CO2 reduction pathways. Nevertheless, critical challenges persist, including data scarcity and quality limitations, computational expense of multiscale modeling, interpretability constraints in black-box algorithms and experimental validation bottlenecks. Future progress requires coordinated efforts to refine data acquisition/curation strategies, optimize algorithm architectures and foster cross-disciplinary convergence between computational catalysis, synthetic chemistry and advanced characterization techniques. Addressing the priorities will unlock the full potential of ML-accelerated catalyst discovery, ultimately advancing the rational design of SACs for efficient CO2-to-methanol conversion.
(4) SACs have demonstrated exceptional catalytic performance in laboratory studies, but their large-scale industrial application faces numerous challenges including the impact of reactor scale-up, the influence of reaction conditions, long-term stability of SACs and efficient recycling and reuse. Detailedly, the mass and heat transfer rates in industrial-scale reactors may change along with operational variations, necessitating synergistic optimization between active site distribution of SACs and large-scale reactor design. Besides, common SACs face significant challenges in post-reaction separation from liquid-phase systems, which can be mitigated through material engineering approaches such as employing magnetic carriers or implementing surface immobilization on membrane substrates. Furthermore, it is imperative to establish comprehensive cost models incorporating critical parameters such as synthesis expenditure, metal atom utilization efficiency and operational lifespan to systematically evaluate the economic viability of SACs. Notably, current SACs implementations demonstrate substantially higher overall costs (approximately 2-10 times greater than traditional heterogeneous catalysts) with limitations in synthetic methodologies and production scalability. However, emerging technological advancements (e.g., ultra-rapid synthesis techniques for energy minimization and breakthrough innovations in low-cost wet chemical methods addressing production bottlenecks) show promising potential to narrow this economic disparity. By incorporating life cycle assessment (LCA), the carbon footprint and cost competitiveness of SACs-driven methanol synthesis can be evaluated, aiming to reduce the carbon footprint and costs. A comprehensive “cradle-to-grave” assessment framework should be established encompassing four critical phases: precursor preparation, SACs synthesis processes, CO2 reduction operations (energy consumption/by-products), and catalyst recycling/reutilization. SACs exhibit significant advantages over conventional catalysts in reducing carbon emission intensity (CEI) and shortening energy payback periods. To enhance sustainability, we advocate a green design paradigm of “renewable carbon-based supports + non-noble metals” for SACs. Promising examples include biomass-derived carbon supports coupled with Cu single-atom sites, which synergistically combine environmental compatibility with catalytic efficiency.
(5) Currently, most catalytic systems rely on large amounts of high-purity CO2. In the future, it is essential to consider direct capture of CO2 from industrial exhaust or ambient air for methanol synthesis. Exploring catalytic conversion technologies coupled with low-concentration industrial exhaust or direct air capture (DAC) and integrating MOFs adsorbents (with CO2/N2 selectivity >200) with bifunctional SACs catalysts, can be a promising approach. Coupling with green hydrogen sources (such as hydrogen produced via water electrolysis) can achieve full-chain decarbonization.
In summary, SACs provide a new paradigm of “atomic economy” for converting CO2 to methanol, offering a revolutionary solution for achieving carbon neutrality goals. The industrial application of SACs requires breakthroughs in three dimensions: material design, technology synergy and system engineering. This holds significant strategic importance for building a green methanol economy and driving the deep decarbonization transformation of the energy and chemical industries.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Jingying Wang: Writing – original draft, Methodology, Conceptualization. Jianhui Zhao: Writing – review & editing, Supervision. Shaopo Wang: Writing – review & editing, Formal analysis. Jingjie Yu: Writing – review & editing, Conceptualization. Ning Li: Writing – review & editing, Supervision.
This work was supported by the National Natural Science Foundation of China (No. 52300170).
Supplementary material associated with this article can be found, in the online version, at doi:
H. Akbar, M.S. Javed, S.T. Iqbal, et al., J. Korean Ceram. Soc. 61 (2024) 367–390. doi: 10.1007/s43207-024-00365-1
J. Ma, N. Sun, X. Zhang, et al., Catal. Today 148 (2009) 221–231.
S. Sorcar, Y. Hwang, J. Lee, et al., Energy Environ. Sci. 12 (2019) 2685–2696. doi: 10.1039/c9ee00734b
E. Gong, S. Ali, C.B. Hiragond, et al., Energy Environ. Sci. 15 (2022) 880–937. doi: 10.1039/d1ee02714j
W. Ma, X. He, W. Wang, et al., Chem. Soc. Rev. 50 (2021) 12897–12914. doi: 10.1039/d1cs00535a
X. Gao, B. Guo, C. Guo, et al., ACS Appl. Mater. Interfaces 12 (2020) 24059–24065. doi: 10.1021/acsami.0c05631
X. Sun, X. Kang, Q. Zhu, et al., Chem. Sci. 7 (2016) 2883–2887.
M. Li, P. Li, K. Chang, et al., Chem. Commun. 51 (2015) 7645–7648.
S. Zhao, S. Guo, C. Zhu, et al., RSC Adv. 7 (2017) 1376–1381.
S. Xu, R. Xu, Y. Zhu, et al., J. Mater. Chem. A 12 (2024) 17628–17641. doi: 10.1039/d4ta01556h
J. Jia, X. Hao, Y. Chang, et al., J. Colloid Interface Sci. 586 (2021) 491–497.
H. Wang, S. Cheng, X. Cai, et al., Catal. Commun. 162 (2022) 106372.
H. Song, J.T. Song, B. Kim, et al., Appl. Catal. B: Environ. 272 (2020) 119049.
Y. Mao, M. Zhang, G. Zhai, et al., Adv. Sci. 11 (2024) 2401933.
J. Li, R. Cai, H. Mu, et al., ACS Catal. 14 (2024) 3266–3277.
S. Wang, X. Bai, Q. Li, et al., Nanoscale Horiz. 6 (2021) 661–668. doi: 10.1039/d1nh00196e
X. Zou, A. Li, C. Ma, et al., Chem. Eng. J. 468 (2023) 143606.
D. Zeng, H. Wang, X. Zhu, et al., Chem. Eng. J. 451 (2023) 138801.
J. Li, Y. He, L. Tan, et al., Nat. Catal. 1 (2018) 787–793. doi: 10.1038/s41929-018-0144-z
G.A. Olah, Angew. Chem. Int. Ed. 44 (2005) 2636–2639. doi: 10.1002/anie.200462121
G.A. Olah, G.K.S. Prakash, A. Goeppert, J. Am. Chem. Soc. 133 (2011) 12881–12898. doi: 10.1021/ja202642y
I. Ganesh, Renew. Sustain. Energy Rev. 31 (2014) 221–257.
L. Liu, M. Li, F. Chen, et al., Small Struct. 4 (2023) 2200188.
X. Yan, C. Duan, S. Yu, et al., Renew. Sustain. Energy Rev. 190 (2024) 114086.
Q. Wang, X. Zheng, J. Wu, et al., Small Struct. 3 (2022) 2200059.
Z. Shang, X. Feng, G. Chen, et al., Small 19 (2023) 2304975.
H. Zhao, X. Liu, C. Zeng, et al., J. Am. Chem. Soc. 146 (2024) 23649–23662. doi: 10.1021/jacs.4c08523
X. Guan, S.S. Mao, S. Shen, ChemNanoMat 7 (2021) 873–880. doi: 10.1002/cnma.202100103
L. Gloag, S.V. Somerville, J.J. Gooding, Nat. Rev. Mater. 9 (2024) 173–189. doi: 10.1038/s41578-023-00633-2
J. Xiao, T. Zhang, Q. Wang, Curr. Opin. Green Sustain. Chem. 37 (2022) 100660.
W. Hu, H. Yang, C. Wang, RSC Adv. 13 (2023) 20889–20908. doi: 10.1039/d3ra03462c
C.B. Hiragond, N.S. Powar, J. Lee, et al., Small 18 (2022) 2201428.
F. Wang, Y. Liu, M. Peng, et al., ACS Catal. 14 (2024) 16434–16458. doi: 10.1021/acscatal.4c06065
Z. Wang, G. Zou, J.H. Park, et al., Sci. China Mater. 67 (2024) 397–423. doi: 10.1007/s40843-023-2698-5
S. Wang, L. Wang, D. Wang, et al., Energy Environ. Sci. 16 (2023) 2759–2803. doi: 10.1039/d3ee00037k
Q. Sun, C. Jia, Y. Zhao, et al., Chin. J. Catal. 43 (2022) 1547–1597.
J. Zhang, W. Cai, F.X. Hu, et al., Chem. Sci. 12 (2021) 6800–6819. doi: 10.1039/d1sc01375k
C. Chen, J. Li, X. Tan, et al., EES Catal. 2 (2024) 71–93. doi: 10.1039/d3ey00150d
K. Atsonios, K.D. Panopoulos, E. Kakaras, Int. J. Hydrog. Energy 41 (2016) 2202–2214.
N.H.M. Dostagir, C. Thompson, H. Kobayashi, et al., Catal. Sci. Technol. 10 (2020) 8196–8202. doi: 10.1039/d0cy01789b
J. Zhong, X. Yang, Z. Wu, et al., Chem. Soc. Rev. 49 (2020) 1385–1413. doi: 10.1039/c9cs00614a
R.P. Ye, J. Ding, W. Gong, et al., Nat. Commun. 10 (2019) 5698.
L.C. Grabow, M. Mavrikakis, ACS Catal. 1 (2011) 365–384. doi: 10.1021/cs200055d
Y. Li, S.H. Chan, Q. Sun, Nanoscale 7 (2015) 8663–8683.
Y. Yang, J. Evans, J.A. Rodriguez, et al., Phys. Chem. Chem. Phys. 12 (2010) 9909–9917. doi: 10.1039/c001484b
Y. Kim, T.S.B. Trung, S. Yang, et al., ACS Catal. 6 (2016) 1037–1044. doi: 10.1021/acscatal.5b02083
J.A. Rodriguez, J. Evans, L. Feria, et al., J. Catal. 307 (2013) 162–169.
L. Liu, X. Wang, S. Lu, et al., N. J. Chem. 46 (2022) 5043–5051. doi: 10.1039/d1nj05938f
Y. Yang, C.A. Mims, D.H. Mei, et al., J. Catal. 298 (2013) 10–17.
Y.F. Zhao, Y. Yang, C. Mims, et al., J. Catal. 281 (2011) 199–211.
W. Cui, F. Wang, X. Wang, et al., Angew. Chem. Int. Ed. 63 (2024) e202407733.
X. Han, T. Xiao, M. Li, et al., ACS Catal. 13 (2023) 13679–13690. doi: 10.1021/acscatal.3c03431
T. Yang, X. Mao, Y. Zhang, et al., Nat. Commun. 12 (2021) 6022.
H. Li, L. Wang, Y. Dai, et al., Nat. Nanotechnol. 13 (2018) 411–417. doi: 10.1038/s41565-018-0089-z
X. Jia, K. Sun, J. Wang, et al., J. Energy Chem. 50 (2020) 409–415.
A. Han, J. Ding, Q. Zhong, Colloid Surf. A 641 (2022) 128535.
S. Zhou, W. Ma, U. Anjum, et al., Nat. Commun. 14 (2023) 5872.
J. Chen, Y. Wang, F. Wang, Y. Li, Angew. Chem. 62 (2023) e202218115.
Y. Shi, Q. Gu, Y. Zhao, et al., Chem. Eng. J. 485 (2024) 150093.
K. Lee, U. Anjum, T.P. Araújo, et al., Appl. Catal. B: Environ. 304 (2022) 120994.
Z. Han, C. Tang, J. Wang, et al., J. Catal. 394 (2021) 236–244.
M.S. Frei, C. Mondelli, R. García-Muelas, et al., Nat. Commun. 10 (2019) 3377.
J. Zhu, F. Cannizzaro, L. Liu, et al., ACS Catal. 11 (2021) 11371–11384. doi: 10.1021/acscatal.1c03170
C. Shen, K. Sun, R. Zou, et al., ACS Catal. 12 (2022) 12658–12669. doi: 10.1021/acscatal.2c03709
T. Len, M. Bahri, O. Ersen, et al., Green Chem. 23 (2021) 7259–7268. doi: 10.1039/d1gc01761f
Y. Chai, B. Qin, B. Li, et al., Natl. Sci. Rev. 10 (2023) nwad043.
L.L. Ling, X. Guan, X. Liu, et al., Natl. Sci. Rev. 11 (2024) nwae114.
A. Rosado, I.-M. Popa, A.A. Markeb, et al., J. Mater. Chem. A 12 (2024) 21758–21771. doi: 10.1039/d4ta03268c
J. Chen, D. Zhang, B. Liu, et al., Angew. Chem. Int. Ed. 63 (2024) e202401168.
N.H.M. Dostagir, C.R. Tomuschat, K. Oshiro, et al., JACS Au 4 (2024) 1048–1058. doi: 10.1021/jacsau.3c00789
R. Gaikwad, A. Bansode, A. Urakawa, J. Catal. 343 (2016) 127–132.
W. Wu, Y. Wang, L. Luo, et al., Angew. Chem. Int. Ed. 61 (2022) e202213024.
D. Xu, X. Hong, G. Liu, Chin. J. Catal. 393 (2022) 207–214. doi: 10.3390/land11020207
T.P. Araújo, J. Morales-Vidal, T. Zou, et al., Adv. Energy Mater. 13 (2023) 2204122.
Y. Chen, H. Li, W. Zhao, et al., Nat. Commun. 10 (2019) 1885.
D. Li, M. Kassymova, X. Cai, et al., Coord. Chem. Rev. 412 (2020) 213262.
W. Tu, Y. Zhou, Z. Zou, Adv. Mater. 26 (2014) 4607–4626. doi: 10.1002/adma.201400087
M. Tahir, N.S. Amin, Renew. Sustain. Energy Rev. 25 (2013) 560–579.
J. Wu, Y. Huang, W. Ye, et al., Adv. Sci. 4 (2017) 1700194.
X. Li, J. Wen, J. Low, et al., Sci. China Mater. 57 (2014) 70–100. doi: 10.1007/s40843-014-0003-1
X. Jiang, L. Zhang, H. Liu, et al., Angew. Chem. Int. Ed. 59 (2020) 23112–23116. doi: 10.1002/anie.202011495
S.K. Kaiser, Z. Chen, D. Faust Akl, et al., Chem. Rev. 120 (2020) 11703–11809. doi: 10.1021/acs.chemrev.0c00576
Z. Li, B. Li, Y. Hu, et al., Small Struct. 3 (2022) 2200041.
W. Liu, L. Cao, W. Cheng, et al., Angew. Chem. Int. Ed. 56 (2017) 9312–9317. doi: 10.1002/anie.201704358
B.B. Sarma, F. Maurer, D.E. Doronkin, et al., Chem. Rev. 123 (2023) 379–444. doi: 10.1021/acs.chemrev.2c00495
J. Wu, H. Lu, X. Zhang, et al., Chem. Commun. 52 (2016) 5027–5029.
D. Adekoya, M. Tahir, N.A.S. Amin, Renew. Sustain. Energy Rev. 116 (2019) 109389.
A. Kumar, P. Raizada, V.K. Thakur, et al., Chem. Eng. Sci. 230 (2021) 116219.
T. Qu, S. Wei, Z. Xiong, et al., Fuel Process. Technol. 251 (2023) 107933.
A. Francis, S.S. Priya, S.Harish Kumar, et al., J. CO2 Util. 47 (2021) 101515.
X. Chang, T. Wang, J. Gong, Energy Environ. Sci. 9 (2016) 2177–2196.
M. Gattrell, N. Gupta, A. Co, J. Electroanal. Chem. 594 (2006) 1–19.
J. Fu, K. Jiang, X. Qiu, et al., Mater. Today 32 (2020) 222–243.
S. Kattel, P.J. Ramírez, J.G. Chen, et al., Science 355 (2017) 1296–1299. doi: 10.1126/science.aal3573
Y. Yang, D. Mei, C.H.F. Peden, et al., ACS Catal. 5 (2015) 7328–7337. doi: 10.1021/acscatal.5b02060
A. Li, E. Kan, S. Chen, et al., Small 18 (2022) 2200073.
G. Wang, C.T. He, R. Huang, et al., J. Am. Chem. Soc. 142 (2020) 19339–19345. doi: 10.1021/jacs.0c09599
M. Ma, Z. Huang, L. Li, et al., Appl. Catal. B: Environ. 330 (2023) 122626.
P. Sharma, S. Kumar, O. Tomanec, et al., Small 17 (2021) 2006478.
C.S. Vennapoosa, S. Varangane, B.M. Abraham, et al., J. Phys. Chem. Lett. 14 (2023) 11400–11411. doi: 10.1021/acs.jpclett.3c02347
L. Su, P. Wang, X. Ma, et al., Angew. Chem. Int. Ed. 60 (2021) 21261–21266. doi: 10.1002/anie.202108937
Z.S. Zhu, S. Zhong, C. Cheng, et al., Chem. Rev. 124 (2024) 11348–11434. doi: 10.1021/acs.chemrev.4c00276
K. Wang, M. Cheng, F. Xia, et al., Small 19 (2023) 2207581.
P. Li, Z. Qi, D. Yan, et al., Angew. Chem. Int. Ed. 63 (2024) e202411000.
M. Ma, Z. Huang, D.E. Doronkin, et al., Appl. Catal. B: Environ. 300 (2022) 120695.
X. Liu, C. Zhu, M. Li, et al., Angew. Chem. Int. Ed. 63 (2024) e202412048.
K. Mori, H. Yamashita, M. Anpo, RSC Adv. 2 (2012) 3165. doi: 10.1039/c2ra01332k
G. Mele, C. Annese, L. D’Accolti, et al., Molecules 20 (2014) 396–415. doi: 10.3390/molecules20010396
I.H. Tseng, W.C. Chang, J. C.S.Wu, Appl. Catal. B: Environ. 37 (2002) 37–48.
K. Kočí, L. Obalová, Z. Lacný, Chem. Pap. 62 (2008) 1–9. doi: 10.2478/s11696-007-0072-x
S. Kaneco, Y. Shimizu, K. Ohta, et al., J. Photochem. Photobiol. A: Chem. 115 (1998) 223–226.
J. Albo, M. Alvarez-Guerra, P. Castaño, A. Irabien, Green Chem. 17 (2015) 2304–2324.
Z. Zhou, S. Sun, Natl. Sci. Rev. 4 (2017) 155–156. doi: 10.1093/nsr/nww083
K. P.Kuhl, T. Hatsukade, E. R.Cave, et al., J. Am. Chem. Soc. 136 (2014) 14107–14113.
G. A. Olah, A. Goeppert, G.K.S. Prakash, et al., J. Org. Chem. 74 (2009) 487–498. doi: 10.1021/jo801260f
A. Liu, M. Gao, X. Ren, et al., J. Mater. Chem. A 8 (2020) 3541–3562. doi: 10.1039/c9ta11966c
X. Feng, Z. Shang, R. Qin, et al., Acta Phys. Chim. Sin. 40 (2024) 2305005.
H. Yang, Y. Wu, G. Li, et al., J. Am. Chem. Soc. 141 (2019) 12717–12723. doi: 10.1021/jacs.9b04907
S. Kong, X. Lv, X. Wang, et al., Nat. Catal. 6 (2023) 6–15.
W. Guo, S. Liu, X. Tan, et al., Angew. Chem. Int. Ed. 60 (2021) 21979–21987. doi: 10.1002/anie.202108635
Q. Zhao, C. Zhang, R. Hu, et al., ACS Nano 15 (2021) 4927–4936. doi: 10.1021/acsnano.0c09755
S. Dutta, S.K. Pati, Phys. Chem. Chem. Phys. 25 (2023) 15788–15797. doi: 10.1039/d3cp00933e
J. Liu, D. Yang, Y. Zhou, et al., Angew. Chem. Int. Ed. 60 (2021) 14473–14479. doi: 10.1002/anie.202103398
J. Yuan, Y. Chen, F. Liu, et al., Catal. Commun. 177 (2023) 106640.
G. Wang, J. Chen, Y. Ding, et al., Chem. Soc. Rev. 50 (2021) 4993–5061. doi: 10.1039/d0cs00071j
L. Hu, X. Sai, X. Liu, et al., ChemCatChem 16 (2024) e202301335.
G. Feng, W. Chen, B. Wang, et al., Chem. Asian J. 13 (2018) 1992–2008. doi: 10.1002/asia.201800637

Figure 2 Mechanism and pathways of photocatalytic CO2-to-methanol conversion with SACs.

Figure 3 Mechanism and pathways of electrocatalytic CO2-to-methanol conversion with SACs.
Table 1. Catalytic activity and reaction parameters of SACs for CO2-to-methanol conversion via thermal hydrogenation.
| SACs | Reaction conditions | Conversion rate (%) | Yield (mmol gcat-1 h-1) | Selectivity (%) | Ref. | |||
| CO2/H2 | GHSV (L h-1 gcat-1) | Pressure (MPa) | Temperature (℃) | |||||
| 2% Pt/Mo-NC | 1/3 | – | 3 | 180 | – | 0.27 | 88.0 | [51] |
| 12.1 wt% Cu/C3N4 | 1/3 N2% =4% | – | 3.2 | 150 | – | 4.20 | 95.5 | [53] |
| 0.2 wt% Pt/MoS2 | 1/3 | – | 3.2 | 150 | – | ≈0.41a | 95.4 | [54] |
| 1/3 | – | 3.2 | 210 | – | ≈37.42a | 93.0 | ||
| 7.5 wt% Pt/MoS2 | 1/3 | – | 3.2 | 150 | – | ≈5.25a | 81.3 | |
| 1/3 | – | 3.2 | 210 | – | ≈46.61a | 76.0 | ||
| Cu/MoS2@SiO2 | 1/4 | 24 | 5 | 260 | 12.7 | ≈13.04a | 72.5 | [57] |
| 0.96 wt% Rh/In2O3 | 1/4 Ar% ≈ 17% | 60 | 5 | 300 | 9.3 | 31.21 | 75.0 | [40] |
| 1/4 Ar% ≈ 17% | 30 | 5 | 270 | 3.7 | 5.67 | 71.0 | ||
| 1/4 Ar% ≈ 17% | 45 | 5 | 270 | 4.2 | 12.17 | 87.0 | ||
| 9.7 wt% Ni/In2O3 | 1/3 N2% = 4% | 21 | 5 | 300 | 18.5 | 17.17 | 54.0 | [55] |
| 1/3 N2% = 4% | 21 | 5 | <225 | – | – | 100.0 | ||
| Pd1/In2O3 | 1/5 | – | 3.3 | 200 | 33.0 | ≈63.62a | 99.4 | [59] |
| 0.58 wt% Pt/In2O3 | 1/3 Ar% = 4% | 54 | 2 | 220 | <1.0 | – | 91.1 | [61] |
| 1/3 Ar% = 4% | 54 | 4 | 300 | >9.0 | 23.72 | 65.0 | ||
| 0.75 wt% Pd/In2O3 | 1/4 | 48 | 5 | 280 | 9.7 | 29.96 | 78.0 | [62] |
| 6 wt% Ni/In2O3 | 1/3 N2% = 20% | 48 | 3 | 250 | – | 7.80 | – | [63] |
| 1 wt% Re/In2O3 | 1/4 | 21 | 5 | 300 | – | 16.85 | 72.1 | [64] |
| Ir1Pd1-In2O3 | 1/3 | – | 3 | 250 | 10.5 | – | 97.0 | [69] |
| 1.0 wt% Mo/TiO2 anatase | 1/3 N2% = 20% | 7.5 | 3 | 275 | 5.2 | 0.35 | 10.2 | [65] |
| 2.9 wt% Mo/TiO2 rutile | 1/3 N2% = 20% | 7.5 | 3 | 275 | 6.8 | 1.09 | 24.2 | |
| 0.83 wt% Cu1/ZnO | 1/3 H2O% = 0.11% | 6 | 3 | 170 | 4.9 | – | 99.1 | [72] |
| 1/3 | 6 | 3 | 170 | 1 | – | 93.5 | ||
| 2% Cu/GaZrOx | 1/3 | 6 | 3 | 300 | – | – | 87.0 | [52] |
| Pd-Cu/ZrO2 | 1/3 | 6 | 1.5 | 260 | ≈5 | 0.51 | 38.0 | [56] |
| Pd-ZnZrOx | 1/4 Ar% = 5% | 24 | 5 | 320 | – | 21.54 | 85.0 | [60] |
| 10% Co/5% In-ZrO2 | 1/4 | 24 | 3 | 300 | – | 4.68 | 86.0 | [70] |
| CuZnZrOx | 1/3 | – | 4.5 | 290 | 9.5 | – | 76.0 | [73] |
| 5 mol% Zn-ZrOx | 1/4 | 24 | 5 | 320-340 | – | 14.36 | 80.0 | [74] |
| Cu@FAU | 1/3 | 12 | 3 | 240 | 11.5 | 12.80 | 89.5 | [66] |
| MOF-808-NaCu | 1/3 | – | 3.5 | 275 | – | 9.55 | 93.0 | [67] |
| 1/3 | – | 3.5 | 250 | 4.1 | – | 93.0 | ||
| NU-1000-NH2/PrS-Cu | 1/3 | 12 | 1 | 280 | – | 3.12 | ≈100 | [68] |
| Pt1@MIL101 | 1/3 | – | 3.2 | 150 | – | 1.08 | 90.3 | [75] |
| Pt@UiO-66-NH2(Co)b | 1/3 | – | 1.5 | 240 | – | 1.91 | 74.8a | [58] |
| a The symbol “≈” in the table indicates methanol yields calculated based on data from the references. b The results from reference are obtained under experimental conditions of a 300 W Xenon lamp with a light intensity of 1.0 W/cm2. |
||||||||
 下载: 导出CSV
下载: 导出CSV
Table 2. Reaction conditions and performance of SACs photocatalytic reduction of CO2 to methanol.
| SACs | Light source | Reaction medium | AQY (%) | Selectivity (%) | Productivity (µmol gcat-1 h-1) | Ref. |
| Pd1/PCN | 300 W xenon lamp (λ > 420 nm) | KHCO3/Na2SO3 | – | 85.1 | 742.4 | [96] |
| Pr1-N4O2/CN | 300 W xenon lamp | ACN/H2O | – | 92.4 | 511.1 | [98] |
| RuSA-mC3N4 | 34 W blue LED light (400-500 nm light density: 22 mW/cm2) | DMF/H2O | – | – | 250.0 | [99] |
| Er1Nd3/CN | 300 W xenon lamp (λ > 420 nm) | TEOA/H2O | – | – | 987.7 | [104] |
| Co/g-C3N4 | 300 W xenon lamp (light density: 1500 mW/cm2) | H2O | – | 96.2 | 235.5 | [105] |
| CuSAs/UiO-66-NH2 | 300 W xenon lamp (λ > 400 nm) | TEOA/H2O | – | – | 5.3 | [97] |
| Cu1@BiOBr | 300 W xenon lamp | H2O | 12.2 | 90 | 627.6 | [103] |
| CuSAs@PCN@UiO-66-NH2 | 300 W xenon lamp (420-800 nm) | TEA/ACN/H2O | – | – | 4150.0 | [106] |
| Ru/CNF(ZnO) | 400 W xenon lamp (400-440 nm) | IPA/ACN/H2O | 1.4 | 85 | 62.3 | [100] |
| Apparent quantum yield (AQY) is the ratio of the number of electrons participating in the reaction to the total number of incident photons at a specific wavelength. | ||||||
下载: 导出CSV
Table 3. Reaction conditions and performance of SACs for electrocatalytic reduction of CO2 to methanol.
| SACs | Electrolyte | Applied potential (V vs. RHE) | Current density (mA/cm2) | FE (%) | Productivity (µmol m-2 s-1) | Ref. |
| CuSAs/TCNFs | 0.1 mol/L KHCO3 | −0.9 | -93.0 | 44.0 | 68.4 | [118] |
| Cu2NCN | 0.5 mol/L KHCO3 | −0.9 | -92.3 | 70.0 | 1600 | [119] |
| Sn1/Ov CuO-90 | [Bmim]BF4 (25 mol%) aqueous solution | −2.0 | 67.0 | 88.6 | – | [120] |
| SA-Cu-MXene | 0.1 mol/L KHCO3 | −1.4 | 21.3 | 59.1 | – | [121] |
| Cu3(HHTQ)2 | 0.1 mol/L KHCO3 | −0.4 | 45.0 | 53.6 | – | [123] |
| CuIn@ZIF-8 | 0.5 mol/L KHCO3 | −0.6 | – | 55.6 | – | [125] |
| Note: Faradaic efficiency (FE) is the ratio of charge utilized for target product formation to the total charge passed. | ||||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
