Received Date:
09 May 2025 Accepted Date:
22 July 2025 Revised Date:
04 July 2025 Available Online:
15 February 2026
Abstract:
Carbon-rich cycloarene macrocycles can adopt multiple atropisomeric forms due to steric hindrance restricting σ-bond rotation. These distinct conformations exhibit variations in cavity structure, electronic properties, and functional site distribution, leading to diverse molecular recognition and self-assembly behaviors. In recent years, research on carbon-rich cycloarene macrocyclic compounds has emerged as a cutting-edge and interdisciplinary focus in the fields of carbon-rich functional molecules and macrocyclic chemistry. This review provides a comprehensive overview of the development of atropisomers in carbon-rich cycloarene macrocycles, spanning their design and synthesis, optoelectronic properties, and supramolecular chemistry.
In recent decades, carbon-rich macrocycles, whose backbones are composed exclusively of sp2− and/or sp-hybridized carbon atoms, have emerged as structurally versatile platforms in supramolecular chemistry. Their unique electronic properties and rigid π-conjugated frameworks enable precise spatial arrangement and strong intermolecular interactions, making them attractive candidates for the development of advanced materials [1-7]. In parallel, atropisomerism, a phenomenon arising from the restricted rotation around σ bonds, has gained significant attention in chemistry. Molecules exhibiting atropisomerism can adopt stable stereoisomeric forms due to steric hindrance, which can be exploited to modulate molecular recognition, assembly behavior, and chiral functionality. Representative examples include calix[4]arenes and pillararenes, which can be locked into specific conformations through the strategic introduction of bulky substituents [8-13]. Each atropisomer exhibits unique cavity structures and functional site distributions, resulting in diverse molecular recognition and self-assembly behaviors [14-34].
In contrast to the extensively studied conventional macrocyclic systems, the rotational isomers of carbon-rich aromatic macrocycles represent a distinct and underexplored class of compounds with unique scientific significance. On the one hand, these systems serve as a conceptual bridge between traditional macrocyclic chemistry and the field of dynamic stereochemistry, offering new perspectives on conformational control and molecular motion. On the other hand, the ability to modulate their conformational dynamics provides a powerful means to influence key functional properties, including optoelectronic behavior, host–guest recognition, and chiroptical responses. These characteristics highlight the potential of controlling macrocyclic atropisomerism as promising strategies for the development of stimuli-responsive smart materials. However, the research on the atropisomerism of carbon-rich cycloarene macrocycles remains underexplored and is still in its early developmental stages. This is primarily due to their intricate topological structures, which present significant challenges in understanding their transformation mechanisms and developing precise regulation strategies.
Herein, this review is structured into three main sections, providing a comprehensive discussion of recent advances in the study of enantiotropic and non-enantiotropic atropisomerism in carbon-rich cycloarene macrocycles. These macrocycles, such as cyclo[n]paraphenylenes and nanobelts, atropisomerism typically originates from conformational restrictions associated with twisted or folded π-conjugated frameworks. These topological distortions lead to significant hindrance in bond rotation, particularly when the macrocycle is fused with sterically demanding aromatic subunits (e.g., chrysene) or bears bulky substituents near the bridging positions between adjacent aromatic rings. Such steric and geometric constraints stabilize distinct atropisomeric conformations, rendering them isolable and functionally tunable. Specifically, this review covers key aspects including synthesis, structural characteristics, optical and coordination properties, supramolecular behavior, and potential applications. Furthermore, it highlights the necessity of future research efforts in molecular design, structure-activity relationship analysis, and the development of stimulus-responsive self-assembly systems. Emphasis is placed on advancing precise regulation strategies and promoting the application of high-performance materials.
2.
Enantiotropic atropisomers of carbon-rich cycloarene macrocycles
2.1
Enantiotropic atropisomers in carbon-rich cycloarenes featuring a single macrocyclic framework
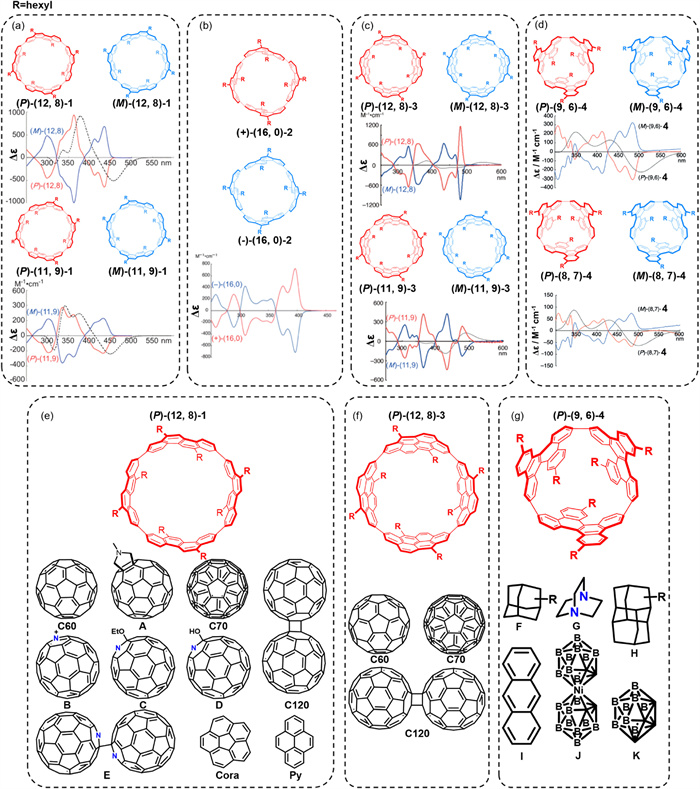
Atropisomerization-based design is an effective strategy for the generation of chiral carbon-rich cycloarene macrocycles. In 2011, Isobe's group employed a bottom-up approach to synthesize macrocycle 1 using 2,8-dibromochrysene as a building block, establishing a constrained model for helical (n,m)-single-walled carbon nanotubes (SWNTs) [35]. High-performance liquid chromatography (HPLC) enabled the successful separation of all rotational isomers of macrocycle 1. Among them, the (12,8)-1 and (11,9)-1 isomers were further resolved into their enantiomers, (P)- and (M)-forms, using chiral HPLC, and their chiral properties were characterized by CD spectroscopy (Fig. 1a). Notably, this study revealed a stereochemical bias during the synthesis: when excess cholesteryl stearate was employed in the reductive elimination step, the resulting macrocycle 1 showed preferential formation of (P)-isomers over (M)-isomers, with enantiomeric excesses (ee) of 17% for (12,8)-1 and 11% for (11,9)-1. This asymmetric induction phenomenon offers valuable insight for the future design of stereoselective synthetic strategies. Over the past decade, Isobe and co-workers have reported a series of structurally defined, chiral carbon-rich macrocycles (2–4) derived from chrysene units via platinum-mediated cyclization [36-38]. Several of these molecules, such as (16,0)-2, (12,8)-3, (11,9)-3, (9,6)-4, and (8,7)-4, exhibited distinct atropisomeric conformations. Their enantiomers were successfully separated using chiral HPLC and their chiroptical properties were confirmed by CD spectroscopy (Figs. 1b–d).
Figure 1
Figure 1.
A series of chiral carbon-rich macrocycles incorporating chrysene motifs and their circular dichroism (CD) spectra: (a) (12,8)-1 and (11,9)-1; (b) (16,0)-2; (c) (12,8)-3 and (11,9)-3; (d) (9,6)-4 and (8,7)-4. Representative guest molecules exhibiting notable host–guest interactions with macrocycles: (e) (12,8)-1, (f) (12,8)-3, and (g) (9,6)-4. Adapted with permission [32-35]. Copyright 2011, Springer Nature Limited; Copyright 2012, American Chemical Society; Copyright 2013, The Royal Society of Chemistry; and Copyright 2019, Wiley-VCH Verlag GmbH & Co. KGaA.
Among these, (12,8)-1 displayed particularly rich supramolecular behavior. It formed strong CH–π interactions with various guest molecules. In 2013, it was demonstrated that (12,8)-1 can co-crystallize with C60 to form a solid-state rotor that undergoes processional and spinning motions at different temperatures. Functionalization of C60 to produce N-methyl fulleropyrrolidine (guest A) enabled the design of a uniaxially molecular rotators bearing (12,8)-1 [39]. Subsequent studies showed that (12,8)-1 also encapsulates nitrogen-containing C60 derivatives (guests B–E) in stable complexes [40]. Additionally, (12,8)-1 formed stable complexes with C70, with a binding constant of log Ka = 9.5, comparable to that of C60 (logKa = 9.6), indicating similar affinity [41].
The host–guest complexation extended to the dumbbell-shaped dimer [60]fullerene (C120), where (12,8)-1 spontaneously formed a 2:1 supramolecular assembly through a two-step binding process. The corresponding association constants were logKa1 = 11.9 and logKa1 = 8.0, with a Gibbs free energy difference (ΔG) of 5 kcal/mol between the two steps [42]. Beyond spherical fullerenes, (12,8)-1 also interacted with planar and bowl-shaped guests. For instance, it formed both 1:1 and 1:2 complexes with corannulene (Cora), where the Ka for the 1:1 complex was 2.94 × 103 L/mol. Single-crystal X-ray diffraction confirmed the 1:2 complex, wherein weak hydrogen bonding facilitated ultrafast guest rotation within the host at gigahertz frequencies [43]. In contrast, binding with the more planar and elliptical guest pyrene (Py) yielded weaker binding (Ka = 41–174 L/mol), and the resulting steric constraints significantly inhibited rotational freedom, enabling design of decelerated molecular rotors [44]. This tunable dynamic behavior illustrates a promising strategy for molecular machine design.
Compared to (12,8)-1, macrocycle (12,8)-3 exhibited narrower guest selectivity. It formed complexes with C60, C70, and C120, with similar binding affinities: logKa = 9.7 for C60 and 9.6 for C70. The stepwise binding with C120 yielded logKa1 = 9.2 and logKa2 = 5.7, both lower than those observed for (12,8)-1 [41,45]. Owing to its deeper cavity, (12,8)-3 promoted the formation of ratchet-like host–guest systems with reduced rotational disorder. Experimental and theoretical investigations have since explored the dynamic motions within these complexes [46-48].
In contrast, macrocycle (9,6)-4 exhibited unique host–guest behavior, forming CH–π complexes with guests containing sp3- and sp2-hybridized carbon centers (guests F–K) [49]. These complexes displayed moderate binding affinities (Ka = 110–183 L/mol) and spontaneously assembled into exfoliable, layered two-dimensional (2D) honeycomb crystals, enabling centimeter-scale single-crystal growth. Recent studies have further elucidated the dynamic behavior of these assemblies using both experimental techniques and computational modeling [50,51].
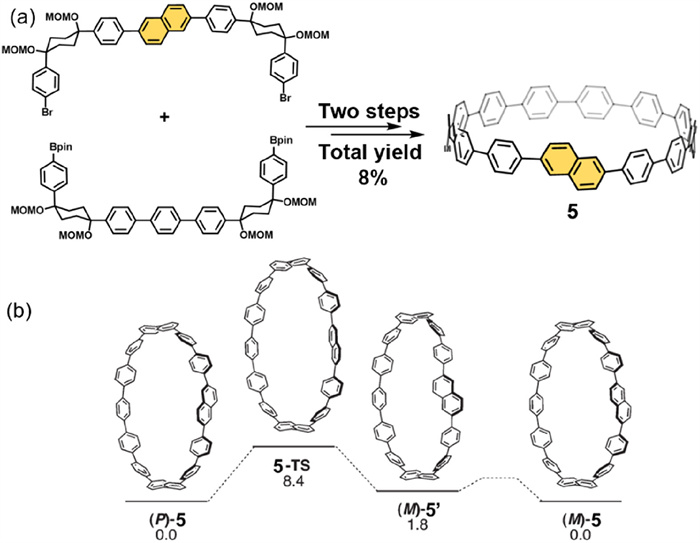
In 2011, Itami's group synthesized the chiral molecule 5 ([13]CPPN) by incorporating a naphthalene unit into cycloparaphenylenes (CPPs) using a [7 + 7] fragment coupling Strategy (Fig. 2a) [52]. However, due to its low racemization barrier (8.4 kcal/mol), a pair of enantiomers separation was not feasible at room temperature (Fig. 2b). Computational studies further revealed that reducing the number of benzene units or increasing the number of acene units would elevate the racemization barrier. These findings suggest that such nanorings have the potential to form stable chiral molecules, providing both experimental and theoretical foundations for future molecular design.
Figure 2
Figure 2.
(a) Synthetic strategy for the preparation of chiral macrocycle 5. (b) Racemization pathway from (P)-5 to (M)-5, with the corresponding activation energy (ΔG‡, kcal/mol). Reproduced with permission [52]. Copyright 2011, American Chemical Society.
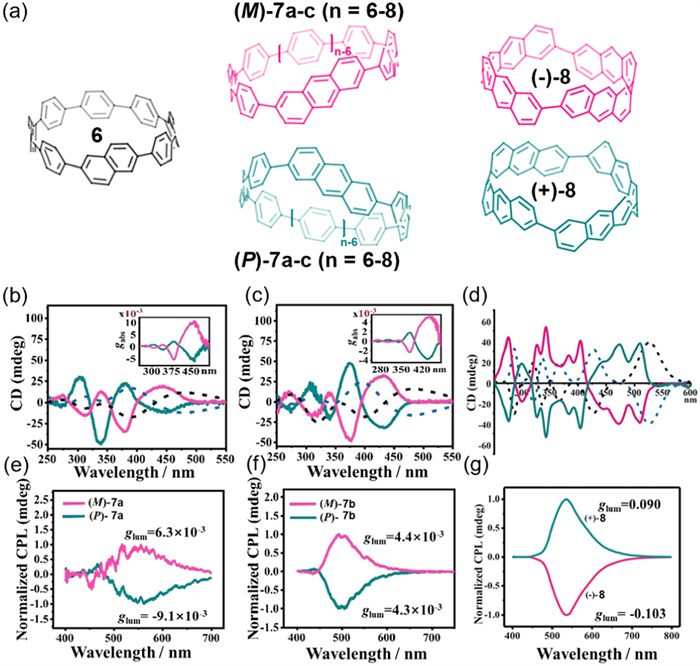
Since 2019, Du's group has synthesized chiral CPPs 6 [53], 7a–c [54], and 8 [55] by incorporating naphthalene or anthracene fragments as structural units (Fig. 3a). Compounds 7a, 7b, and 8 were successfully resolved into their enantiomers by chiral HPLC at room temperature and exhibited strong chiral signals in both circular dichroism (CD) and circularly polarized luminescence (CPL) spectra (Figs. 3b–g). Notably, compound 8 displayed an exceptionally high luminescence asymmetry factor (|glum|) up to 0.10, demonstrating excellent chiral luminescence properties. The high magnitude of |glum| observed in this system offers valuable insights for the rational design of next-generation chiral luminescent materials. However, the host–guest recognition properties of this series of macrocycles remain largely unexplored, representing a promising direction for future research.
Figure 3
Figure 3.
(a) A series of cycloparaphenylenes (CPPs) (6, 7a-c, 8) incorporating naphthalene or anthracene fragments. (b-d) Circular dichroism (CD) spectrum of (P/M)-7a, (P/M)-7b and (±)-8, respectively. (e-g) Circularly polarized luminescence (CPL) spectra of (P/M)-7a, (P/M)-7b and (±)-8, respectively. Reproduced with permission [54,55]. Copyright 2022, Wiley-VCH GmbH. Copyright 2020, Wiley-VCH Verlag GmbH & Co. KGaA.
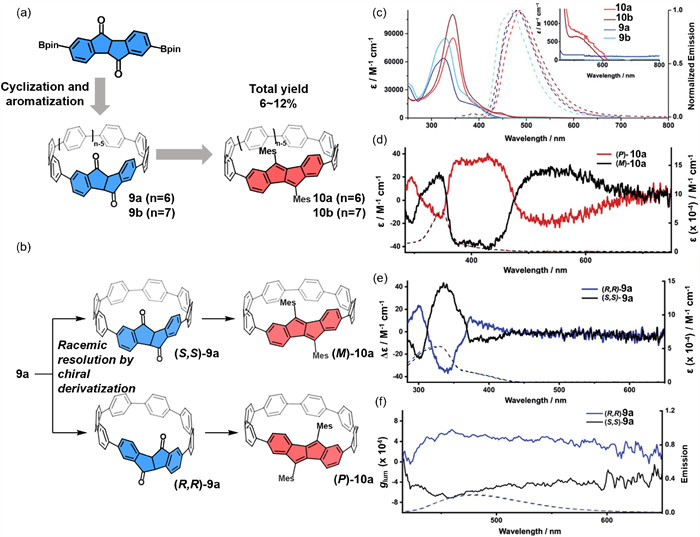
In 2021, Esser's team reported diketone [n]CPPs (9a, n = 6; 9b, n = 7) and DBP[n]CPPs (10a, n = 6; 10b, n = 7) with diketone core (Fig. 4a) [56]. The UV–vis absorption and fluorescence emission spectra of 9a and 9b were highly similar, as were those of 10a and 10b Notably, DBP[n]CPPs (10a and 10b) exhibited a slight red shift in both spectra compared to diketone [n]CPPs (9a and 9b) (Fig. 4c). Moreover, the authors successfully resolved a pair of enantiomers of 9avia chiral derivatization, achieving an enantiomeric excess (ee) up to 99% (Fig. 4b). The chiroptical properties of the resolved enantiomers were further characterized using CD and CPL spectroscopy (Figs. 4c–f). Compound 9a exhibited a high fluorescence quantum yield (ΦF = 57%) with a |glum| value of approximately 4 × 10–4 at its emission peak. This synthetic strategy enables the scalable production of enantiomerically pure nanohoops, thereby facilitating their integration into advanced applications across materials science and chirality research.
Figure 4
Figure 4.
(a) Synthetic strategy for diketone-functionalized [n]cyclopara phenylenes (CPPs) 9a (n = 6) and 9b (n = 7), as well as dibenzo[a,e]pentalene-embedded [n]CPPs (DBP[n]CPPs) 10a (n = 6) and 10b (n = 7). (b) A pair of enantiomers resolution of 9a through chiral derivatization of the carbonyl groups to access enantiopure nanohoops 10. (c) Absorption (10–5 to 10–6 mol/L) and emission spectra (10–6 to 10–7 mol/L) of 9a-b and 10a-b in CH2Cl2. (d) CD spectra of (P/M)-10a. (e) CD spectrum of (R,R/S,S)-9a. (f) CPL spectra of (R,R/S,S)-9a. Reproduced with permission [56]. Copyright 2021, The Royal Society of Chemistry.
In the same year, Esser's group enantioselectively synthesized the chiral conjugated nanohoop 11 from enantiopure precursors (R-/S-diketone) and characterized its chiroptical properties [57]. CD spectroscopy revealed a pronounced Cotton effect across the 300–700 nm range (Fig. 5), confirming its strong chiral response. Nanohoop 11, characterized by its cyclic geometry and extended π-conjugated surface, is particularly well-suited for encapsulating guest molecules such as C60. NMR titration experiments combined with DFT calculations reveal that nanohoop 11 is capable of binding up to two C60 molecules, with Ka in the range of 102–103 L/mol. This binding capacity is attributed to its relatively large internal diameter (~2.5 nm), which accommodates multiple fullerene units.
Figure 5
Figure 5.
(a) Schematic representation of the synthesis of nanohoop (±)-11via macrocyclization of an enantiomerically pure diketone precursor. (b) CD spectra of (±)-11. Reproduced with permission [57]. Copyright 2021, The Authors.
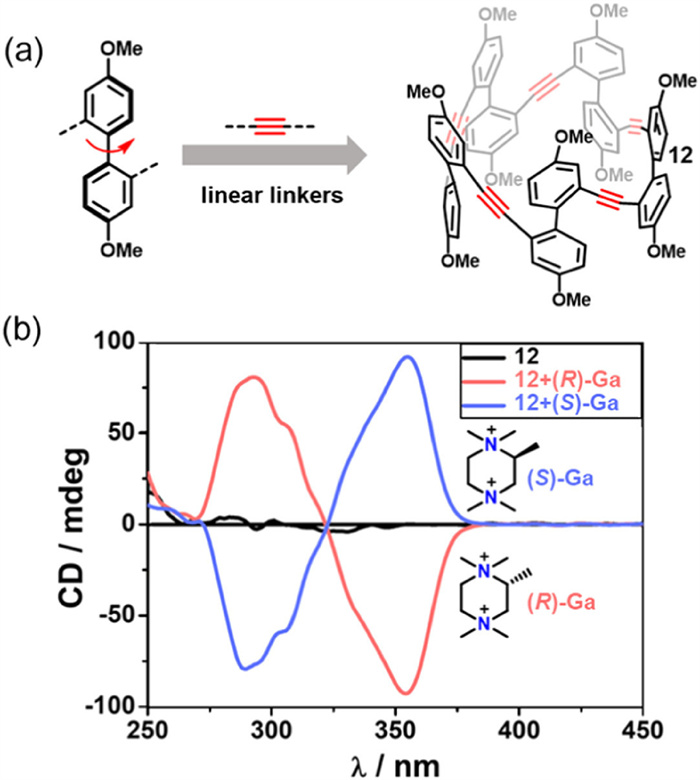
In recent years, BINOL-derived macrocycles have also been reported, showing their unique optoelectronic properties and host-guest characteristics [58]. In 2022, Cai’s group reported the synthesis of a flexible tubular macrocycle (12) exhibiting five-fold symmetry, constructed from 4,4′-dimethoxybiphenyl building blocks (Fig. 6a) [59]. Macrocycle 12 features a fully conjugated backbone and emits strong blue fluorescence with a ΦF value of 56%, indicating its potential for use in luminescent materials and fluorescence-based sensing platforms. The flexible biphenyl segments enable dynamic conformational inversion in solution. Upon addition of chiral guest molecules such as (S)-Ga or (R)-Ga, 12 exhibits induced CD signals, highlighting its chiral responsiveness and potential for enantioselective sensing (Fig. 6b). In the context of host–guest chemistry, macrocycle 12 combines a deep cavity with high conformational adaptability, enabling it to accommodate a wide range of neutral and cationic electron-deficient guests. Its cavity can adapt to diverse molecular geometries, from planar guests like tetracyanoquinodimethane (TCNQ) to spherical fullerenes such as C60, with binding constants spanning from 103 L/mol to 107 L/mol. Notably, the binding constant with CoCp2+ reaches 4.79 × 107 L/mol, while that with C60 is 2.25 × 104 L/mol. These results underscore the versatility of 12 as a supramolecular host, providing a valuable platform for the development of functional materials. Its modular synthetic strategy and multifunctional properties lay the groundwork for the future design of advanced, functionalized macrocyclic systems.
Figure 6
Figure 6.
(a) Synthetic strategy for macrocycle 12. (b) CD spectra of 12 alone and in the presence of chiral guest molecules (R)-Ga and (S)-Ga, demonstrating chiroptical responsiveness. Reproduced with permission [59]. Copyright 2022, American Chemical Society.
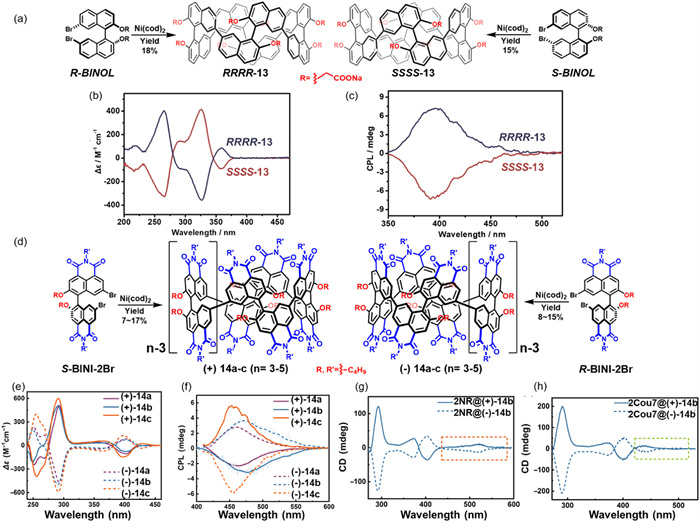
In 2023, Cai and co-workers reported the synthesis of water-soluble chiral macrocycles RRRR/SSSS-13, constructed from enantiopure 1,1′-bi-2,2′-naphthol (BINOL) precursors (Fig. 7a) [60]. Macrocycle 13 features a conjugated, electron-rich framework with a deep hydrophobic cavity and displays strong fluorescence emission (ΦF = 34%) as well as significant CPL in aqueous solution (|glum| = 1.4 × 10–3) (Figs. 7b and c).
Figure 7
Figure 7.
(a) Synthetic route to water-soluble chiral macrocycles RRRR/SSSS-13. (b) CD spectra of RRRR/SSSS-13 in water (2.5 × 10–6 mol/L). (c) CPL spectra of RRRR/SSSS-13 in water (2.5 × 10–6 mol/L). (d) Synthetic strategy for tubular chiral macrocycles (±)-14a–c (n = 3–5). CD (e) and CPL spectra (f) of (+)- and (−)-14a–c in CHCl3 (1.0 × 10–5 mol/L). (g) CD spectra of 2NR@(+)-14b and 2NR@(−)-14b in hexane (14b: 40 µmol/L; NR: 80 µmol/L; light path length: 2.0 mm). (h) CD spectra of 2Cou7@(+)-14b and 2Cou7@(−)-14b in hexane (14b: 40 µmol/L; Cou7: 140 µmol/L; light path length: 2.0 mm). Reproduced with permission [60,61]. Copyright 2023, Wiley‐VCH GmbH; Copyright 2025, Chinese Chemical Society.
Owing to its structural features, 13 demonstrates excellent binding affinity for hydrophobic, positively charged substrates. Remarkably, it exhibits binding constants up to 1010 L/mol for achiral guests and enantioselectivity ratios (KSSSS/KRRRR) of up to 18.7 for chiral analytes, both of which represent benchmark values for chiral macrocycles operating in aqueous environments. The combination of high binding strength, pronounced enantioselectivity, synthetic accessibility, and superior optical activity positions macrocycle 13 as a highly promising candidate for applications in analytical sensing, chiral separation, and molecular recognition.
Building upon their previous design strategies, the same group recently developed a new class of chiral tubular macrocycles, 14a–c (n = 3–5), based on repeating dicarboxylic acid imide units (Fig. 7d) [61]. These molecules exhibit excellent optical properties in nonpolar solvents. In particular, 14a displays a ΦF of 75% in cyclohexane. CD and CPL spectroscopic analyses confirmed the presence of strong chiroptical signals across the series, with solvent polarity exerting a notable influence on CPL activity. Notably, 14c exhibited a |glum| value of 2.4 × 10–3 and a circularly polarized luminescence brightness (BCPL) of 119 L mol⁻1 cm−1 for its enantiomer (Figs. 7e and f). Although macrocycles 14a–c showed lower guest binding affinities (Ka ≈ 105 L/mol) compared to 13, their supramolecular behavior revealed unique characteristics. For instance, 14b was capable of simultaneously binding two planar dye molecules, Nile red (NR) and Coumarin 7 (Cou7), in a 1:2 stoichiometry, with a cumulative binding constant of up to 1012 M-2. This interaction facilitated chirality transfer from the host to the non-chiral guests, as confirmed by induced CD and CPL signals (Figs. 7g and h). These results suggest that this class of macrocycles can function as effective chiral hosts for modulating the optical activity of bound guests. Future structural modifications, such as the incorporation of hydrophilic substituents, could enable their application in aqueous-phase molecular recognition and artificial transmembrane channel design.
2.2
Enantiotropic atropisomers in carbon-rich cycloarenes featuring a double macrocyclic framework
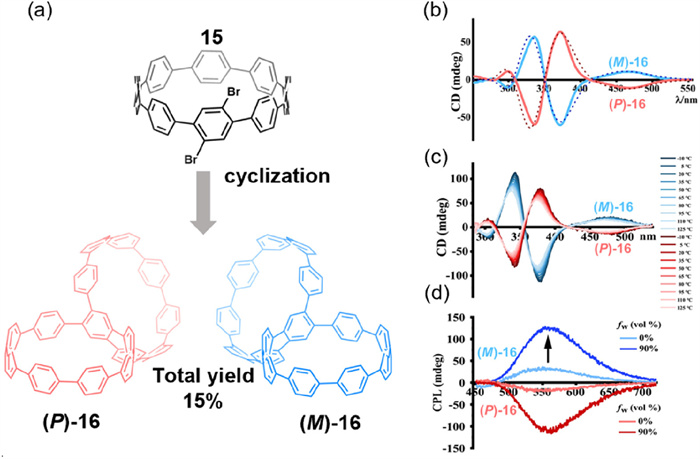
The enantiotropic atropisomers of cycloarene macrocycles and their derivatives can also adopt bismacrocyclic structures. In 2022, building on the design of SCPP10 [62], Du and colleagues synthesized the chiral dual-state emission bismacrocycle 16 from precursor 15 [63] and successfully resolved its enantiomers via chiral HPLC (Fig. 8a). CD spectroscopy revealed that the enantiopure isomer exhibited a |gabs| value of 1.2 × 10–2, indicating strong chiroptical activity in the ground-state electronic transition (Figs. 8b and c). This pronounced signal is consistent with a high kinetic barrier to racemization, reflecting excellent configurational stability of the atropisomer. In solution, its |glum| value was 6.9 × 10–3, while in the aggregated state, the CPL signal was significantly enhanced (|glum| = 1.9 × 10–2) (Fig. 8d). This enhancement surpasses that observed in many small molecules exhibiting aggregation-induced emission (AIE) properties, highlighting its potential for high-performance chiral optoelectronic applications. However, its host–guest recognition and self-assembly behaviors remain largely unexplored, thereby limiting its further development as a functional supramolecular material.
Figure 8
Figure 8.
(a) Synthetic route to chiral macrocycle 16 featuring a 1,2,4,5-tetraphenylbenzene (TPB) core. (b) Experimental (solid lines) and theoretical (dashed lines) CD spectra for (P)/(M)-16 in CH2Cl2. (c) Temperature-dependent CD spectra of (P)/(M)-16 from −10 ℃ to 125 ℃ at 15 ℃ intervals. (d) CPL spectra of (P)/(M)-16 in THF solution and in a 90:10 H2O/THF solution (excited at 355 nm). Reproduced with permission [63]. Copyright 2022, The Author(s).
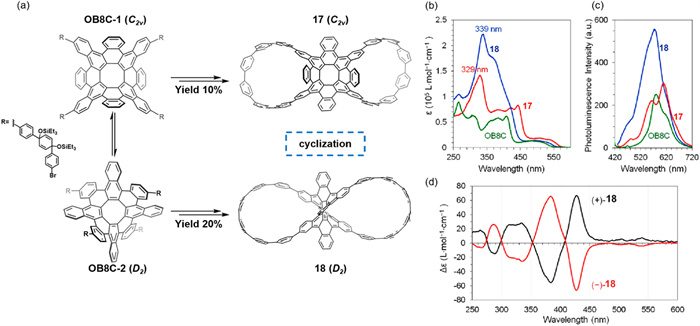
In 2023, Miao’s group reported two negatively curved nanographenes, compound 17 (C2V symmetry) and compound 18 (D2 symmetry), constructed through the conformationally controlled cyclization of an octabenzo[8]circulene (OB8C) core [64]. Their study revealed that the rapid interconversion of two key intermediates significantly contributes to the conformational diversity of the target products (Fig. 9a). Notably, a pair of enantiomers of 18 were successfully separated. Compared to 17, isomer 18 exhibited a higher molar absorption coefficient and enhanced fluorescence emission intensity (Figs. 9b and c). CD spectroscopy revealed pronounced Cotton effects in the 250–600 nm range (Fig. 9d). Notably, the host–guest recognition behavior of the distinct conformational isomers remains unexplored, leaving a promising direction for future investigation. Overall, this study provides valuable insights into the rational design of multi-conformational nanographenes with negative curvature, highlighting their potential in supramolecular chemistry and molecular recognition.
Figure 9
Figure 9.
(a) Curved nanographene 17 and 18 resulting from OB8C precursors with conformational interconversion. (b) UV–vis absorption spectra of 17, 18, and OB8C (each concentration as 2.5 × 10–6 mol/L in CH2Cl2). (c) Photoluminescence spectra of 17, 18, and OB8C (each concentration as 2.5 × 10–6 mol/L in CH2Cl2; 17 and 18 excited at 420 nm, OB8C excited at 410 nm). (d) CD spectra of the two enantiomers of 18 (6.0 × 10–5 mol/L in CH2Cl2). Reproduced with permission [64]. Copyright 2023, The Authors.
In 2024, Cong and colleagues synthesized all-carbon conjugated helical nanotubes 20 [65] via an axial covalent bonding strategy, using site-specifically functionalized cycloparaphenylene 19 as the building block (Fig. 10a). Compared to [9]CPP, 20 exhibits a significantly higher molar absorption coefficient (Fig. 10b). Especially, 20 demonstrates strong chiral properties, with the circularly polarized luminescence brightness (BCPL) of its enantiomer reaching 1470 L mol-1 cm-1, marking one of the highest BCPL values reported for chiral organic small molecules (Usually within the order of 101–102 in organic molecules) (Fig. 10c) [65]. The diacetylene moiety provides a versatile platform for post-synthetic functionalization, enabling the incorporation of additional functional groups or responsive units. However, the potential applications of this system in molecular recognition and artificial channel design remain to be fully explored. Overall, this study presents a novel bottom-up strategy for constructing conjugated nanotubes from cycloparaphenylene (CPP) building blocks, offering new avenues for the development of functional nanostructured materials.
Figure 10
Figure 10.
(a) Helical nanotubes 20 prepared by dimerization of [9]cycloparaphenylene 19. (b) UV–vis absorption (solid lines) and fluorescence emission (dashed lines) spectra of [9]CPP, 19, and 20 (5.0 × 10⁻⁶ mol/L) in CH2Cl2, respectively. (c) electronic circular dichroism (ECD) (solid lines) and CPL (dashed lines) spectra of the enantiomers of 20 (5.0 × 10–6 mol/L in CH2Cl2). Reproduced with permission [65]. Copyright 2024, Wiley-VCH GmbH.
2.3
Enantiotropic atropisomers in carbon-rich cycloarenes featuring a multi macrocyclic framework
Efforts to integrate carbon-rich macrocycles into the construction of chiral carbon materials have recently emerged. In 2019, Du’s group reported the first synthesis of long π-conjugated segments of single-chirality single-walled carbon nanotubes (SWCNTs, 21) via one-dimensional nickel-catalyzed polymerization of difunctionalized CPPs (Fig. 11) [66]. This polymer not only exhibits enhanced photophysical properties, including improved absorption and emission characteristics, but also demonstrates efficient charge transport for both electrons and holes. These findings establish a foundation for developing solution-phase synthesis strategies for single-chirality carbon nanotubes.
Figure 11
Figure 11.
Design of a long π-extended poly(para-phenylene)-based polymeric segment of armchair [8,8]single-walled carbon nanotube 21. Reproduced with permission [66]. Copyright 2019, American Chemical Society.
In 2022, Isobe’s group employed triangular phenine units as vertices to construct a minimal chiral cage representing a diamond-twin network (23) via a coupling reaction [67]. Single-crystal X-ray diffraction analysis confirmed the solid-state structure of compound 23, while theoretical calculations suggested that it undergoes dynamic chiral inversion in solution. To enhance its stereochemical stability, rigid stereogenic groups were introduced, yielding OMe-23 (Fig. 12a). This modification enabled chiral resolution, and enantiomerically resolved CD spectra were recorded in the 250–350 nm range (Figs. 12b and c). However, the supramolecular functions of the cavity, such as molecular recognition and guest encapsulation, remain largely unexplored. Nevertheless, this study presents a novel cavity-directed design strategy for constructing chiral carbon-rich materials, offering new opportunities in supramolecular chemistry and functional material development.
Figure 12
Figure 12.
(a) Ni(cod)2-mediated coupling of macrocycle MC-22 to form the chiral diamond-twin network (±)-OMe-23. (b) Chiral resolution of OMe-23 using a chiral column (R)-1-naphthylglycine (SUMICHIRAL OA-2500, 4.6 × 250 mm) for chiral HPLC analysis (eluent = 0.5% 1-butanol/hexane; flow rate = 1.0 mL/min, 40 ℃) (stereoisomers are labeled with the CD sign at 300 nm). (c) CD spectra of the enantiomers of OMe-23 in CHCl3 at 25 ℃. Reproduced with permission [67]. Copyright 2022, The Author(s). Published by PNAS.
On the other hand, carbon-rich macrocycles can also adopt Möbius topologies, leading to inherently chiral spatial configurations. The unique topological features of Möbius macrocycles have garnered significant interest in recent years due to their fundamental and potential functional relevance in molecular design. Research on this molecular architecture has been reviewed and further developed across several studies [68].
In 2022, Itami and co-workers reported the synthesis of a Möbius carbon nanobelt, MCNB-24 (Fig. 13a), via a Z-selective Wittig reaction followed by a nickel-mediated intramolecular homocoupling process [69]. The UV–vis absorption spectrum of (25,25)MCNB displayed peaks at 389 and 409 nm, along with a very weak absorption at 477 nm. Fluorescence emission was observed at 480, 513, and 551 nm, with a ΦF value of 10% (Fig. 13b). The Möbius topology imparts inherent chirality to the molecule, and its topological enantiomers were separated by chiral HPLC and characterized via CD spectroscopy. The two stereoisomers were tentatively assigned as the P and M enantiomers (Fig. 13c). However, further investigation into additional chiroptical properties and host–guest recognition capabilities of this unique molecular structure was not pursued in this study.
Figure 13
Figure 13.
(a) Molecular structure of Möbius carbon nanobelt MCNB-24. (b) UV–vis absorption (solid line) and fluorescence emission (dashed line) spectra of MCNB-24 in CH2Cl2. (c) CD spectra of MCNB-24 enantiomers in CH2Cl2 separated by chiral column chromatography (CHIRALPAK-IE), with tentative assignment of (M) and (P) configurations. Reproduced with permission from [69]. Copyright 2022, The Author(s), published by Springer Nature.
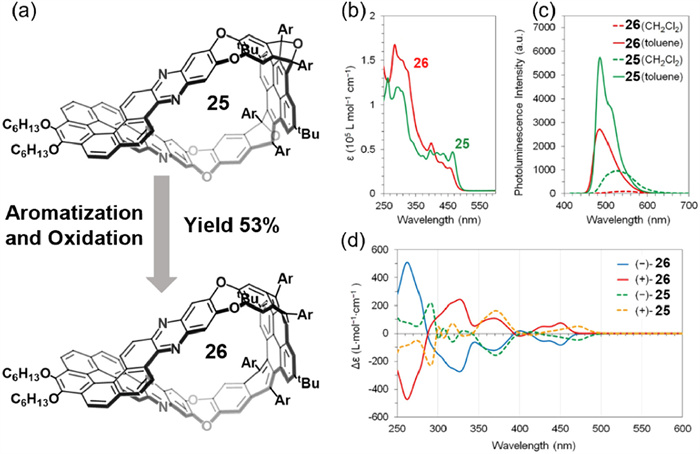
In 2025, Miao and co-workers reported the synthesis of a nanobelt (25) featuring a norbornene framework, constructed via aromatic nucleophilic substitution between a helicene-derived unit (based on [7]helicene) and a C-shaped segment derived from pyrene [70]. Compound 25 was subsequently converted into a twisted Möbius nanobelt (26) through sequential reductive aromatization and DDQ-mediated oxidation (Fig. 14a). The resulting molecule exhibited a writhed topology, which endowed it with unique photophysical characteristics. Both 25 and 26 displayed pronounced sensitivity to solvent polarity, with a ΦF value of 2.9% in CH2Cl2 and 24.3% in toluene (Figs. 14b and c). However, these nanobelts were photochemically unstable, undergoing decomposition upon light exposure. Notably, enantiopure 26 could be obtained by employing enantiopure helicene precursors. The compound exhibited strong chiroptical activity, with CD (|gabs| = 4 × 10–3) and CPL (|glum| = 1 × 10–3), highlighting its potential for chiral photonic applications (Fig. 14d). This work introduces a novel molecular design strategy that expands the toolbox for creating advanced chiral optoelectronic materials based on topologically nontrivial carbon-rich frameworks.
Figure 14
Figure 14.
(a) Synthesis of nanobelt 26via reductive aromatization and oxidation of precursor 25. (b) UV–vis absorption spectra of 25 and 26 in CH2Cl2 (1 × 10–5 mol/L, λex = 396 nm). (c) Photoluminescence spectra of 25 and 26 in CH2Cl2 and toluene (1 × 10–5 mol/L). (d) CD spectra of 25 and 26 in CH2Cl2. Reproduced with permission [70]. Copyright 2025, American Chemical Society.
Chiral carbon-rich macrocycles can be constructed by linking aromatic ring moieties and carbon atoms, exhibiting topological or atropisomeric chirality. In recent years, numerous studies have reported the synthesis of various chiral cycloarene macrocycles, including spirobifluorene-based chiral macrocycles and alkyne-linked bismacrocycles, with promising applications in chiral recognition and luminescent materials [71-87]. Moreover, these macrocycles can serve as key intermediates for constructing novel topologically chiral systems, such as figure-8-shaped chiral carbon nanoribbons [88,89], catenanes and rotaxanes [90,91], offering a promising pathway for the structural innovation of carbon-rich materials.
3.
Non-enantiotropic atropisomers of carbon-rich cycloarene macrocycles
Non-enantiotropic atropisomers, also referred to as non-canonical atropisomers, are conformational isomers that arise from the concerted rotation of multiple σ bonds within macrocyclic frameworks possessing non-helical, conventional topologies. These atropisomers exhibit stereochemical behavior that differs fundamentally from classical axially chiral molecules, such as biaryl systems [91-96], in which chirality originates from restricted rotation around a single σ bond.
In 2016, Müllen's group first synthesized and separated two cyclohexa-1,3-pyrenylene 27 and 28 [97]. Experimental and theoretical studies identified two plausible conformational structures, revealing that isomer 27 undergoes thermal conversion to the more thermodynamically stable isomer 28 upon heating to 473 K in [D8]sulfolane (Fig. 15a). However, the absence of comprehensive NMR and spectroscopic studies has limited the understanding of their dynamic behavior.
Figure 15
Figure 15.
Temperature-responsive conformational transitions of pyrene-containing macrocycles. (a) Isomer 27 converts to 28 upon heating at 473 K. (b) Isomer 29a (Cs symmetry) transforms into 29b (D3d symmetry) after heating at 533 K for 48 h. (c) Isomer 30a (C2 symmetry) isomerizes to 30b (C2 symmetry) at 533 K within 1 h. (d) Isomers 31a (D2d symmetry) and 31b (Cs symmetry) are interconvertible at 533 K, and both can further convert to 31c (D4d symmetry) after heating at 533 K for 24 h.
In 2023, Aratani, Yamada, and colleagues reported a series of [n]Cyclo-4,10-pyrenylenes (29 (n = 6), 30 (n = 7), and 31 (n = 8)) (Figs. 15b-d) [98]. They systematically investigated the atropisomeric distribution and conformational interconversion mechanisms in macrocycles of different sizes and refined NMR and spectroscopic characterization, providing deeper insight into their structural dynamics.
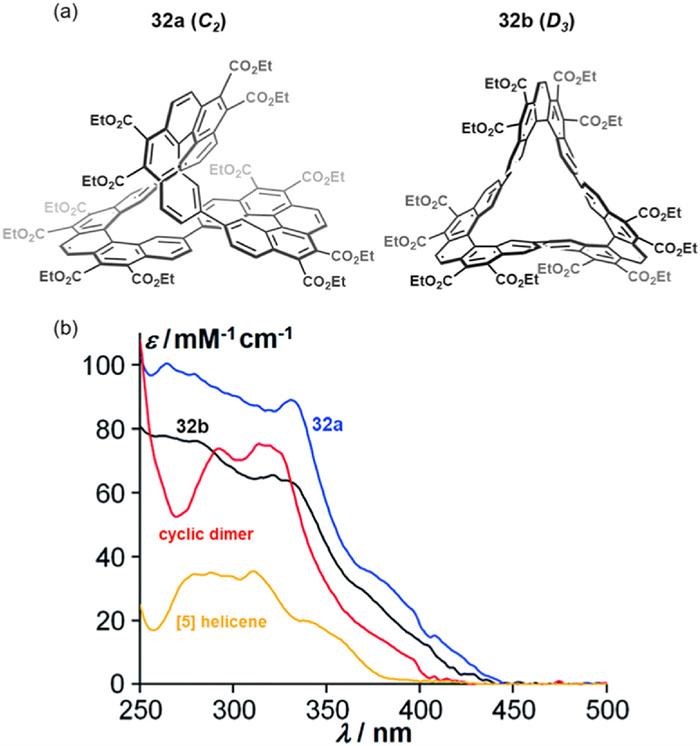
In 2018, Herges, Durola, and colleagues synthesized chiral non-enantiotropic triple helicene macrocycles 32a (Cs symmetry) and 32b (D3 symmetry) from a common precursor molecule (Fig. 16a) [99]. UV-visible absorption spectroscopy revealed a significantly higher molar absorption coefficient relative to the [5]helicene core, while the atropisomers exhibited similar absorption peak profiles (Fig. 15b).
Figure 16
Figure 16.
(a) Structures of isomers 32a (C2 symmetry) and 32b (D3 symmetry). (b) Absorption spectra of a single [5]helicene tetraester (yellow), the corresponding cyclic dimer (red), and isomers 32a (blue) and 32b (black). Reproduced with permission [99]. Copyright 2018, The Royal Society of Chemistry.
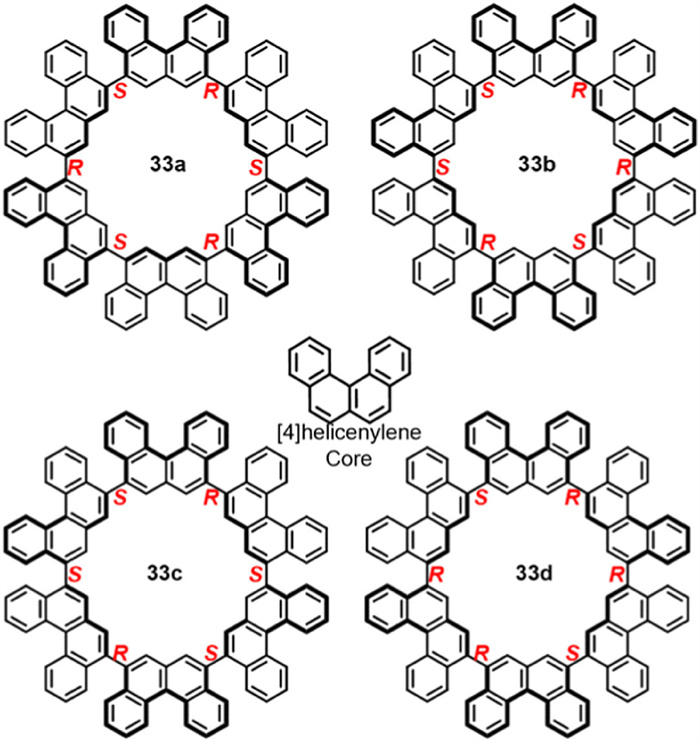
In 2021, Sato and Isobe synthesized [6]cyclo[4]helicenylenes ([6]CH) [100]. Four conformations (33a–d) were identified through HPLC separation and X-ray single-crystal diffraction, among which 33c and 33d were enantiomeric pairs (Fig. 17). Upon heating the isomer mixture to 473 K, the thermodynamically favored atropisomer 33a was obtained in 12% yield.
Figure 17
Figure 17.
Four atropisomers 33a-d of [6]cyclo[4]helicenylenes ([6]CH).
However, the kinetics of isomer transformation remained unexamined in the aforementioned studies. Furthermore, variations in the supramolecular chemistry of atropisomers were not investigated.
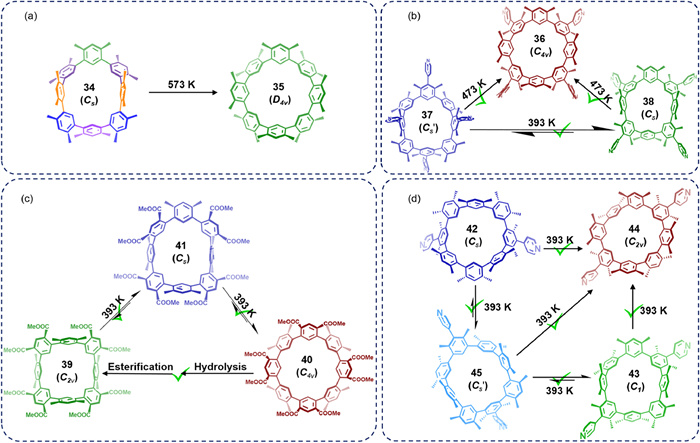
Since 2019, Gong's group has systematically investigated the atropisomerism of cyclometa-phenylenes (CMPs). In 2019, they reported two atropisomers, 34 (Cs symmetry) and 35 (D4d symmetry), of the novel all-hydrocarbon macrocycle cyclo[8](1,3-(4,6-dimethyl)benzene) (CDMB-8) and demonstrated complete conversion of 34 to 35 at 573 K (Fig. 18a) [101].
Figure 18
Figure 18.
(a) Thermal annealing achieves quantitative conversion of isomer 34 (Cs symmetry) to 35 (D4d symmetry) at 573 K. (b) Schematic diagram of the reversible conversion of isomers 37 (Cs' symmetry) and 38 (Cs symmetry) at 393 K, and the quantitative conversion of isomers 37 (Cs' symmetry) and 38 (Cs symmetry) to 36 (C4v symmetry) at 473 K. (c) Schematic diagram of the interconversion between isomers 39 (C2v symmetry), 40 (C4v symmetry) and intermediate 41 (Cs symmetry) at 393 K, and the efficient conversion of 40 (C4v symmetry) to 39 (C2v symmetry) under chemical reaction. (d) Schematic diagram of the interconversion between isomers 42 (Cs symmetry), 43 (C1 symmetry), 44 (C2v symmetry) and intermediate 45 (Cs' symmetry) at 393 K.
In 2023, the group synthesized the pyridine-substituted cyclo[8](1,3-(4,6-dimethyl)benzene derivative CP4, which exists in three rigid atropisomeric forms: 36 (C4v symmetry), 37 (Cs' symmetry), and 38 (Cs symmetry) [102]. NMR studies revealed that these atropisomers undergo reversible or irreversible interconversion under temperature regulation (Fig. 18b). Further theoretical calculations confirmed that these conformational transitions proceed via well-defined transition states.
Building on these findings, in 2024, the group synthesized an ester-substituted cyclo[8]-meta-phenylenes derivative, OC-4 [103]. By tuning the reaction conditions, they selectively obtained two rigid atropisomers, 39 (C2v symmetry) and 40 (C4v symmetry). During thermodynamic investigations, they captured the conversion intermediate 41 (Cs symmetry) and found that the activation energy for atropisomerization was significantly reduced through this intermediate, enabling interconversion between atropisomers. Furthermore, via controlled chemical modifications, the group achieved efficient conversion from the most thermodynamically stable state 40 to the metastable state 39 and 41 (Fig. 18c).
Based on previous exploration of CP4, a cyclo[8]-meta-phenylenes derivative, they recently reported that CP2, modified with two pyridine substituents, is similar to CP4 and also features three rigid atropisomers: 42 (Cs symmetry), 43 (C1 symmetry), and 44 (C2v symmetry) [104]. However, in contrast to the isomer transformation of CP4, NMR studies of CP2 revealed that its atropisomers can interconvert through an intermediate, 45 (Cs' symmetry) (Fig. 18d). Thermal transformation experiments and theoretical calculations indicated that the intermediate process of CP2 has a lower transition energy barrier compared to the transition state process of CP4. As a result, conformational transformation in CP2 can occur at a relatively low temperature (393 K). These studies offer new strategies for optimizing stimulus-responsive behavior in atropisomeric macrocycles.
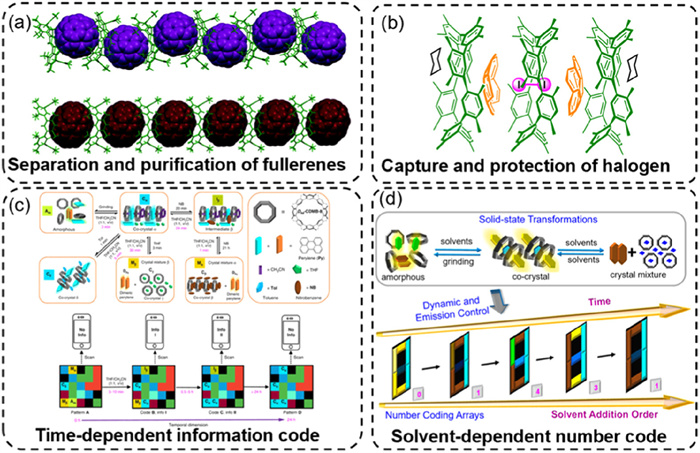
The distinct properties of atropisomers enabled the development of stimuli-responsive supramolecular self-assemblies with potential applications. The atropisomers 34 and 35, reported by Gong's group, exhibited distinct host-guest interactions. Notably, 35 functions as a fullerene acceptor, facilitating the separation and purification of fullerenes from fullerene mixtures or carbon soot extracts, whereas 34 lacked these capabilities (Fig. 19a) [101]. In 2021, they synthesized a self-assembled multicomponent capsule structure, (((35)3⊃(Cora)2)⊃I2) [105], in cyclohexane via the synergistic interactions between 35, corannulene (Cora), and I2 (Fig. 19b). Within this capsule, I2 was encapsulated inside the (35)3⊃(Cora)2) shell, exhibiting remarkable stability. No evidence of I2 escape was observed, even at elevated temperatures (up to 418 K). Similarly, the encapsulated I2 remained unreactive toward alkaline and reductive aqueous solutions, such as saturated NaOH or Na2S2O3. They propose that this multicomponent supramolecular structure offers a promising strategy for the encapsulation and stabilization of small high-energy molecules and reactive intermediates.
Figure 19
Figure 19.
(a) Separation and purification of fullerenes using 35. (b) Emergent self-assembly of a multicomponent capsule structure (((35)3⊃(Cora)2)⊃I2) in cyclohexane through the synergistic interaction between 35, corannulene (Cora), and I2. (c) Schematic diagram of the time-dependent dynamic 4D code system. (d) Specific number coding arrays. Reproduced with permission from [101,105-107]. Copyright 2019, The Royal Society of Chemistry; Copyright 2020, The Author(s); Copyright 2021, American Chemical Society; Copyright 2023, American Chemical Society.
For the development of stimulus-responsive materials, 35 and a series of guest molecules (e.g., perylene) can form fluorescent host-guest complexes [106,107]. By varying the solvent environment, these materials undergo transitions between co-crystalline, crystalline mixture, and amorphous states. These phase transformations induce fluorescence changes with distinct time-dependent behaviors. These tunable fluorescence properties enable the creation of a time-dependent dynamic 4D code system (Fig. 19c) and specific number coding arrays (Fig. 19d), facilitating dynamic information encoding and automated optical reading. This system holds potential applications in gas-induced fluorescence sensing, high-density information storage, and anti-counterfeiting technologies.
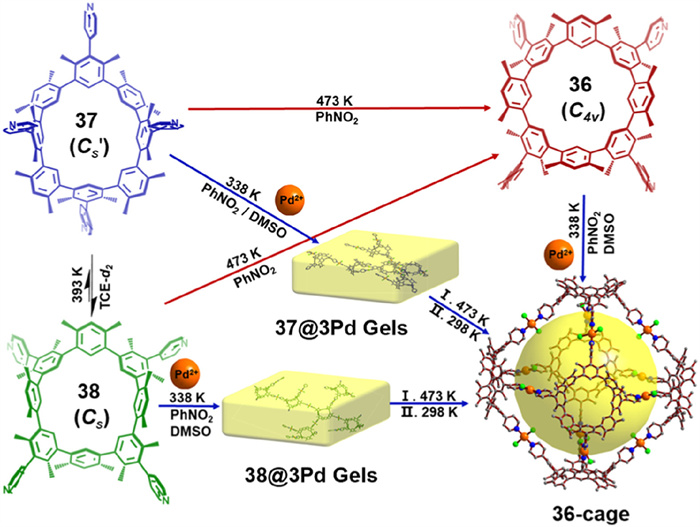
Extending beyond individual molecular behavior, atropisomerism can also govern supramolecular self-assembly. In 2023, they elucidated the conformational interconversion of CP4 atropisomers and investigated their supramolecular properties. The studies revealed that distinct atropisomers assemble into structurally unique Pd2+-coordinated systems. X-ray single-crystal diffraction confirmed that atropisomers 37 (Cs' symmetry) and 38 (Cs symmetry) form network Pd2+-coordinated assemblies, resulting in yellow gels (37@3Pd and 38@3Pd). In contrast, atropisomer 36 (C4v symmetry) assembled into a three-dimensional Pd2+-coordinated cage structure (36-cage). Thermal annealing enabled precise control over macrocyclic atropisomer structures, allowing modulation of the self-assembly process and facilitating the synthesis of nanoscale metal-organic cages (Fig. 20) [102].
Figure 20
Figure 20.
Schematic diagram illustrating the heating regulation from macrocycle to further metal–organic cage assembly via both stepwise and in situ pathways. Isomers 37 and 38 coordinate with Pd2+ to form gels at temperatures below 338 K, which can be converted into metal–organic molecular cages upon heating at 473 K. The coordination of 36 with Pd2+ at 338 K directly forms the same cage, 36-cage. Reproduced with permission [102]. Copyright 2023, The Author(s).
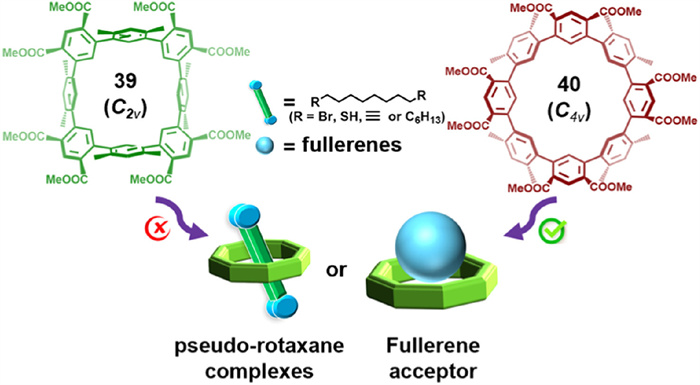
In 2024, they reported OC-4, displaying supramolecular behavior analogous to CDMB-8. Atropisomer 40 (C4v symmetry), with higher molecular symmetry, exhibited enhanced host-guest interactions compared to atropisomer 39 (C2v symmetry). X-ray single-crystal diffraction confirmed that 40 readily forms host-guest complexes with linear molecules and fullerenes (Fig. 21), demonstrating potential applications in fullerene separation and pseudo-rotaxane assembly [103].
Figure 21
Figure 21.
Schematic diagram of complexes formed by 40 with linear guests or fullerenes, which can be used in fullerene separation and the construction of pseudo-rotaxane complexes. Reproduced with permission [103]. Copyright 2024, The Author(s).
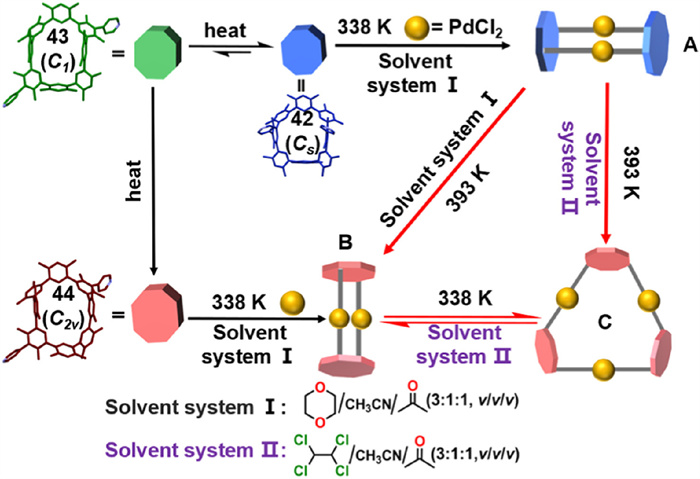
In 2025, they reported the synthesis of CP2, which exhibits supramolecular behavior similar to CP4. The metastable isomers 42 (Cs symmetry) and 43 (C1 symmetry), as well as the thermodynamically stable 44 (C2v symmetry), independently react with Pd2+ in the presence of 1,4-dioxane to yield M2L2 rectangular molecular cages, labeled A or B, respectively. In contrast, replacing 1,4-dioxane with 1,1′,2,2′-tetrachloroethane leads to the formation of a unique M3L3 hexagonal molecular cage (C) from isomer 44 (C2v symmetry). Notably, solvent exchange enables the reversible transformation between M2L2 cage B and M3L3 cage C. Furthermore, under the cooperative effects of temperature variation and solvent changes, M2L2 cage A can transition into either cage B or cage C, enabling a multipathway self-assembly system (Fig. 22). This multipathway self-assembly system offers a strategic approach for precisely controlling the formation of smart materials [104].
Figure 22
Figure 22.
Schematic diagram illustrating the temperature-induced interconversion among the three atropisomers enables the selective formation of distinct molecular cages. Reproduced with permission [104]. Copyright 2025, The Author(s). Published by PNAS.
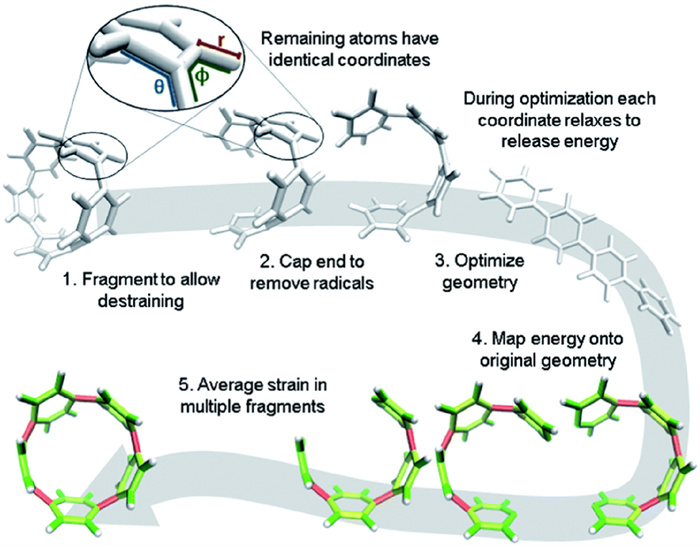
In 2020, Jasti's group developed StrainViz, a computational method that correlates strain energy with molecular reactivity [108]. This approach enables the localization and quantification of strain, aiding the prediction of atropisomer formation and serving as a valuable tool in atropisomer research (Fig. 23).
Figure 23
Figure 23.
Workflow for strain analysis. The coordinates in each molecular fragment relax to release strain energy, which is quantified per coordinate (r: bond length, q: angle, f: torsional angle). Copied with permission [108]. Copyright 2020, The Royal Society of Chemistry.
In 2022, Wu's group reported the synthesis of trimeric macrocycles trans-46 and cis-47 [109]. These atropisomeric structures displayed distinct aromatic characteristics and open-shell radical properties across various oxidation states. Oxidation by AgSbF6 readily yielded the corresponding dications, producing 462+·2SbF6− and 472+·2SbF6−, respectively. Both experimental and theoretical analyses revealed that these dications exhibited distinct degrees of electronic coupling, with 462+·2SbF6− adopting a singlet ground state, whereas 472+·2SbF6− stabilized a triplet state to relieve strain. Notably, 462+·2SbF6− demonstrated pronounced global antiaromaticity, while 472+·2SbF6− exhibited no global aromaticity (Fig. 24). This study further enriched the current understanding of aromaticity in conjugated molecules, offering new insights into their electronic structures and stability.
Figure 24
Figure 24.
The structure of trimeric macrocycles trans-46 and cis-47, and their respective cations 462+·2SbF6− and 472+·2SbF6−. The red arrows indicate the spins. The spin ground states and (anti) aromaticity of individual macrocycles are summarized below the structures.
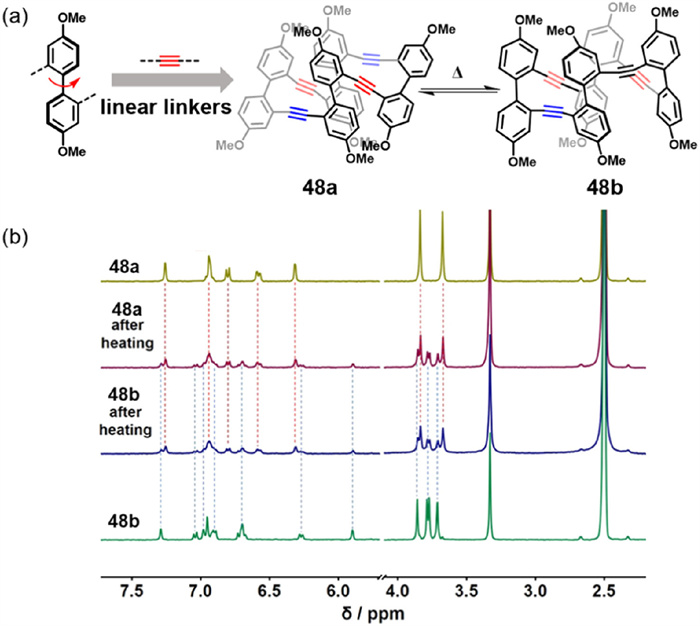
In 2022, Cai and colleagues reported the synthesis of non-enantiotropic atropisomers 48a and 48b, which both exhibited bright blue fluorescence with a fluorescence quantum yield (ΦF) of 53% (Fig. 25a) [59]. To explore the conformational interconversion between these atropisomers, variable-temperature 1H NMR spectroscopy was employed. Upon heating to 90 ℃ for 2 h, a dynamic equilibrium was established, resulting in a 1:1 mixture of 48a and 48b (Fig. 25b). This molecular design strategy facilitates the simultaneous generation of enantiomers and diastereomers, enabling the comparative study of host–guest recognition across distinct conformational states.
Figure 25
Figure 25.
(a) Synthetic strategy for macrocycles 48a and 48b. (b) 1H NMR spectra (400 MHz, DMSO–d6, 298 K) of 48a and 48b (1.0 mmol/L): spectra shown for pure 48a, pure 48b, and samples of each compound after heating at 90 ℃ for 2 h and rapidly cooling to 298 K. The spectra indicate that both macrocycles undergo thermally induced conformational interconversion, reaching an equilibrium mixture containing approximately equal amounts of 48a and 48b Reproduced with permission [59]. Copyright 2022, American Chemical Society.
In the past decade, enantiotropic atropisomers of carbon-rich cycloarenes and their macrocyclic derivatives have seen significant advancements. These atropisomers adopt distinct topological structures, leading to diverse properties in spatial functional site distribution, electronic configurations, and energy gaps. Such variations contribute to differences in chiroptical responses and host-guest recognition behaviors. This progress has provided new strategies for tuning the structures and properties of macrocycles and for designing novel carbon-rich materials.
In parallel, non-enantiotropic atropisomers of carbon-rich cycloarenes have gained increasing research interest due to their distinct conformations and associated physicochemical properties. However, existing studies have primarily centered on the synthesis of molecular frameworks and fundamental physicochemical characterization. Investigations into their atropisomeric behavior remain in an early stage, with challenges persisting in their selective synthesis. The mechanistic understanding of their interconversion is still underexplored, and precise control strategies have yet to be established. Furthermore, their potential applications in supramolecular chemistry remain largely unexplored.
Future efforts should prioritize the precise construction of enantiopure atropisomers with enhanced efficiency, selectivity, and scalability. Integrating experimental insights with theoretical modeling, we aim to systematically elucidate atropisomerization mechanisms and governing principles. Additionally, developing predictive theoretical frameworks for material behavior will facilitate the transition from fundamental research to real-world applications.
Finally, by leveraging atropisomerism, we aim to integrate additional molecular components to design stimulus-responsive supramolecular self-assembled materials. Establishing guiding principles for their formation will enable the exploration of their applicability in smart devices, biomedical technologies, energy systems, and environmental remediation.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Y.D. Yang, X.L. Chen, J. Liang, et al., J. Am. Chem. Soc. 145 (2023) 14010–14018. doi: 10.1021/jacs.3c03727
[108]
C.E. Colwell, T.W. Price, T. Stauch, R. Jasti, Chem. Sci. 11 (2020) 3923–3930. doi: 10.1039/d0sc00629g
[109]
S. Wu, Y. Ni, Y. Han, et al., J. Am. Chem. Soc. 144 (2022) 23158–23167. doi: 10.1021/jacs.2c10859
Figure 1
A series of chiral carbon-rich macrocycles incorporating chrysene motifs and their circular dichroism (CD) spectra: (a) (12,8)-1 and (11,9)-1; (b) (16,0)-2; (c) (12,8)-3 and (11,9)-3; (d) (9,6)-4 and (8,7)-4. Representative guest molecules exhibiting notable host–guest interactions with macrocycles: (e) (12,8)-1, (f) (12,8)-3, and (g) (9,6)-4. Adapted with permission [32-35]. Copyright 2011, Springer Nature Limited; Copyright 2012, American Chemical Society; Copyright 2013, The Royal Society of Chemistry; and Copyright 2019, Wiley-VCH Verlag GmbH & Co. KGaA.
Figure 2
(a) Synthetic strategy for the preparation of chiral macrocycle 5. (b) Racemization pathway from (P)-5 to (M)-5, with the corresponding activation energy (ΔG‡, kcal/mol). Reproduced with permission [52]. Copyright 2011, American Chemical Society.
Figure 4
(a) Synthetic strategy for diketone-functionalized [n]cyclopara phenylenes (CPPs) 9a (n = 6) and 9b (n = 7), as well as dibenzo[a,e]pentalene-embedded [n]CPPs (DBP[n]CPPs) 10a (n = 6) and 10b (n = 7). (b) A pair of enantiomers resolution of 9a through chiral derivatization of the carbonyl groups to access enantiopure nanohoops 10. (c) Absorption (10–5 to 10–6 mol/L) and emission spectra (10–6 to 10–7 mol/L) of 9a-b and 10a-b in CH2Cl2. (d) CD spectra of (P/M)-10a. (e) CD spectrum of (R,R/S,S)-9a. (f) CPL spectra of (R,R/S,S)-9a. Reproduced with permission [56]. Copyright 2021, The Royal Society of Chemistry.
Figure 5
(a) Schematic representation of the synthesis of nanohoop (±)-11via macrocyclization of an enantiomerically pure diketone precursor. (b) CD spectra of (±)-11. Reproduced with permission [57]. Copyright 2021, The Authors.
Figure 6
(a) Synthetic strategy for macrocycle 12. (b) CD spectra of 12 alone and in the presence of chiral guest molecules (R)-Ga and (S)-Ga, demonstrating chiroptical responsiveness. Reproduced with permission [59]. Copyright 2022, American Chemical Society.
Figure 7
(a) Synthetic route to water-soluble chiral macrocycles RRRR/SSSS-13. (b) CD spectra of RRRR/SSSS-13 in water (2.5 × 10–6 mol/L). (c) CPL spectra of RRRR/SSSS-13 in water (2.5 × 10–6 mol/L). (d) Synthetic strategy for tubular chiral macrocycles (±)-14a–c (n = 3–5). CD (e) and CPL spectra (f) of (+)- and (−)-14a–c in CHCl3 (1.0 × 10–5 mol/L). (g) CD spectra of 2NR@(+)-14b and 2NR@(−)-14b in hexane (14b: 40 µmol/L; NR: 80 µmol/L; light path length: 2.0 mm). (h) CD spectra of 2Cou7@(+)-14b and 2Cou7@(−)-14b in hexane (14b: 40 µmol/L; Cou7: 140 µmol/L; light path length: 2.0 mm). Reproduced with permission [60,61]. Copyright 2023, Wiley‐VCH GmbH; Copyright 2025, Chinese Chemical Society.
Figure 8
(a) Synthetic route to chiral macrocycle 16 featuring a 1,2,4,5-tetraphenylbenzene (TPB) core. (b) Experimental (solid lines) and theoretical (dashed lines) CD spectra for (P)/(M)-16 in CH2Cl2. (c) Temperature-dependent CD spectra of (P)/(M)-16 from −10 ℃ to 125 ℃ at 15 ℃ intervals. (d) CPL spectra of (P)/(M)-16 in THF solution and in a 90:10 H2O/THF solution (excited at 355 nm). Reproduced with permission [63]. Copyright 2022, The Author(s).
Figure 9
(a) Curved nanographene 17 and 18 resulting from OB8C precursors with conformational interconversion. (b) UV–vis absorption spectra of 17, 18, and OB8C (each concentration as 2.5 × 10–6 mol/L in CH2Cl2). (c) Photoluminescence spectra of 17, 18, and OB8C (each concentration as 2.5 × 10–6 mol/L in CH2Cl2; 17 and 18 excited at 420 nm, OB8C excited at 410 nm). (d) CD spectra of the two enantiomers of 18 (6.0 × 10–5 mol/L in CH2Cl2). Reproduced with permission [64]. Copyright 2023, The Authors.
Figure 11
Design of a long π-extended poly(para-phenylene)-based polymeric segment of armchair [8,8]single-walled carbon nanotube 21. Reproduced with permission [66]. Copyright 2019, American Chemical Society.
Figure 12
(a) Ni(cod)2-mediated coupling of macrocycle MC-22 to form the chiral diamond-twin network (±)-OMe-23. (b) Chiral resolution of OMe-23 using a chiral column (R)-1-naphthylglycine (SUMICHIRAL OA-2500, 4.6 × 250 mm) for chiral HPLC analysis (eluent = 0.5% 1-butanol/hexane; flow rate = 1.0 mL/min, 40 ℃) (stereoisomers are labeled with the CD sign at 300 nm). (c) CD spectra of the enantiomers of OMe-23 in CHCl3 at 25 ℃. Reproduced with permission [67]. Copyright 2022, The Author(s). Published by PNAS.
Figure 13
(a) Molecular structure of Möbius carbon nanobelt MCNB-24. (b) UV–vis absorption (solid line) and fluorescence emission (dashed line) spectra of MCNB-24 in CH2Cl2. (c) CD spectra of MCNB-24 enantiomers in CH2Cl2 separated by chiral column chromatography (CHIRALPAK-IE), with tentative assignment of (M) and (P) configurations. Reproduced with permission from [69]. Copyright 2022, The Author(s), published by Springer Nature.
Figure 14
(a) Synthesis of nanobelt 26via reductive aromatization and oxidation of precursor 25. (b) UV–vis absorption spectra of 25 and 26 in CH2Cl2 (1 × 10–5 mol/L, λex = 396 nm). (c) Photoluminescence spectra of 25 and 26 in CH2Cl2 and toluene (1 × 10–5 mol/L). (d) CD spectra of 25 and 26 in CH2Cl2. Reproduced with permission [70]. Copyright 2025, American Chemical Society.
Figure 15
Temperature-responsive conformational transitions of pyrene-containing macrocycles. (a) Isomer 27 converts to 28 upon heating at 473 K. (b) Isomer 29a (Cs symmetry) transforms into 29b (D3d symmetry) after heating at 533 K for 48 h. (c) Isomer 30a (C2 symmetry) isomerizes to 30b (C2 symmetry) at 533 K within 1 h. (d) Isomers 31a (D2d symmetry) and 31b (Cs symmetry) are interconvertible at 533 K, and both can further convert to 31c (D4d symmetry) after heating at 533 K for 24 h.
Figure 16
(a) Structures of isomers 32a (C2 symmetry) and 32b (D3 symmetry). (b) Absorption spectra of a single [5]helicene tetraester (yellow), the corresponding cyclic dimer (red), and isomers 32a (blue) and 32b (black). Reproduced with permission [99]. Copyright 2018, The Royal Society of Chemistry.
Figure 18
(a) Thermal annealing achieves quantitative conversion of isomer 34 (Cs symmetry) to 35 (D4d symmetry) at 573 K. (b) Schematic diagram of the reversible conversion of isomers 37 (Cs' symmetry) and 38 (Cs symmetry) at 393 K, and the quantitative conversion of isomers 37 (Cs' symmetry) and 38 (Cs symmetry) to 36 (C4v symmetry) at 473 K. (c) Schematic diagram of the interconversion between isomers 39 (C2v symmetry), 40 (C4v symmetry) and intermediate 41 (Cs symmetry) at 393 K, and the efficient conversion of 40 (C4v symmetry) to 39 (C2v symmetry) under chemical reaction. (d) Schematic diagram of the interconversion between isomers 42 (Cs symmetry), 43 (C1 symmetry), 44 (C2v symmetry) and intermediate 45 (Cs' symmetry) at 393 K.
Figure 19
(a) Separation and purification of fullerenes using 35. (b) Emergent self-assembly of a multicomponent capsule structure (((35)3⊃(Cora)2)⊃I2) in cyclohexane through the synergistic interaction between 35, corannulene (Cora), and I2. (c) Schematic diagram of the time-dependent dynamic 4D code system. (d) Specific number coding arrays. Reproduced with permission from [101,105-107]. Copyright 2019, The Royal Society of Chemistry; Copyright 2020, The Author(s); Copyright 2021, American Chemical Society; Copyright 2023, American Chemical Society.
Figure 20
Schematic diagram illustrating the heating regulation from macrocycle to further metal–organic cage assembly via both stepwise and in situ pathways. Isomers 37 and 38 coordinate with Pd2+ to form gels at temperatures below 338 K, which can be converted into metal–organic molecular cages upon heating at 473 K. The coordination of 36 with Pd2+ at 338 K directly forms the same cage, 36-cage. Reproduced with permission [102]. Copyright 2023, The Author(s).
Figure 21
Schematic diagram of complexes formed by 40 with linear guests or fullerenes, which can be used in fullerene separation and the construction of pseudo-rotaxane complexes. Reproduced with permission [103]. Copyright 2024, The Author(s).
Figure 22
Schematic diagram illustrating the temperature-induced interconversion among the three atropisomers enables the selective formation of distinct molecular cages. Reproduced with permission [104]. Copyright 2025, The Author(s). Published by PNAS.
Figure 23
Workflow for strain analysis. The coordinates in each molecular fragment relax to release strain energy, which is quantified per coordinate (r: bond length, q: angle, f: torsional angle). Copied with permission [108]. Copyright 2020, The Royal Society of Chemistry.
Figure 24
The structure of trimeric macrocycles trans-46 and cis-47, and their respective cations 462+·2SbF6− and 472+·2SbF6−. The red arrows indicate the spins. The spin ground states and (anti) aromaticity of individual macrocycles are summarized below the structures.
Figure 25
(a) Synthetic strategy for macrocycles 48a and 48b. (b) 1H NMR spectra (400 MHz, DMSO–d6, 298 K) of 48a and 48b (1.0 mmol/L): spectra shown for pure 48a, pure 48b, and samples of each compound after heating at 90 ℃ for 2 h and rapidly cooling to 298 K. The spectra indicate that both macrocycles undergo thermally induced conformational interconversion, reaching an equilibrium mixture containing approximately equal amounts of 48a and 48b Reproduced with permission [59]. Copyright 2022, American Chemical Society.

 DownLoad:
DownLoad:








 下载:
下载:


























