
Scheme 1.
Representative bioactive peptides and this review.

Natural peptides generally play significant roles in human physiology, such as actions as neurotransmitters, growth factors, hormones and ion channel ligands [1-3]. Peptides often have selective and effective binding to specific cell surface receptors initiating intracellular effects, including ion channels or G protein-coupled receptors [3]. Compared to small molecule drugs, peptides display higher affinity and specificity to receptors and show lower toxicity [4]. During the past decades, peptides are widely used in medicine and biotechnology, and therapeutic peptide studies gain rapid development (Scheme 1a). Currently, there are >80 commercially available peptide drugs approved by US Food and Drug Administration (FDA) on the global market [5]. And, >140 peptide medicines are in clinical trials while >500 therapeutic peptides are in preclinical research. For instance, the peptide-based drug Lupron™ is employed to treat prostate cancer, obtaining global sales of more than US$2.3 billion in 2011 [6]. However, using naturally occurring peptides as therapeutic medicines often encounters some intrinsic weaknesses, such as metabolic instability and low membrane permeability [7,8]. Thus, a series of strategies has been developed to modify natural amino acids and peptides. Inserting unnatural amino acids into peptides or directly modifying natural peptides is highly popular for medicinal and biological chemistry as well as industry. This generally leads to improved bioactivity and pharmacokinetics [9,10]. Additionally, peptides conjugated with drugs, polymers or fluorophores are critical for a variety of biological and therapeutic applications. Peptides labelled with fluorophores are applied for realtime tracking of cells, metabolites and biomolecules under physiological conditions [11-13]. Peptides conjugated to drugs are used for selective target therapy, such as antibody-drug conjugates (ADCs) [14-16]. Peptides ligation to polymers are employed for increasing their circulation half-lives and stability [17-19]. Among them, chemo- and site-selective functionalizations of amino acids and peptides are essential in contemporary biochemistry and pharmaceutical chemistry, as the ability to fine-tune structural features, which is beneficial to modulate physicochemical and biological properties [20-22]. Therefore, classical condensations and (cyclo)additions are firstly employed in the derivatizations of amino acids and peptides [22,23]. With the advancement of synthesis technology, transition metal-catalyzed cross-coupling reactions have emerged as important strategies for amino acid and peptide modifications, largely extending the chemical scope [21,24].
Over the past decades, transition metal-catalyzed C—H activation is becoming a powerful tool for the construction of C—C and C-X (X = N, O, S, P, Si…) bonds in a high atom- and step-economical manner [25-32]. Compared to transition metal-catalyzed cross-coupling, this approach avoids prefunctionalization and triggers the transformations at inert C—H bonds. Recently, transition metal-catalyzed C—H activation has been successfully employed in the diversifications of amino acids and peptides [30,31], such as C—H arylation [33-35], olefination [36], allylation [37], alkynylation [38] and alkylation [39-41]. Through meticulous design, an array of novel cyclopeptides is synthesized via transition metal-catalyzed C—H activation and subsequent intramolecular macrocyclization [42-45]. These have been summarized in several excellent reviews, and are outside the scope of this review [42,43]. As domino reactions can largely increase the complexity and diversity of products, they have been widely utilized in the synthesis of heterocycles, biologically active molecules and natural products [46]. Complex domino reactions recently have been developed to synthesize functionalized amino acids and peptides bearing diverse heterocycles, largely enriching the structural complexity and diversity, as well as the physical/chemical properties and biological activities. To the best of our knowledge, there is currently no review dedicated specifically to the synthesis of unnatural amino acids and peptides via transition metal-catalyzed C—H activation/annulation. In this review, we aim to provide a comprehensive summary and documentation of reported methods employed for the preparation of unnatural amino acids and peptides with various heterocycles, through transition metal-catalyzed C—H activation/annulation (Scheme 1b).
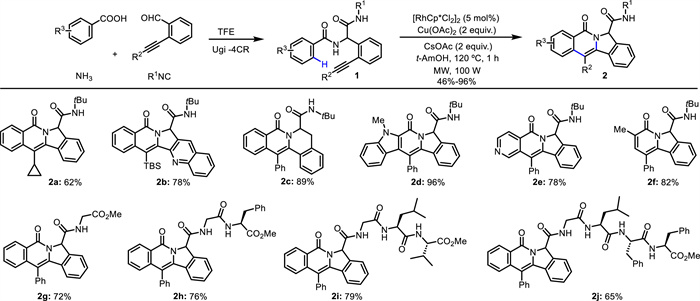
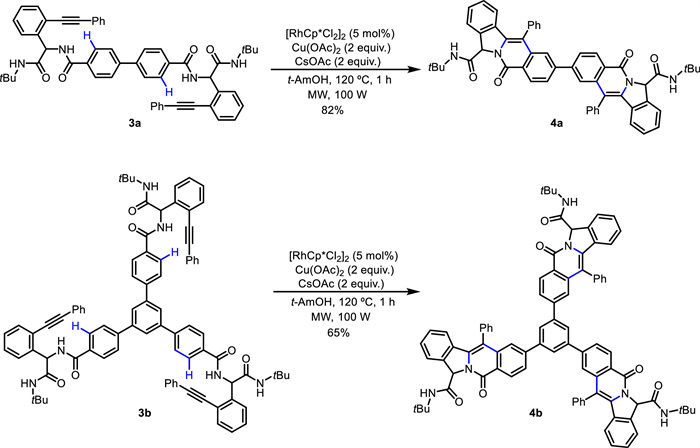
In 2017, Tian and Van der Eycken developed an efficient Rh(Ⅲ)-catalyzed intramolecular annulation of alkyne-tethered benzamides for the synthesis of indolizinones and quinolizinones via C—H activation [47]. Subsequently, they employed this strategy in the modification of peptidomimetics and oligopeptides via the combination of the Ugi reaction and microwave irradiation (Scheme 2) [48]. Through C—H activation and subsequent intramolecular annulation with alkyne without installing a directing group, diversifications of peptidomimetics and oligopeptides were chemoselectively achieved, resulting in indolizinone and quinolizinone scaffolds in a rapid and step-economical manner. Diverse heteroaryl peptidomimetics involving furan, thiophene, pyrrole, benzofuran, benzothiophene, indole (2d), pyridine (2e) and thiazole, were well tolerated. α-Substituted acrylamides (2f) were compatible with this reaction, while β-substituted and α,β-disubstituted acrylamides did not work. This method could be applied to efficiently construct rosettacin (2b), oxypalmatime (2c) and camptothecin derivatives. Moreover, double and triple C—H activation/annulation were successfully performed to give the di- and trimers (4a and 4b) (Scheme 3). Additionally, this method was compatible with water and showed high chemoselectivity when adding extra N-unprotected or N-protected amino acids.
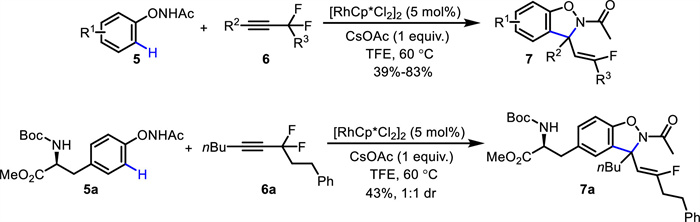
Later on, Zhou and Yi reported a redox-neutral [4 + 1] annulation of N-phenoxy amides 5 with α,α-difluoromethylene alkynes 6 via Rh(Ⅲ)-catalyzed C—H activation, leading to Z-configured monofluoroalkenyl dihydrobenzo[d]isoxazoles 7 [49]. This approach was further employed in the late-stage C—H modification of complex bioactive compounds including an amino acid (7a) (Scheme 4).
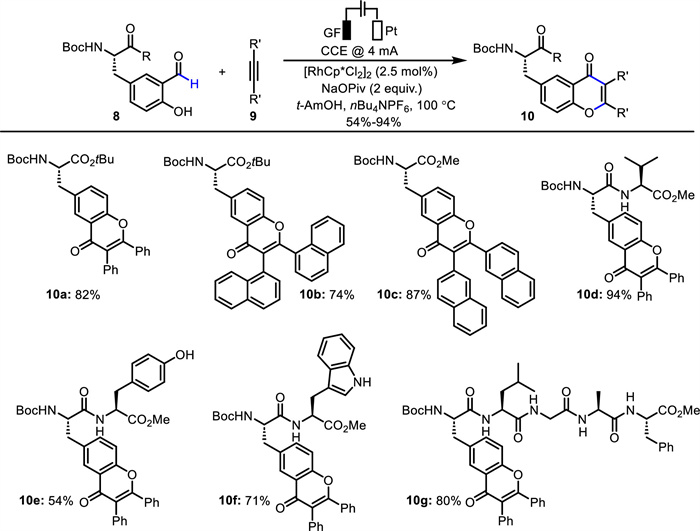
Ackermann and co-workers developed a series of elegant works employing C—H activation triggered by Ru, Mn or Co catalyst for the modification of peptides [37,50,51]. Recently, they presented a rhodaelectro-catalyzed approach to chromones via a formyl C—H activation and subsequent alkyne insertion/reductive elimination process (Scheme 5) [52]. Notably, this method could be employed for the functionalization of amino acids and peptides 8 containing a hydroxy–benzaldehyde moiety. Also, a pentapeptide was well tolerated in this reaction (10g). The obtained amino acids could undergo further photoelectrochemical oxidation to give polycyclic products with enhanced fluorescence.
Later on, Song and Gong reported a Ru(Ⅱ)-catalyzed C—H activation of α-substituted or α,α-disubstituted glycine ester phosphinamides 11, leading to ortho-alkenylated phosphinamides 13 (Scheme 6) [53]. When a Rh(Ⅲ) catalyst was employed, ortho-C-H activation of α-unsubstituted glycine ester phosphinamide 14 and subsequent annulation with alkynes 15 occurred, resulting in various phosphaisoquinolin-1-ones 16. With the assistance of microwave irradiation, this reaction could be efficiently finished in 1 h.

In 2022, Sakhuja and Bajaj described a Rh(Ⅲ)-catalyzed C—H activation/annulation reaction between vinylated tyrosines 17 and internal alkynes 15, leading to diverse oxepine-mounted unnatural tyrosines 18 (Scheme 7) [54]. Notably, this [5 + 2] cyclization could be employed in a series of dipeptides and tripeptides bearing a tyrosine residue (18g-18i). When amino acid/peptide-appended alkynes were used, the chemical ligation was achieved (18e and 18f).

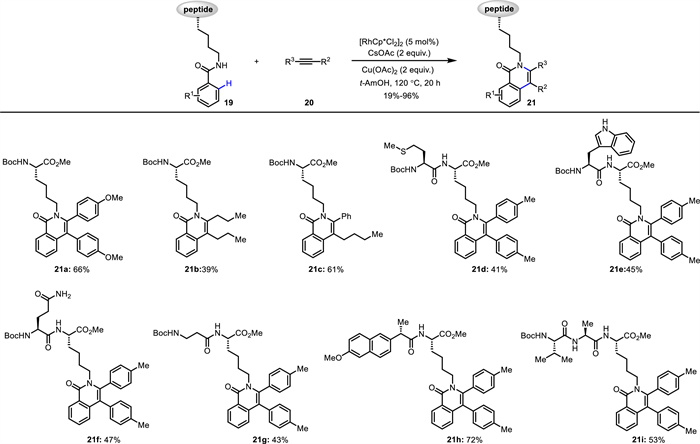
In 2023, Song, Cai and Van der Eycken developed a Rh(Ⅲ)-catalyzed C—H activation/annulation reaction for the diversification of Lys-based peptides 19 (Scheme 8) [55]. This method was performed under racemization-free conditions, owning high atom- and step-economy, and excellent chemo- and site-selectivity. This reaction featured a broad scope including peptides bearing unprotected Trp (21e) and Tyr, free Ser and Gln (21f), and Met (21d) residues, in situ resulting in an array of peptide-isoquinolone conjugates. The obtained peptide-isoquinolone conjugates exhibited maximum emission wavelength up to 460 nm. Interestingly, biological activity experiments suggested that the peptide-isoquinolone conjugates possess good antifungal activity towards crop and forest pathogenic fungi.
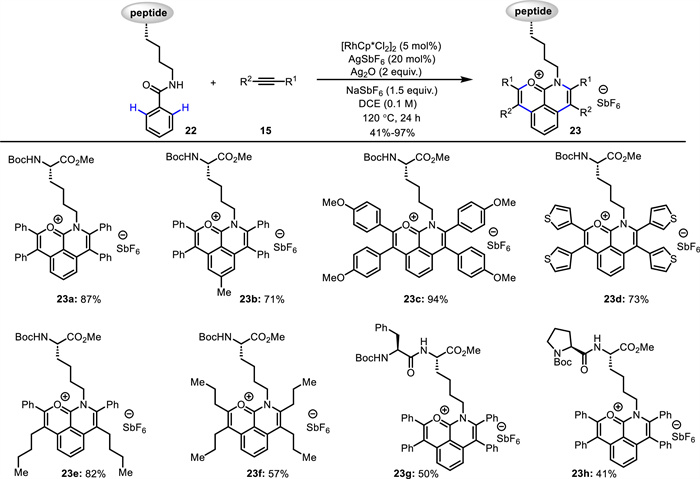
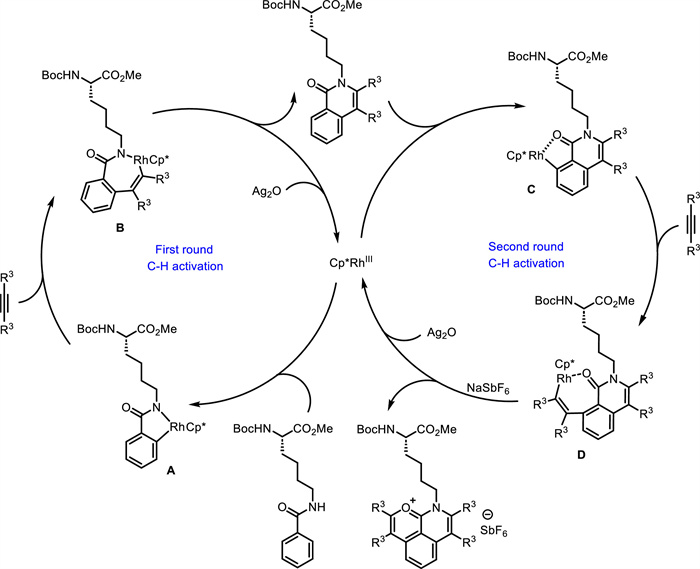
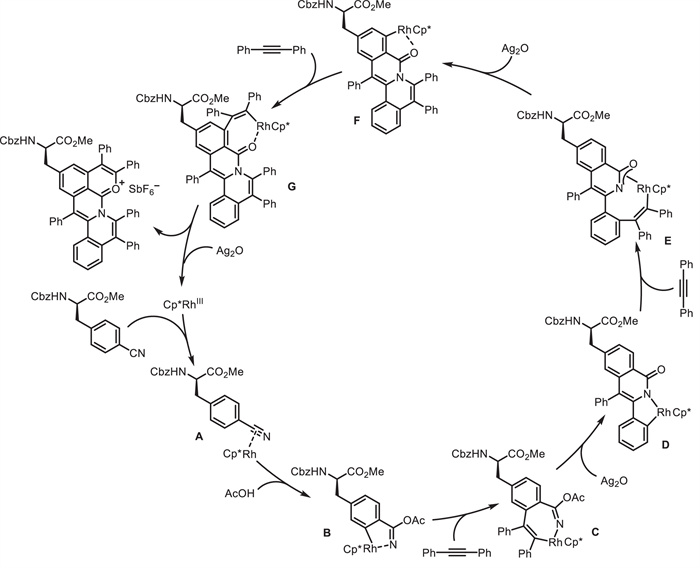
Later on, they used Ag2O as oxidant, AgSbF6 and NaSbF6 as additives, and DCE as solvent, leading to double C—H activation products. This Rh(Ⅲ)-catalyzed double C—H activation/annulation reaction was successfully employed in the synthesis of fluorescent amino acids and peptides 23 (Scheme 9) [56]. Through the coupling of Lys-based amino acids or peptides 22 with alkynes 15, the fluorescent tricyclic-fused aromatic hydrocarbon oxonium ions 23 were in situ generated. The robustness of this protocol was reflected by broad substrate scope, high atom- and step-economy, as well as high chemo- and site-selectivity. Notably, unsymmetrical double C—H activation/annulation using two different alkynes could be achieved via sequential two steps, producing novel fluorescent amino acids and peptides. The obtained fluorescent oxonium ions owned tunable fluorescence emission and low cytotoxicity, and could specifically target lysosomes of living cells. As proposed (Scheme 10), a five-membered rhodacycle intermediate A is formed via Rh(Ⅲ)-catalyzed C—H activation. Following alkyne insertion and subsequent reductive elimination, the mono C—H activation product is formed along with a Rh(Ⅰ) complex, which is then oxidized by Ag₂O to regenerate the active Rh(Ⅲ) species. Second Rh(Ⅲ)-catalyzed C—H activation affords intermediate C, followed by alkyne insertion and reductive elimination delivering the final product and Rh(Ⅰ) species, which is oxidized by Ag2O to regenerate the Rh(Ⅲ) catalyst.
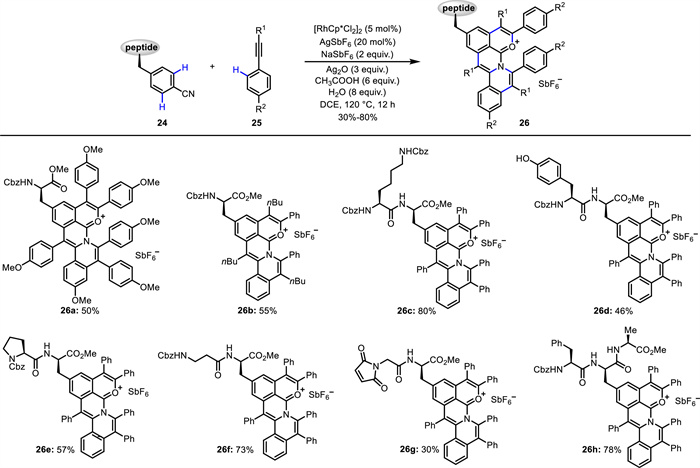
Very recently, Song and Cai disclosed a Rh(Ⅲ)-catalyzed triple C—H activation/annulation for peptide modification (Scheme 11) [57]. An array of fluorescent peptides 26 was synthesized from Phe-based peptides 24 and alkynes 25 via in situ formation of polycyclic aromatic hydrocarbon oxonium ions. Mechanistic experiments indicated that the triple C—H activation/annulation is successive rather than step-by-step. This method exhibited broad substrate scope, high atom- and step-economy, and excellent chemo- and site-selectivity. The fluorescent peptides showed maximum emission wavelength up to 628 nm and low cell cytotoxicity. A dipeptide bearing maleimide residue was tolerated to give the corresponding fluorescent product 26g, which could be employed in bioconjugation with protein BSA. Notably, fluorescent amino acids and peptides could specifically target lysosomes and mitochondria of live mammalian cells. According to the mechanism (Scheme 12), intermediate A is initially formed via the coordination of the Rh(Ⅲ) center with the CN group. Nucleophilic attack of AcOH to A and rapid Rh(Ⅲ)-catalyzed C—H activation prior to the dissociation of Rh(Ⅲ) species, give five-membered rhodacycle B. Subsequent alkyne insertion affords seven-membered rhodacycle C. Reductive elimination of C generates Rh(Ⅰ)-N species, which undergoes rapid oxidization to form Rh(Ⅲ)-N intermediate prior to the dissociation of Rh(Ⅰ) species, followed by Rh(Ⅲ)-catalyzed C—H activation forming five-membered rhodacycle D. The second alkyne insertion yields intermediate E, which involves coordination between the Rh(Ⅲ) center and the oxygen atom. Subsequent reductive elimination produces a Rh(Ⅰ) complex that coordinates with the carbonyl group. This suffers from rapid oxidization affording a Rh(Ⅲ) intermediate prior to dissociation of the Rh(Ⅰ) species, followed by Rh(Ⅲ)-catalyzed C—H activation generating a five-membered rhodacycle F. The third alkyne insertion and reductive elimination deliver the final product and Rh(Ⅰ) intermediate, which is oxidized by Ag2O to regenerate the Rh(Ⅲ) species.
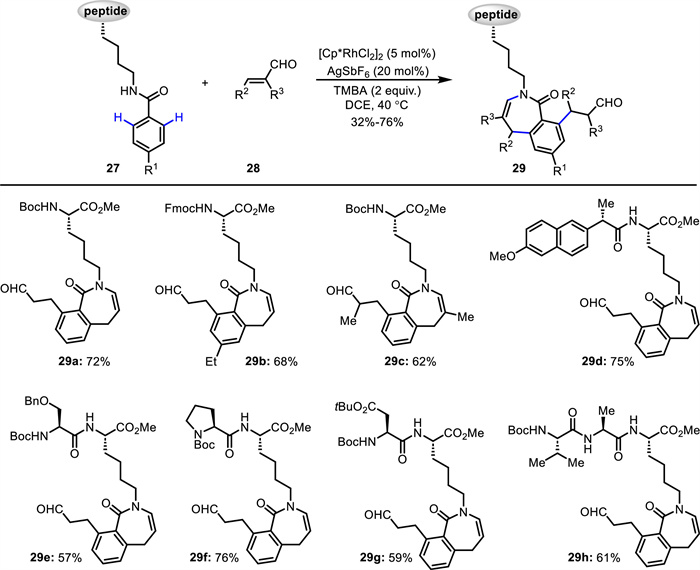
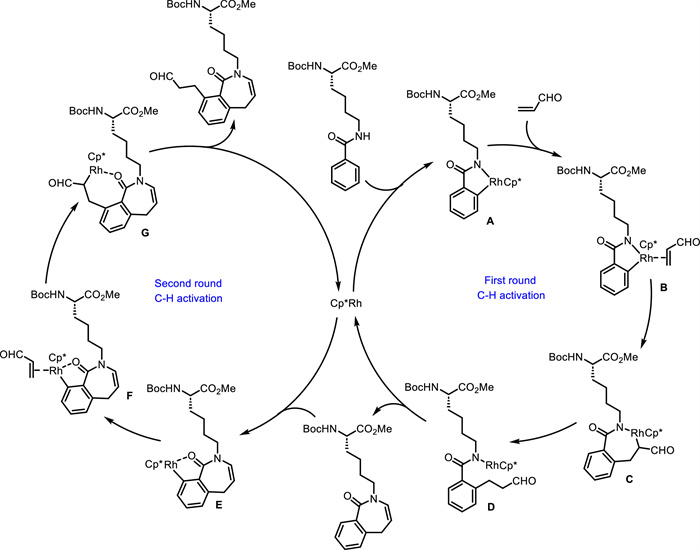
Based on their previous works [55,56], Song, Cai and Van der Eycken reported an efficient method for synthesizing peptide-benzazepine conjugates 29 from Lys-based peptides 27 and acroleins 28 (Scheme 13) [58]. This Rh(Ⅲ)-catalyzed double C—H activation was performed under mild reaction conditions, featuring broad scope, high atom- and step-economy as well as excellent chemo- and site-selectivity. α-Me- (29c), Et- or Bn-substituted acroleins worked well to afford double C—H activation products in good yield, while α-acroleins bearing a long aliphatic chain gave a mixture of mono and double C—H activation products. 2-Phenylacrolein was not compatible with the standard conditions, probably because of the much easier polymerization. β-Me-substituted acrolein reacted smoothly to afford a mixture of mono and double C—H activation products, while a β-Et-substituted acrolein only delivered the mono C—H activation product. Both trans-cinnamaldehyde and vinyl ketone were not compatible, probably because of the poor reactivity. The synthetic practicality of this method was further demonstrated by scale-up experiments and product transformations including various late-stage aldehyde-based ligations. The preliminary bioactivity experiments displayed that peptide-benzazepine conjugates possess good antifungal activities toward crop and forest pathogenic fungi. As proposed (Scheme 14), initially, a five-membered rhodacycle A is formed via Rh(Ⅲ)-catalyzed C—H activation. Subsequent coordination of the Rh(Ⅲ) center with the alkene affords intermediate B, followed by alkene insertion forming a seven-membered rhodacycle C. Protonation of C generates intermediate D, which undegoes intramolecular cyclization and dehydration to produce the mono C—H activation product and regenerate Rh(Ⅲ) species. Next, a five-membered rhodacycle E is formed via Rh(Ⅲ)-catalyzed C—H activation. The coordination of E with a second alkene generates intermediate F, followed by alkene insertion and protonation delivering the final product and regenerating the Rh(Ⅲ) catalyst.
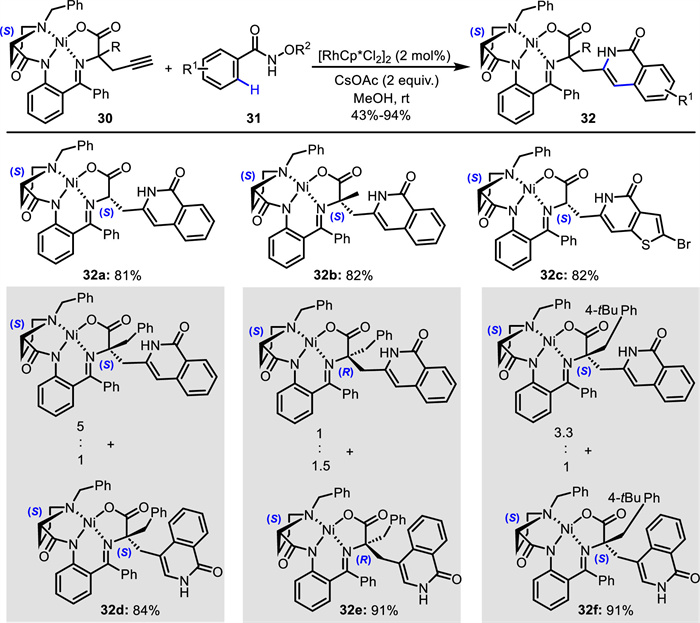
In addition to the design and modification of C—H activation partners, the adjustment of alkynes is also carried out. For example, Larionov and Loginov disclosed an asymmetric synthesis of artificial amino acids 32 bearing an isoquinolone skeleton (Scheme 15) [59]. Through Rh(Ⅲ)-catalyzed C—H activation/annulation of aryl hydroxamates 31 with diverse chiral propargylglycine Ni(Ⅱ) complexes 30 derived from Gly, Ala and Phe, a series of Ni(Ⅱ) complexes 32 comprising an isoquinolone moiety was efficiently synthesized in methanol at room temperature. Notably, for Phe-derived chiral propargylglycine Ni(Ⅱ) complexes (32d-32f), the simultaneous formation of 3- and 4-substituted isoquinolone regioisomers was observed. Moreover, the obtained Ni(Ⅱ) complexes could undergo decomposition using 6 mol/L HCl, offering the corresponding unnatural α-substituted and α,α-disubstituted amino acids having an isoquinolone scaffold in an enantiopure form.
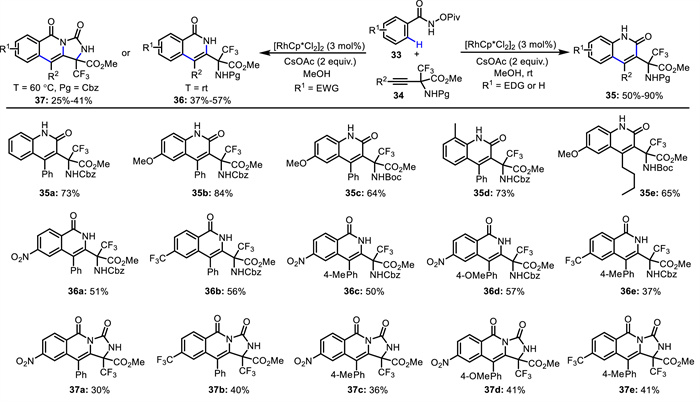
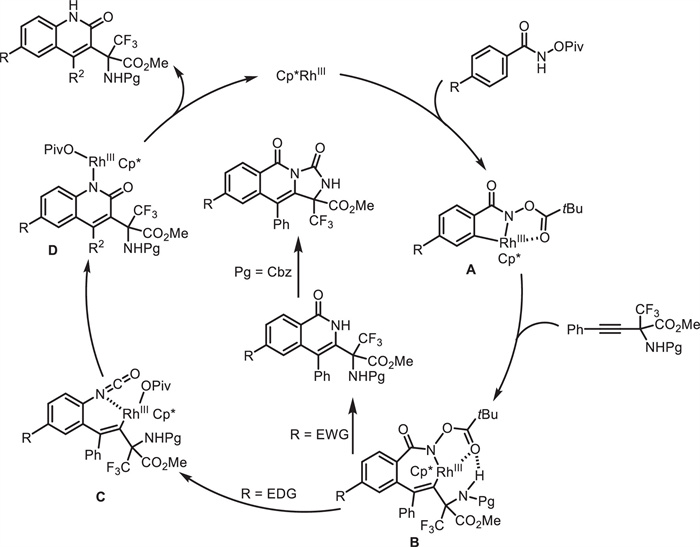
In 2021, Osipov and co-workers developed a convenient method for the synthesis of quinolone-containing amino acid derivatives (Scheme 16) [60]. By using internal alkynes 34 bearing an CF3-containing α-amino acid moiety, N-pivaloyloxy-arylamides 33 with an electron-donating group (EDG) underwent a Rh(Ⅲ)-catalyzed C—H activation and subsequent Lossen rearrangement/annulation process, resulting in 2-quinolone-containing α-CF3-amino carboxylates 35. When N-pivaloyloxy-arylamides 33 with an electron-withdrawing group (EWG) were used, isoquinolone-containing α-CF3-amino carboxylates 36 were obtained. When protecting group (Pg) = Cbz and the reaction temperature was increased from room temperature to 60 ℃, α-CF3-amino carboxylates 37 bearing a tricyclic core were prepared. As proposed (Scheme 17), Rh(Ⅲ)-catalyzed ortho-C-H bond activation of the N-pivaloyloxy amide generates rhodacycle A. Subsequent alkyne insertion proceeds in a regioselective manner, facilitated by intramolecular hydrogen bonding, to afford intermediate B. When R = EDG, Lossen rearrangement occurs to generate intermediate C. Intramolecular addition and protonation deliver the rearranged product and release the active Rh(Ⅲ) catalyst. When R = EWG, sequential reductive elimination, oxidative addition into the N—O bond and protonation give the isoquinolone product. When Pg = Cbz, further intramolecular cyclization occurs to afford tricyclic product.
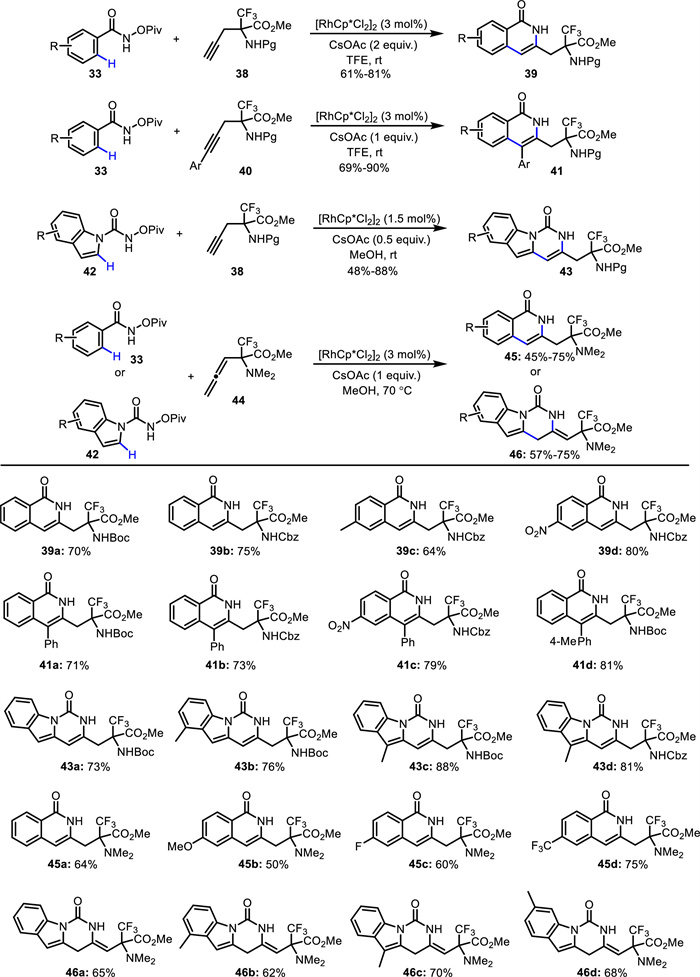
Subsequently, they synthesized a series of new terminal and internal α-amino acid-containing alkynes (38 and 40) bearing one more carbon, and employed them in [4 + 2] cycloaddition reactions (Scheme 18) [61-63]. Through Rh(Ⅲ)-catalyzed C—H activation of N-pivaloyloxy-arylamides 33 or N-(pivaloyloxy)-indole-1-carboxamides 42, diverse CF3-substituted amino acid derivatives bearing an isoquinolone (39 and 41) or pyrimido[1,6-a]indolone (43) core were efficiently constructed with excellent regioselectivity. Besides alkyne-tethered amino acids, allenyl-containing α-amino carboxylates 44 were also tolerated, resulting in an array of unnatural α-amino acids (45 and 46) [64].
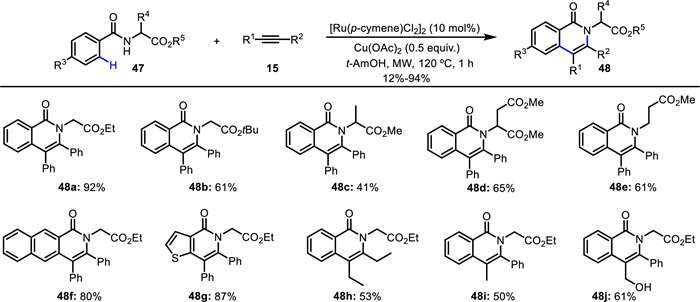
In 2018, Sharma and Van der Eycken reported a Ru(Ⅱ)-catalyzed C—H activation/annulation using an α-amino acid ester (47) as a directing group (Scheme 19) [65]. Under microwave irradiation, a wide range of unnatural amino acids 48 bearing an isoquinolone moiety were efficiently synthesized in 1 h. Diverse symmetrical diaryl- and dialkyl-substituted alkynes as well as unsymmetrical disubstituted alkynes (15) were well tolerated in this reaction. This method was successfully applied to the synthesis of an oxyavicine derivative.
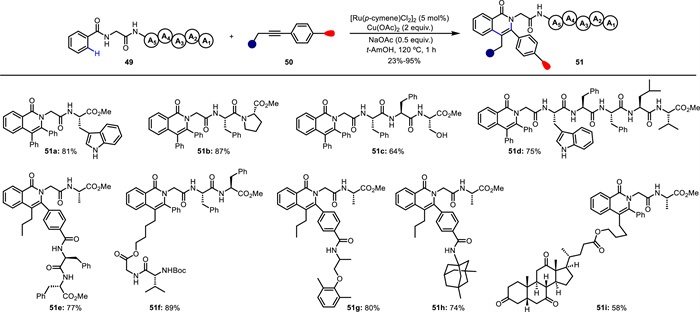
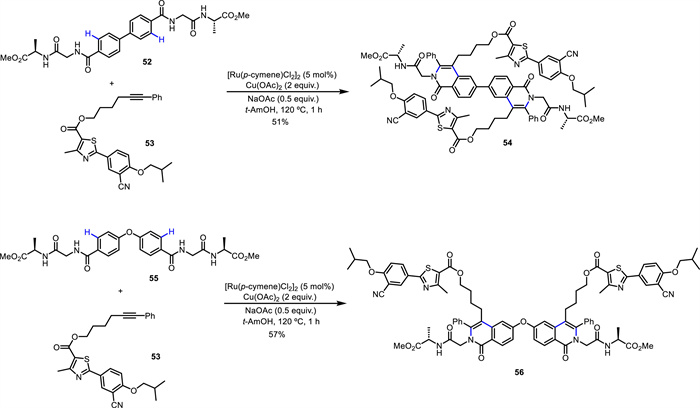
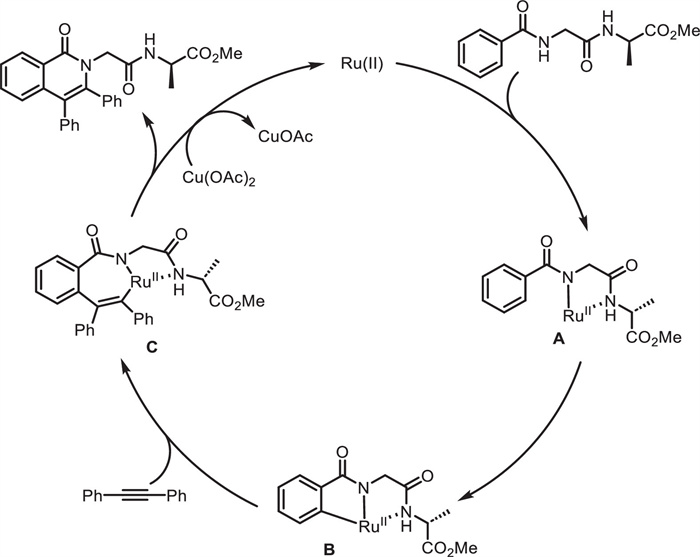
Subsequently, the same group developed an efficient and chemoselective method for the modification of peptide backbone [66]. Through a Ru(Ⅱ)-catalyzed C—H activation/annulation sequence, diverse functionalized isoquinolone scaffolds 51 were constructed in a rapid and step-economical manner (Scheme 20). Under racemization-free conditions, this method was employed in the synthesis of peptide conjugated to drugs (51g and 51h), natural products (51i) and other peptide (51e and 51f) fragments with excellent chemo- and regioselectivity. This reaction gave access to the preparation of novel peptide-pharmacophore conjugates. Notably, a double C—H activation/annulation process was successfully achieved to give biaryl and biaryl-ether dimeric peptide conjugates (54 and 56) (Scheme 21), generating molecular complexity in a short sequence. Moreover, the obtained unnatural peptides showed good fluorescence properties with maximum emission wavelength up to 479 nm. Importantly, mechanistic experiments indicated that amide bonds of the peptide backbone serve as the bidentate directing group to enable Ru(Ⅱ)-catalyzed C—H activation. As proposed (Scheme 22), Ru(Ⅱ) catalyst firstly coordinates with the NH-deprotonated peptide, leading to a five-membered intermediate A. Subsequent C—H bond cleavage gives the tridentate coordinated intermediate B. Squential alkyne insertion and reductive elimination deliver the final product and Ru(0), which is reoxidized by Cu(OAc)2 to regenerate the Ru(Ⅱ) species, closing the catalytic cycle.
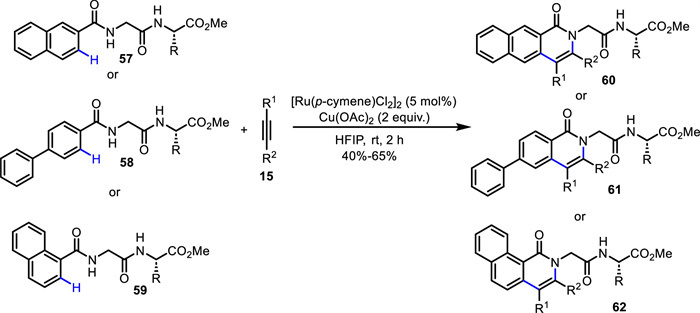
Following the same line, Sharma and co-workers disclosed the construction of novel benzoisoquinolone amino acid/peptide derivatives 60–62 via a Ru(Ⅱ)-catalyzed C—H activation/annulation process at room temperature (Scheme 23) [67]. Naphthamide (60 and 62) and 1,4-biphenylamide (61) amino acid as well as peptides were well tolerated. 1,4-Diphenylbuta-1,3-diyne was also a productive coupling partner. The obtained products displayed good fluorescence properties with maximum emission wavelength up to 455 nm. Notably, the benzoisoquinolinones showed cell internalization to the cell nucleus without any important cytotoxicity to human cell lines HEK293T.
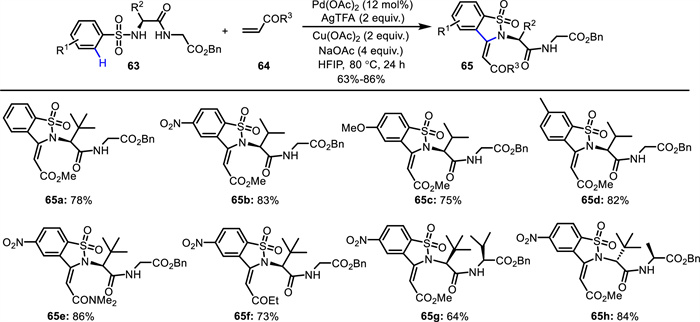
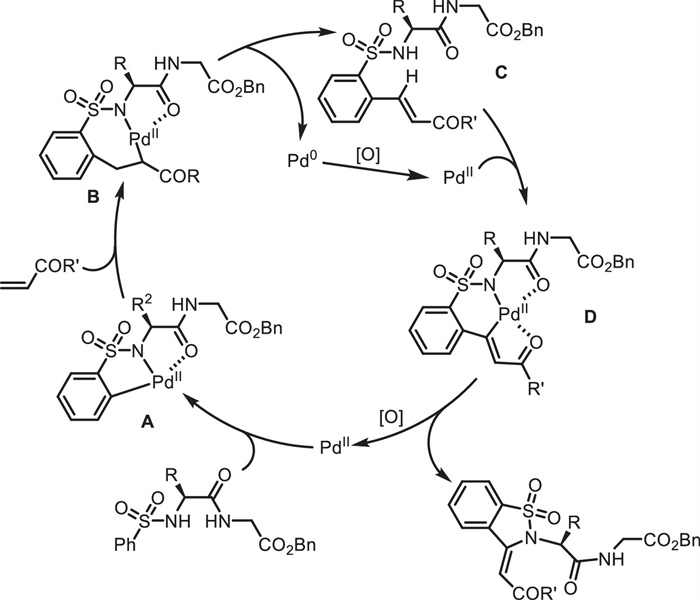
On the other hand, Wang and co-workers described a peptide-guided method for the functionalization of peptidosulfonamides 63 via Pd(Ⅱ)-catalyzed C—H activation (Scheme 24) [68]. In this approach, the peptide backbone served as an internal directing group and facilitated site-selective C—H activation/annulation of benzosulfonamides 63, efficiently leading to benzosultam-peptidomimetics 65. According to the mechanism (Scheme 25), Pd(Ⅱ)-catalyzed ortho-C-H bond activation directed by the N-sulfonated peptide occurs, giving a tridentate coordinated intermediate A. Subsequent alkene insertion and β-H elimination afford olefination intermediate C and the Pd(0) complex, which is reoxidized to regenerate the Pd(Ⅱ) species. Second Pd(Ⅱ)-catalyzed C—H activation at the newly generated olefin produces Pd-centered ternary fused-ring intermediate D. Reductive elimination of D delivers the final cyclic product and the Pd(0) complex, which is reoxidized to regenerate the Pd(Ⅱ) species for the next cycle.
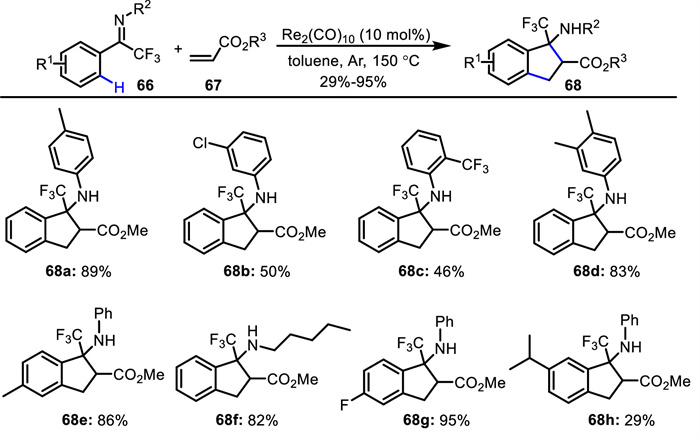
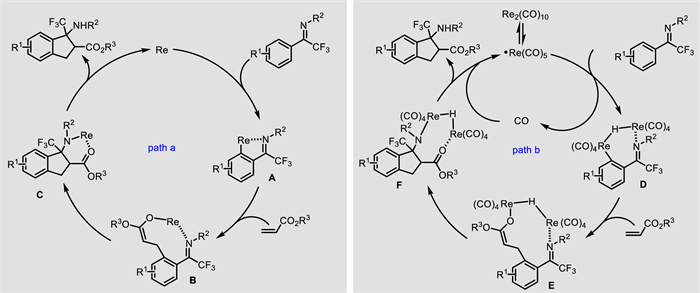
Besides the synthesis of unnatural α-amino acids, the strategies concerning constructing unnatural β-amino acids have also been explored. In 2020, Zhang and Xiong presented a [3 + 2] annulation of trifluoromethylated ketimines 66 with acrylates 67, leading to a series of β-CF3 β-amino esters 68 with an indane backbone (Scheme 26) [69]. This rhenium-catalyzed C—H activation showed broad substrate scope and a gram scale synthesis was elaborated. According to the experimental results, two plausible catalytic cycles were proposed (Scheme 27). On the one hand (path a), rhenium-catalyzed C—H activation occurs to give a five-membered intermediate A. Subsequent alkene insertion affords rhenium enolate B, followed by intramolecular Mannich reaction and proto-demetalation delivering the final product and regenerating the active rhenium catalyst. On the other hand (path b), Re2(CO)10 undergoes homolytic Re-Re bond cleavage to give the Re(CO)5 radical, which interacts with the ketimine to generate dinuclear Re complex D via CO dissociation and C—H bond cleavage. Subsequent alkene insertion, intramolecular Mannich reaction and reductive elimination deliver the final product and regenerate the active rhenium catalyst.
Natural peptides are usually not be used as direct therapeutics due to their intrinsic weaknesses, such as poor physical and chemical stability, and a short circulating plasma half-life. Unnatural peptides are often essential for academic and industry circles, because of the enhanced pharmacokinetics and bioactivity in contrast with the natural ones. Thus, concise and efficient accesses to unnatural amino acids and peptides have been investigated. Among them, chemo- and site-selective methods are the key points in modern chemistry, due to the capacity to fine-tune structural characters that can help modulate biological and physicochemical properties. Compared to classical condensations and (cyclo)additions as well as transition metal-catalyzed cross-coupling reactions, transition metal-catalyzed C—H activation shows great advantages in the modifications of amino acids and peptides. Without substrate prefunctionalizations, diverse transformations can be performed in a high atom- and step-economical fashion. Recently, transition metal-catalyzed C—H activation/annulation has been employed in the late-stage derivatizations of amino acids and peptides. For Rh-catalyzed C—H activation/annulation, by using amide as directing group, an array of structurally novel amino acids and peptides with different kinds of heterocycles is achieved, such as bearing quinolone, isoquinolone, benzazepine, indolizinone, quinolizinone, pyrimido[1,6-a]indolone and polycyclic aromatic hydrocarbon oxonium ion scaffolds. Employing N-phenoxy amide or phosphinamide as directing group gives amino acids bearing a dihydrobenzo[d]isoxazole or a phosphaisoquinolin-1-one moiety, respectively. Utilizing phenol as directing group affords amino acids and peptides bearing a chromone core. For other transition metal-catalyzed C—H activation/annulation, such as Ru, Pd and Re, by using amide or sulfonamide as directing group, amino acids and peptides bearing isoquinolone or benzosultam scaffolds are respectively generated. Employing imine as directing group produces amino acids bearing an indane moiety.
Despite major advances, some limitations still remain. Firstly, the directing groups are mainly limited to amide-type groups, largely restricting the reaction types. Exploring other kinds of directing groups is necessary for the C—H activation/annulation process, resulting in novel reactions. Secondly, except alkyne and alkene, other coupling partners are seldom investigated. Expanding the scope of the coupling partners would deliver structurally novel unnatural amino acids and peptides. Thirdly, enantioselective C—H activation/annulation has not yet been reported for the construction of enantiopure unnatural amino acids and peptides. Especially, when the generated heterocycles own an extra chiral center, it will be more difficult to achieve the control over diastereoselectivity. Therefore, developing effective strategies for enantioselective C—H activation/annulation is demanded, which is also important for new peptide drug discovery. Finally, developing mild and efficient transition metal-catalyzed C—H activation/annulation for the synthesis of unnatural amino acids and peptides is another direction. We hope this review will be beneficial for researchers to better understand the chemistry behind transition metal-catalyzed C—H activation/annulation for the construction of unnatural amino acids and peptides.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Xiao Tang: Writing – original draft. Erik V. Van der Eycken: Writing – review & editing, Conceptualization. Liangliang Song: Writing – review & editing, Writing – original draft, Supervision, Funding acquisition, Conceptualization.
This work was supported by the Natural Science Foundation of Jiangsu Province (No. BK20220409) and the National Natural Science Foundation of China (No. 22401153). The authors acknowledge the FWO [Fund for Scientific Research-Flanders (Belgium)] for financial support (recipient Erik V. Van der Eycken) and the Research Council of the KU Leuven (recipient Erik V. Van der Eycken). This paper has been prepared with the support of the “RUDN University Strategic Academic Leadership Program” (recipient Erik V. Van der Eycken).
H. Buchwald, R.B. Dorman, N.F. Rasmus, et al., Surg. Obes. Relat. Dis. 10 (2014) 780–786.
C. Giordano, M. Marchiò, E. Timofeeva, G. Biagini, Front. Neurol. 5 (2014) 63.
K. Fosgerau, T. Hoffmann, Drug Discov. Today 20 (2015) 122–128.
S.R. Gracia, K. Gaus, N. Sewald, Future Med. Chem. 1 (2009) 1289–1310. doi: 10.4155/fmc.09.97
M. Muttenthaler, G.F. King, D.J. Adams, P.F. Alewood, Nat. Rev. Drug Discov. 20 (2021) 309–325. doi: 10.1038/s41573-020-00135-8
A.A. Kaspar, J.M. Reichert, Drug Discov. Today 18 (2013) 807–817.
A. Henninot, J.C. Collins, J.M. Nuss, J. Med. Chem. 61 (2018) 1382–1414. doi: 10.1021/acs.jmedchem.7b00318
F. Albericio, H.G. Kruger, Future Med. Chem. 4 (2012) 1527–1531. doi: 10.4155/fmc.12.94
A.F.B. Räder, M. Weinmüller, F. Reichart, et al., Angew. Chem. Int. Ed. 57 (2018) 14414–14438. doi: 10.1002/anie.201807298
D.J. Craik, D.P. Fairlie, S. Liras, D. Price, Chem. Biol. Drug Des. 81 (2013) 136–147. doi: 10.1111/cbdd.12055
T. Sun, X. Guan, M. Zheng, X. Jing, Z. Xie, ACS Med. Chem. Lett. 6 (2015) 430–433. doi: 10.1021/acsmedchemlett.5b00041
A. Vázquez-Romero, N. Kielland, M.J. Arévalo, et al., J. Am. Chem. Soc. 135 (2013) 16018–16021. doi: 10.1021/ja408093p
K.M. Marks, G.P. Nolan, Nat. Methods 3 (2006) 591–596. doi: 10.1038/nmeth906
P. Agarwal, C.R. Bertozzi, Bioconjugate Chem. 26 (2015) 176–192. doi: 10.1021/bc5004982
Y. Wang, A.G. Cheetham, G. Angacian, et al., Adv. Drug Deliver. Rev. 110-111 (2017) 112–126.
P. Hoppenz, S. Els-Heindl, A.G. Beck-Sickinger, Front. Chem. 8 (2020) 571.
I.W. Hamley, Biomacromolecules 15 (2014) 1543–1559. doi: 10.1021/bm500246w
M.A. Gauthier, H.A. Klok, Chem. Commun. (2008) 2591–2611. doi: 10.1039/b719689j
S. Dehn, R. Chapman, K.A. Jolliffe, S. Perrier, Polym. Rev. 51 (2011) 214–234. doi: 10.1080/15583724.2011.566404
J.N. deGruyter, L.R. Malins, P.S. Baran, Biochemistry 56 (2017) 3863–3873. doi: 10.1021/acs.biochem.7b00536
C. Zhang, E.V. Vinogradova, A.M. Spokoyny, S.L. Buchwald, B.L. Pentelute, Angew. Chem. Int. Ed. 58 (2019) 4810–4839. doi: 10.1002/anie.201806009
H. Jiang, W. Chen, J. Wang, R. Zhang, Chin. Chem. Lett. 33 (2022) 80–88.
J. Li, J. Chen, Q. Hu, Z. Wang, X.F. Xiong, Chin. Chem. Lett. 36 (2025) 110126.
M. Jbara, S.K. Maity, A. Brik, Angew. Chem. Int. Ed. 56 (2017) 10644–10655. doi: 10.1002/anie.201702370
J. He, M. Wasa, K.S.L. Chan, Q. Shao, J.Q. Yu, Chem. Rev. 117 (2017) 8754–8786. doi: 10.1021/acs.chemrev.6b00622
G. Rouquet, N. Chatani, Angew. Chem. Int. Ed. 52 (2013) 11726–11743. doi: 10.1002/anie.201301451
G. Song, F. Wang, X. Li, Chem. Soc. Rev. 41 (2012) 3651–3678. doi: 10.1039/c2cs15281a
S.K. Sinha, S. Guin, S. Maiti, et al., Chem. Rev. 122 (2022) 5682–5841. doi: 10.1021/acs.chemrev.1c00220
B. Liu, A.M. Romine, C.Z. Rubel, K.M. Engle, B.F. Shi, Chem. Rev. 121 (2021) 14957–15074. doi: 10.1021/acs.chemrev.1c00519
B.B. Zhan, M.X. Jiang, B.F. Shi, Chem. Commun. 56 (2020) 13950–13958. doi: 10.1039/d0cc06133f
H.R. Tong, B. Li, G. Li, G. He, G. Chen, CCS Chem. 3 (2021) 1797–1820.
Q. Zhang, X.S. Yin, K. Chen, S.Q. Zhang, B.F. Shi, J. Am. Chem. Soc. 137 (2015) 8219–8226. doi: 10.1021/jacs.5b03989
Z. Bai, Q. Chen, J. Gu, et al., ACS Catal. 11 (2021) 15125–15134. doi: 10.1021/acscatal.1c05030
W. Gong, G. Zhang, T. Liu, R. Giri, J.Q. Yu, J. Am. Chem. Soc. 136 (2014) 16940–16946. doi: 10.1021/ja510233h
B. Li, X. Li, B. Han, et al., J. Am. Chem. Soc. 141 (2019) 9401–9407. doi: 10.1021/jacs.9b04221
Z. Bai, C. Cai, Z. Yu, H. Wang, Angew. Chem. Int. Ed. 57 (2018) 13912–13916. doi: 10.1002/anie.201807953
M.M. Lorion, N. Kaplaneris, J. Son, R. Kuniyil, L. Ackermann, Angew. Chem. Int. Ed. 58 (2019) 1684–1688. doi: 10.1002/anie.201811668
N. Kaplaneris, J. Son, L. Mendive-Tapia, et al., Nat. Commun. 12 (2021) 3389.
B.B. Zhan, Y. Li, J.W. Xu, et al., Angew. Chem. Int. Ed. 57 (2018) 5858–5862. doi: 10.1002/anie.201801445
X. Chen, F. Ye, X. Luo, et al., J. Am. Chem. Soc. 141 (2019) 18230–18237. doi: 10.1021/jacs.9b09127
A. Schischko, N. Kaplaneris, T. Rogge, et al., Nat. Commun. 10 (2019) 3553.
W. Wang, M.M. Lorion, J. Shah, A.R. Kapdi, L. Ackermann, Angew. Chem. Int. Ed. 57 (2018) 14700–14717. doi: 10.1002/anie.201806250
Z. Bai, H. Wang, Synlett 31 (2020) 199–204.
X. Zhang, G. Lu, M. Sun, et al., Nat. Chem. 10 (2018) 540–548. doi: 10.1038/s41557-018-0006-y
L. Mendive-Tapia, S. Preciado, J. García, et al., Nat. Commun. 6 (2015) 7160.
L. Bai, X. Jiang, Chem. Catal. 3 (2023) 100752.
L. Song, G. Tian, Y. He, E.V. Van der Eycken, Chem. Commun. 53 (2017) 12394–12397.
L. Song, G. Tian, A. Blanpain, L. Van Meervelt, E.V. Van der Eycken, Adv. Synth. Catal. 361 (2019) 4442–4447. doi: 10.1002/adsc.201900550
H. Gao, M. Sun, H. Zhang, et al., Org. Lett. 21 (2019) 5229–5233. doi: 10.1021/acs.orglett.9b01831
A. Schischko, H. Ren, N. Kaplaneris, L. Ackermann, Angew. Chem. Int. Ed. 56 (2017) 1576–1580. doi: 10.1002/anie.201609631
Z. Ruan, N. Sauermann, E. Manoni, L. Ackermann, Angew. Chem. Int. Ed. 56 (2017) 3172–3176. doi: 10.1002/anie.201611118
M. Stangier, A.M. Messinis, J.C.A. Oliveira, H. Yu, L. Ackermann, Nat. Commun. 12 (2021) 4736.
X.H. Li, J.F. Gong, M.P. Song, Chem. Asian J. 17 (2022) e202101158.
N.D. Kharat, C.K. Mahesha, K. Bajaj, R. Sakhuja, Org. Lett. 24 (2022) 6857–6862. doi: 10.1021/acs.orglett.2c02820
L. Song, Z. Lv, Y. Li, et al., Org. Lett. 25 (2023) 2996–3000. doi: 10.1021/acs.orglett.3c00766
L. Song, Z. Zhang, Z. Lv, et al., ACS Catal. 13 (2023) 13569–13576. doi: 10.1021/acscatal.3c03783
Z. Zhang, T. Wan, Q. Quan, et al., Org. Lett. 26 (2024) 10915–10920. doi: 10.1021/acs.orglett.4c04081
Q. Quan, Y. Li, Z. Zhang, et al., Org. Lett. 27 (2025) 482–487. doi: 10.1021/acs.orglett.4c04498
M.A. Arsenov, N.V. Stoletova, T.y.F. Savel’yeva, et al., Org. Biomol. Chem. 20 (2022) 9385–9391. doi: 10.1039/d2ob01970a
D.A. Petropavlovskikh, D.V. Vorobyeva, I.A. Godovikov, et al., Org. Biomol. Chem. 19 (2021) 9421–9426. doi: 10.1039/d1ob01711j
D.V. Vorobyeva, A.S. Bubnova, I.A. Godovikov, A.A. Danshina, S.N. Osipov, Asian J. Org. Chem. 12 (2023) e202200485.
D.V. Vorobyeva, D.A. Petropavlovskikh, I.A. Godovikov, F.M. Dolgushin, S.N. Osipov, Molecules 27 (2022) 8488. doi: 10.3390/molecules27238488
D.V. Vorobyeva, D.A. Petropavlovskikh, I.A. Godovikov, S.E. Nefedov, S.N. Osipov, Eur. J. Org. Chem. 2021 (2021) 1883–1890. doi: 10.1002/ejoc.202100040
D.V. Vorobyeva, A.S. Bubnova, I.A. Godovikov, A.F. Smol’yakov, S.N. Osipov, Asian J. Org. Chem. 14 (2025) e202400476.
N. Sharma, V. Bahadur, U.K. Sharma, et al., Adv. Synth. Catal. 360 (2018) 3083–3089. doi: 10.1002/adsc.201800458
L. Song, G.M. Ojeda-Carralero, D. Parmar, et al., Adv. Synth. Catal. 363 (2021) 3297–3304. doi: 10.1002/adsc.202100323
M.K. Gupta, A. Panda, N.K. Sharma, J. Org. Chem. 89 (2024) 7485–7494. doi: 10.1021/acs.joc.4c00048
J. Tang, H. Chen, Y. He, et al., Nat. Commun. 9 (2018) 3383.
T. Hu, Y. Xu, S. Zhang, H.Y. Xiong, G. Zhang, Org. Lett. 22 (2020) 8866–8871. doi: 10.1021/acs.orglett.0c03239

Scheme 9 Rh(Ⅲ)-catalyzed double C—H activation/annulation of Lys-based amino acids and peptides.

Scheme 10 Proposed mechanism for the formation of tricyclic-fused aromatic hydrocarbon oxonium ions.

Scheme 16 Rh(Ⅲ)-catalyzed C—H activation and subsequent Lossen rearrangement/annulation.

Scheme 17 Proposed mechanism for Rh(Ⅲ)-catalyzed C—H activation and subsequent Lossen rearrangement/annulation.

Scheme 20 Ru(Ⅱ)-catalyzed C—H activation for the synthesis of peptide-pharmacophore conjugates.

Scheme 21 Double C—H activation/annulation for the synthesis of dimeric peptide conjugates.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:



















