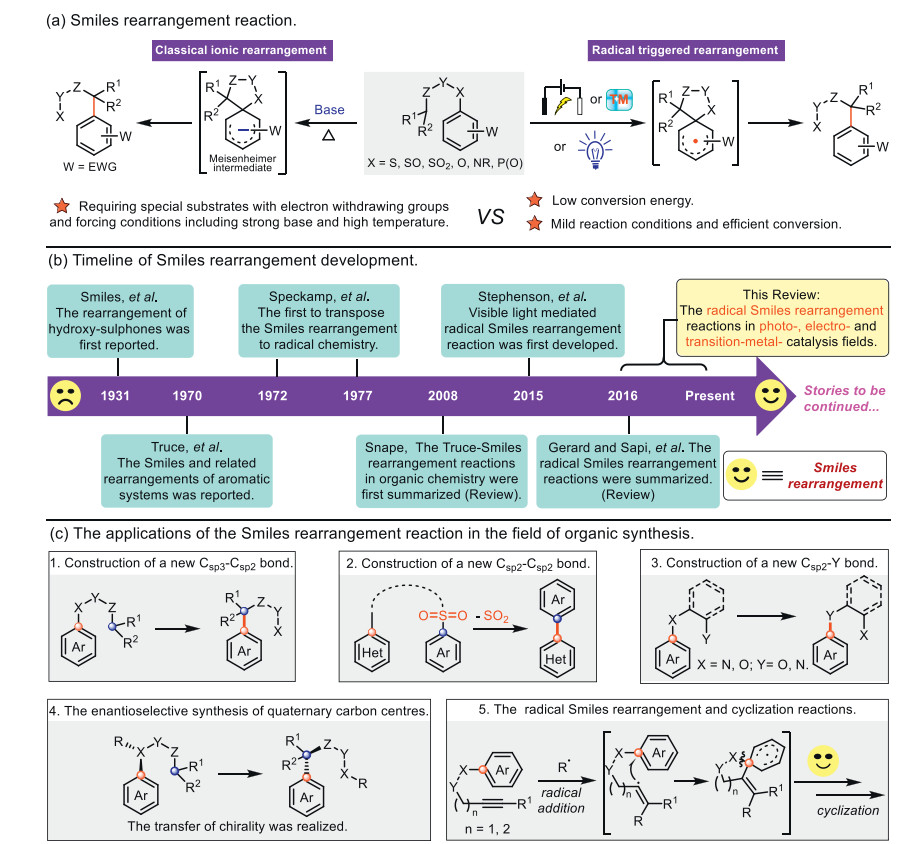
Scheme 1.
Background of the Smiles rearrangement.
Advances in radical Smiles rearrangement
Nianhua Luo , Jiayi Jiang , Muhammad Suleman , Zhaowen Liu , Shuping Huang , Wei Xiao , Jie Wu , Jiapian Huang

The advent of new chemical methodologies has en-riched the repertoire of organic chemists, enabling the reconstruction of organic molecules with remarkable diversity through rearrangement reactions. These transformations offer the ability to convert readily accessible "easy-to-prepare" precursors into complex and challenging "difficult-to-make" molecules, showcasing the transformative power of modern synthetic approaches [1]. In particular, the Smiles rearrangement is a reaction that has found widespread application in organic chemistry [2]. Owing to the nearly universal and distinctive chemical properties of aromatic systems, aryl migration and rearrangement techniques provide a valuable opportunity for structural modification. These methods enable the formation of new Csp2-Csp3 or Csp2-Y (Y = O, N, S, CO, etc.) bonds through step- and atom-economical pathways [3]. The classical ionic-type Smiles rearrangement involves intramolecular nucleophilic ipso substitution and is highly dependent on electronic and steric factors. Often, this rearrangement requires specially designed substrates with electron-withdrawing groups (EWGs) and harsh reaction conditions, such as the use of strong bases and elevated temperatures, to achieve efficient transformations (Scheme 1a, left). In contrast, the radical-triggered Smiles rearrangement has gained considerable attention as a complementary pathway that addresses the limitations of the ionic-type mechanism (Scheme 1a, right). In the original Smiles rearrangement mechanism, the ipso attack is followed by the migration of aryl units, which are activated and transferred from one heteroatom to a more nucleophilic heteroatom. A notable variation of this reaction, the Truce-Smiles rearrangement, employs a carbanion as the nucleophile instead of a heteroatom. Furthermore, the electrophilic aromatic ring in this variation does not require additional activation, which simplifies the process. This variation is particularly significant as it exclusively generates new carbon–carbon bonds [4].
The Smiles rearrangement has a rich history spanning nearly a century (Scheme 1b). In 1931, the first reported example of hydroxy-sulphone rearrangement was pioneered by the Smiles group, marking the beginning of this transformative reaction in organic chemistry [5]. In 1970, Truce and co-workers explored a versatile Smiles rearrangement capable of forming new carbon–carbon bonds by utilizing a carbon-based nucleophile and a pre-activated electrophilic arene. Later, Speckamp made a pioneering contribution by adapting the Smiles rear-rangement to radical chemistry, opening new avenues for synthetic transformations [6,7]. Since then, radical Smiles rearrangement-based methodologies have been recognized as highly efficient tools for synthetic chemists, enabling the construction of biaryl compounds with enhanced precision and versatility [8]. Entering the 21st century, the Truce-Smiles rearrangement continued to demonstrate its remarkable contributions to the construction of carbon–carbon bonds. In 2008, Snape independently provided a comprehensive summary of Truce-Smiles rearrangement reactions, tracing their development since the 1930s [4]. Owing to the versatile reactivity of radical chemical transformations, photocatalysis has provided countless opportunities for rearrangement reactions. Notable examples include the Brook rearrangement [9], the New-man-Kwart rearrangement [10], the 1,2-boron shift [11], the Cope rearrangement [12], and the Truce-Smiles rearrangement [13]. For instance, in 2015, Stephenson and colleagues were the first to describe a visible light-induced radical Smiles rearrangement of difluorobromo arylsulfonates, successfully synthesizing a variety of aryl- and heteroaryl-difluoroethanol derivatives [14]. In 2016, Gérard and Sapi were the first to comprehensively summarize and present the broader applications of Csp3-centered radical-promoted and Csp2 radical-assisted Smiles rearrangements, covering advancements from 2012 to 2016 [15]. Since then, the field of radical Smiles rearrangement has experienced a remarkable surge in reports, driven by rapid advancements in photocatalysis and electrocatalysis—particularly in the realm of asymmetric radical Smiles rearrangement. Given this rapid progress, we find it timely to compile and analyze the current state-of-the-art research in radical Smiles rearrangement while also highlighting potential avenues for future development.
The organization of this review is centered on categorizing the reported studies based on their underlying reaction mechanism or catalytic mode. Accordingly, we have broadly classified these reactions into four distinct categories (2 Radical addition to alkynes and subsequent radical Smiles rearrangement, 3 Radical addition to alkenes and subsequent radical Smiles rearrangement, 3.1 Intermolecular radical Smiles rearrangement induced by N-centered radicals, 3.2 Intermolecular radical Smiles rearrangement induced by C). In particular, the applications of radical Smiles rearrangement, as illustrated in Scheme 1c, primarily include: (a) Construction of new Csp3-Csp2 bonds, (b) Formation of new Csp2-Csp2 bonds, (c) Generation of new Csp3-Y (Y = O or N) bonds, (d) Synthesis of quaternary carbon stereocenters, (e) Applications in cyclization reactions. Within each subfield, we provide a general overview of the reaction formula, emphasizing the fundamental principles that govern C–C bond formation, as well as the specific challenges associated with each methodology. Despite our best efforts to categorize these reactions based on their underlying mechanisms, overlap between subfields and the lack of sufficient experimental evidence may complicate clear classifications. In this review, we aim to offer readers a comprehensive and in-depth perspective on the diverse array of catalytic tools available in this rapidly evolving field.
Alkynes, among the simplest functional groups in organic chemistry, provide a versatile platform for designing cascade reactions, allowing multiple new bonds to form at their two energy-rich carbon atoms. These alkyne units can be likened to tightly wound springs, storing high energy that, when released, drives reactivity. However, fully harnessing their potential requires a deep understanding of stereo-electronic control in alkyne activation. Despite their apparent simplicity, alkynes are far from a blank slate in terms of reactivity. The relatively high electronegativity of sp-hybridized carbons makes them inherently electrophilic, influencing their chemical behavior in a distinct and predictable manner [16]. Due to these factors, common reactive intermediates, such as cations, anions, and radicals, generated from similarly substituted alkynes exhibit distinctly different thermodynamic tendencies. Consequently, alkynes are more likely to serve as reaction partners for electron-rich species, reinforcing their electrophilic nature. Fundamentally, alkynes can function as energy-rich alternatives to other functional groups, unlocking reactions that would otherwise be thermodynamically unfavorable. Moreover, the intrinsic features of alkyne reactivity lead to unique substituent effects on their stability and interactions with electrophiles, nucleophiles, and radicals. The properties of reactive intermediates derived from alkynes are governed by similar principles. For example, vinyl anions generated from alkynes tend to be relatively stable, reacting with nucleophiles through hydride addition. However, vinyl cations formed through reactions with electrophiles via protonation are highly unstable, significantly limiting their synthetic utility. In contrast, vinyl anions, despite their enhanced stability, can present challenges in cascade transformations by creating thermodynamic traps that hinder subsequent steps in domino reactions. In other words, while vinyl radicals are relatively easy to generate, they are also highly reactive, readily engaging in transformations, even with otherwise inactive aromatic systems [17].
Due to the reversible nature of vinyl radical formation via intermolecular radical addition to alkynes, radical acceptors have a greater likelihood of persisting until the productive step occurs. This mechanism enables precise control over selectivity in radical transformations. Conversely, although the formation of alkyl radicals through intermolecular radical addition to alkenes is faster than the generation of vinyl radicals from alkynes, vinyl radicals exhibit significantly lower stability compared to their alkyl counterparts [18]. From this perspective, the selective activation of alkynes, particularly in the presence of alkenes and other sensitive functional groups, presents intriguing opportunities and challenges. Due to the unique properties of vinyl radicals, their strategic incorporation via alkynyl group introduction enables the formation of reactive alkenyl radical intermediates through radical addition. These intermediates can then undergo diverse radical transformations, facilitating the synthesis of a broad array of target molecules. Notably, vinyl radicals serve as crucial intermediates in radical Smiles rearrangement reactions.
Phthalazine structures are commonly found in numerous bioactive molecules, many of which exhibit important properties, such as antibacterial [19] or antitumor [20] activities (Scheme 2a). To construct this skeletal molecule, Belmont and Brachet et al. reported two significant advancements: A visible light-induced radical Smiles [21] rearrangement in 2016 (Scheme 2c) and "phospho-Smiles" (Scheme 2d) rearrangement cascade reaction in 2021 [22]. The authors propose that the catalytic cycle begins with the deprotonation of sulfonohydrazone (1 or 3) by a base, followed by photo-oxidation via an excited Ru photocatalyst, leading to the formation of an N-centered radical intermediate 2b. This radical undergoes intramolecular addition onto the alkyne via a 6-exo-dig cyclization, generating the C-centered vinyl radical 2c, which then participates in a (phospho-)Smiles rearrangement to form intermediate 2d. In cases where X = P(O)Ar, intermediate 2c can undergo a back-electron transfer (BET) process with the photocatalyst, yielding the vinyl anion 2g. Subsequent protonation of 2g by the solvent or ethanol results in the hydroamination by-product. Next, rearomatization of 2d leads to the formation of either an N-centered or C-centered radical intermediate (2e or 2e'). Finally, reduction of these radicals generates the corresponding anionic species, which can be protonated by the solvent to afford the final product 2, while simultaneously regenerating the photocatalyst to complete the catalytic cycle. In this transformation, the formation of the alkenyl radical intermediate plays a crucial role in ensuring the success of the radical Smiles rearrangement.
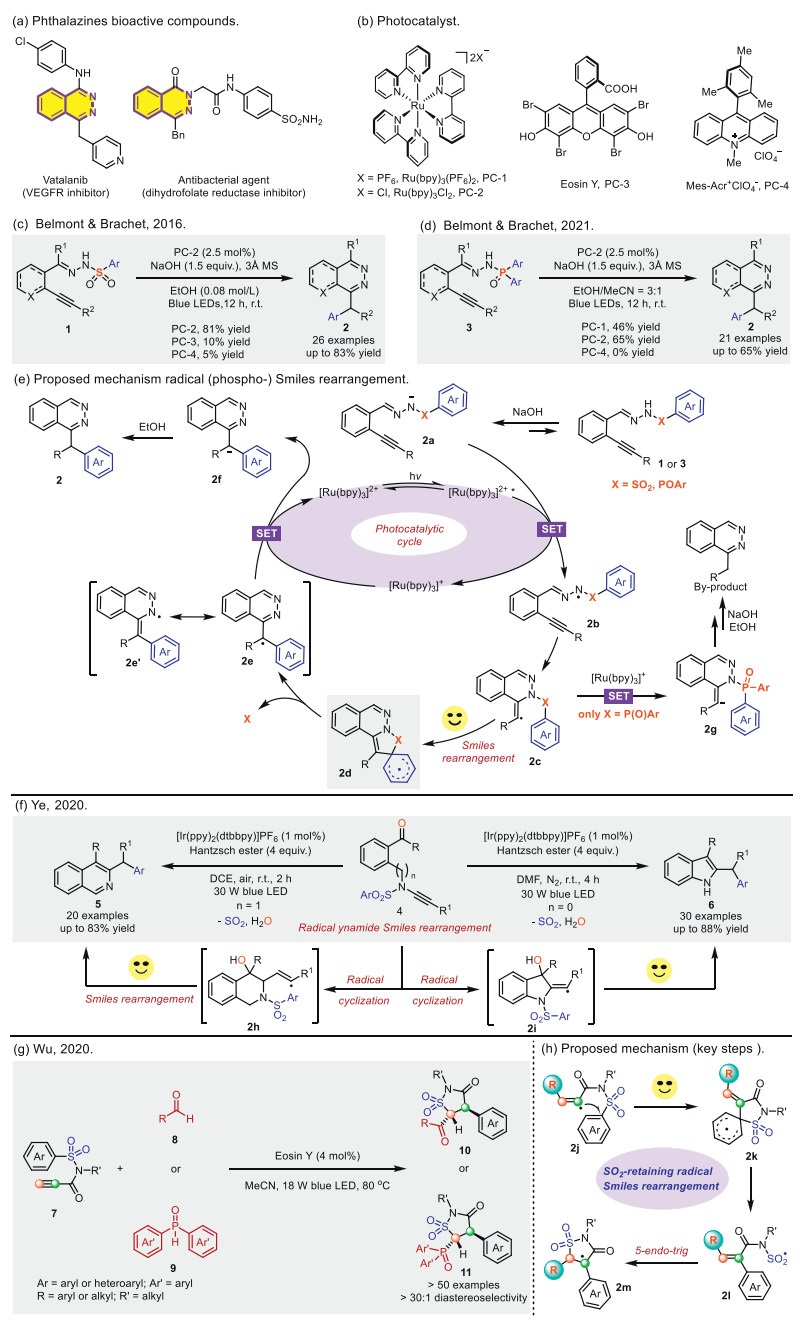
Similarly, the Ye group [23] developed a novel photoredox-catalyzed radical Smiles rearrangement triggered regioselective ketyl−ynamide coupling reaction to give functionalized indoles and isoquinolines (Scheme 2f). This methodology is the first radical Smiles rearrangement of ynamides. Additionally, a variety of valuable 2-benzhydrylindoles and 3-benzhydrylisoquinolines are obtained with broad substrate scope in good yields under mild reaction conditions. Mechanistic studies revealed that the reaction probably proceeds via key ketyl-ynamide radical coupling and undergoes an ynamide radical Smiles rearrangement process.
Almost simultaneously, Wu and colleagues [24] delineated a visible-light-induced radical Smiles rearrangement using Eosin Y as a hydrogen atom transfer (HAT) photocatalyst (Scheme 2g). In contrast to the previous examples, where the alkyne and radical precursors (aldehydes 8, phosphine oxides 9) are in the same molecule, here alkyne (N-(hetero)arylsulfonyl propiolamides 7) is not connected to the radical precursors while the radical precursors serve as an intermolecular partner in this cascade. A key step in the proposed mechanism is illustrated in Scheme 2h. The excited Eosin Y undergoes a hydrogen atom transfer (HAT) with aldehyde 8, generating an acyl radical that subsequently undergoes radical addition onto N-arylsulfonyl propiolamide 7, leading to the formation of vinyl radical species 2j. Following this, a Smiles rearrangement occurs, producing sulfonyl radical 2l via a five-membered ring transition state 2k. Intermediate 2l then undergoes a 5-endo-trig cyclization, yielding the α-carbonyl benzylic radical 2m. Finally, 2m facilitates the regeneration of Eosin Y through a reverse HAT process, while simultaneously forming the isothiazolidinone product (10 or 11). Of particularly note, the authors cannot exclude the possibility of a SET process between radical 2m and Eosin Y − H, one of the strongest arguments from the DFT calculations indicated that the SET pathway was impossible, because the relative free energy barrier was 10 kcal/mol higher than that of the reverse HAT pathway.
Alkynes are often viewed as chemically intriguing due to their complex relationship between thermodynamics and kinetics. While they possess high-energy properties, making many of their reactions more exergonic than comparable reactions with alkenes, their kinetic stability is notably greater. As a result, alkynes frequently react more slowly than alkenes, despite their higher energy levels. Additionally, studies on the rate of intermolecular radical addition indicate that reactions with alkenes occur more rapidly than those involving alkynes, further highlighting their distinct reactivity profile [25]. Compared to the previously discussed examples, when both alkynes and alkenes coexist within the same molecule, radical precursors preferentially attack the alkenes, initiating the radical cascade reaction.
In 2017, Li and co-wokers [26] reported an unprecedented in situ SO2-capture triggered by alkyl radicals, leading to the formation of the alkylsulfonyl radicals. These reactive species could subsequently add to 1,8-enynes, undergoing Smiles rearrangement to generate alkylsulfonylated conjugated heterocyclic amides (Scheme 3a). Similarly, Jr and colleagues [27] described a photo-induced difluoromethylation of conjugated arylsulfonylated amides using CF2HSO2Cl as the CF2H radical source (Scheme 2b). The proposed mechanism for these reactions is depicted in Scheme 3c. In Li's work, an alkyl radical is generated through the SET between the FeⅡ salt and the alkyl peroxide. Following the addition of the methyl radical to 12, the Smiles-type rearrangement and subsequent radical cyclization lead to the formation of a methylation by-product, accompanied by the release of SO2. Subsequently, the in situ capture of SO2 by the alkyl radical generates the methyl-sulfonyl radical, which can undergo radical addition onto 12, forming the α-carbonyl carbon radical intermediate 3a. Similar to Li's work, Jr's study was initiated by the photo-excited catalyst via single-electron transfer (SET) to CF2HSO2Cl, leading to the loss of both Cl⁻ and SO2 and the generation of a CF2H radical. This radical could then add to the conjugated amide 12, forming intermediate 3a. Subsequently, 3a undergoes ipso-cyclization, yielding the spirocyclic key intermediate 3b This intermediate then undergoes rearomatization by expelling an SO2, ultimately furnishing the amidyl radical intermediate 3c through a Smiles rearrangement process. The intramolecular cyclization of 3c leads to the formation of the vinyl radical intermediate 3d, which rapidly undergoes further cyclization to yield the tetracyclic intermediate 3e. Next, 3e is oxidized by a high-valent Fe or Ir catalyst, generating the carbocation species 3f while simultaneously regenerating the catalyst. Finally, the rearomatization of 3f results in the formation of the final product (13 or 14).

The seminal report on intermolecular radical Smiles rearrangement and cyclization of dienynes was published by Li, Yang and Liang et al. in 2021 and described a Cu-catalyzed [3 + 2]/[3 + 2] carboannulation of dienynes and arylsulfonyl chlorides to yield cyclopenta[a]indene-fused quinolinones in high diastereoselectivity (Scheme 3d) [28]. On the basis of mechanistic studies, the authors propose a catalytic cycle consistent with Scheme 3e, in which 4-arylsulfonyl chloride and CuⅠ species undergo SET to form an arylsulfonyl radical and CuⅡ complex, which proceeds through subsequent radical addition/annulation, Smiles rearrangement and finally deprotonation to form the cyclized product 17.
In another report showcasing the fascination of intermolecular radical Smiles rearrangement and annulation of 1, n-enynes, Davies and Shu et al. [29] disclosed a simple methodology to synthesize pyrrolo[1,2-a]indole, pyrido[1,2-a]indole, and azepino[1,2-a]indole skeletons using a CuⅠ- or Ir-initiated radical Smiles rearrangement cascade of ene-ynamides (Scheme 3f). On the basis of the DFT calculations, the authors suggest that the catalytic mechanism commences with radical addition of 1, n-enynes 18 and radical precursors to form vinyl radical species 3o. Subsequently, a Smiles rearrangement would provide five-membered spiral ring intermediate 3p, which could quickly release SO2 to give N-centered radical intermediate 3q or 3q'. Finally, oxidation of 3q (or 3q') by CuⅡ complex (or IrⅣ species) followed by cyclization and deprotonation would give rise to the final product 19 (or 19′) and regenerate the CuⅠ catalyst (or IrⅢ catalyst).
It is well known that silicon atoms often impart unique biological and physicochemical properties to their corresponding silicon-containing organic compounds [30]. Thus, sila-molecules are commonly found in pharmaceuticals and materials chemistry, where they contribute to unique structural and functional properties [31]. On the other hand, vinylsilanes have been regard as an important source of alkene in Hiyama coupling, which could easily incorporate C=C double bonds in a molecule with expelling silyl units [32]. Additionally, the significant difference in electronegativity between carbon (2.35) and silicon (1.64) [33] along with silyl β-effect [34] often contributes to the high reactivity of these compounds. It is essential to develop vinylsilane-based coupling-cyclization strategies, as they may provide a valuable platform for constructing complex silacycles. In order to assemble silicon-containing polycyclic arenes, Zeng, Ke and Su [35] in 2021 jointly developed a visible-light-mediated radical cascade cyclization of ortho-alkynylaryl vinylsilanes and arylsulfonyl azides, giving rise to polycyclic benzosilole skeletons with broad substrate scope, albeit yields are not excellent (up to 74%) (Scheme 3g). This methodology features a unique combination of S-N/C-S bond cleavages and radical Smiles rearrangement. Of particular note, these silaarenes establish foreseeable potential in luminescent material fields.
The alkenes, as one of the fundamental and ubiquitous starting materials, can be directly afforded from petroleum or simply prepared by a one-step synthesis. The alkenes can be roughly classified into terminal and internal alkenes. As an excellent radical receptor, alkenes provide a versatile platform for designing radical transformations, allowing the formation of two new bonds at their energy-rich carbon atoms. Among terminal alkene radical additions, relatively stable radical species can be generated in situ through the attack of a radical precursor at the terminal carbon of the C=C double bond, followed by the installation of an additional functionality. As a result, the intrinsic reactivity of alkenes plays a crucial role in determining their stability and unique addition patterns when interacting with various radicals.
N-centered radicals, including N-radicals and N-radical ions, are versatile reaction intermediates that can undergo radical addition to various π systems to build new C–N bonds. C–N bonds are fundamental functional groups in organic compounds, playing a crucial role in their structural and functional diversity. Notably, approximately 84% of organic small molecules contain at least one C–N bond, highlighting their significance in chemical and biological systems [36]. For instance, pharmaceuticals incorporating arylethylamine motifs, each containing at least one C–N bond, are widely found in drugs such as agomelatine, baclofen, and many others. These compounds play a crucial role in modulating pain sensation and are commonly used in the treatment of heart disease and cancer (Scheme 4a) [37]. N-centered radicals have been established as valuable reactive species, finding widespread applications in C–N bond-forming reactions over the past few decades [38]. In particular, N-centered radicals frequently serve as key intermediates in alkene aminoarylation. A general representation of alkene aminoarylation via nitrogen-centered radicals is shown in Scheme 4b. Under visible light irradiation, sulfonamides 23 generate N-centered sulfo(ⅰ)namidyl radicals 4a, which subsequently add to an alkene, forming the β-aminoalkyl radical intermediate. This intermediate then undergoes a classical radical Smiles rearrangement, yielding the desired aminoarylation product 24.
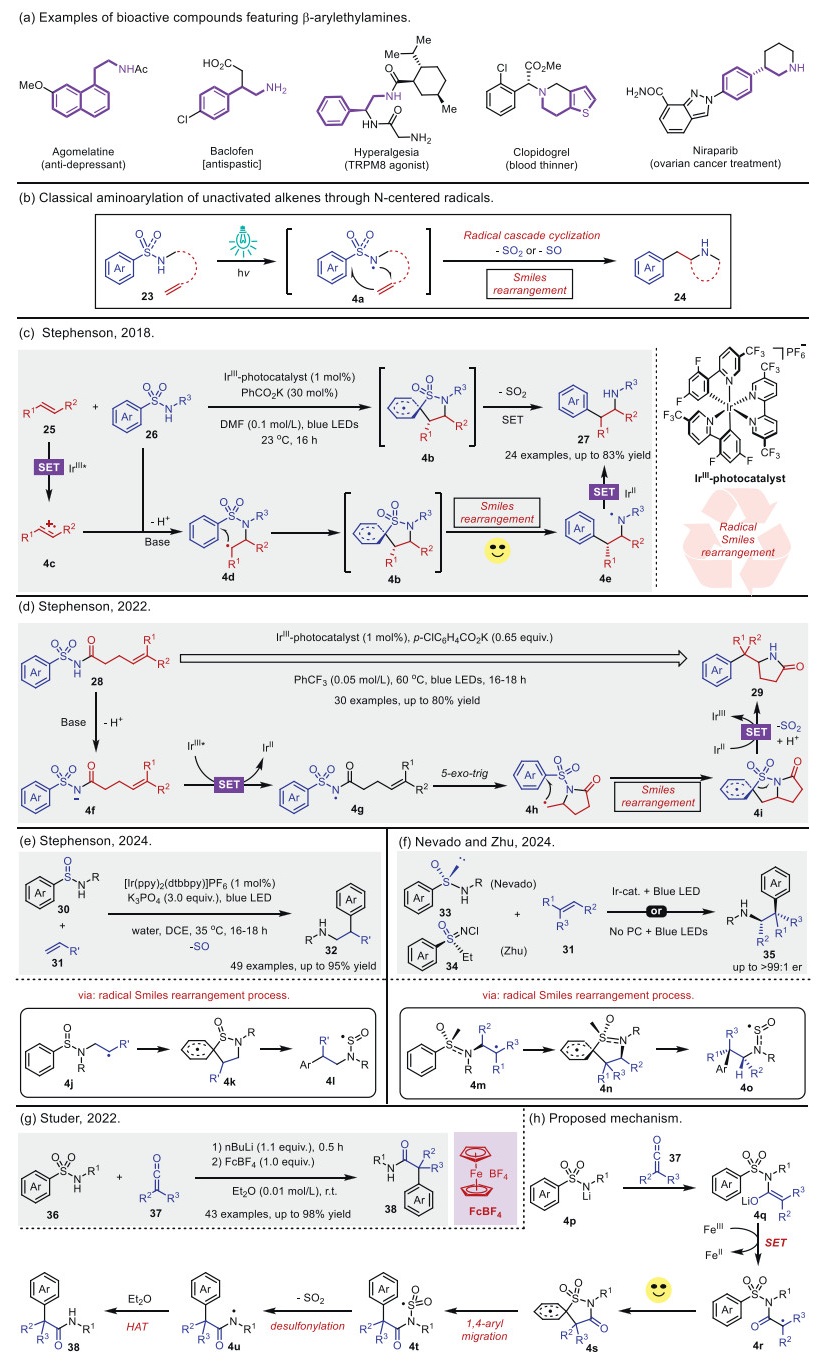
In 2018, Stephenson and co-workers [39] published a photocatalytic methodology for the radical addition of arylsulfonylacetamides 26 onto alkenes 25 with thorough anti-Markovnikov regioselectivity and good diastereoselectivity to produce 2,2-diarylethylamines 27 (Scheme 4c). In this transformation, single-electron oxidation of alkene enables C—N bond formation to give a key five-membered ring intermediate 4b poised for the Smiles rearrangement-type 1,5-aryl shift. This reaction showcases broad functional group compatibilities and proceeds smoothly at mild reaction conditions with catalytic quantities of IrⅢ-photocatalyst, albeit with mild yields. The authors propose that the catalytic cycle begins with a single-electron oxidation process between a photo-excited catalyst (*IrⅢ) and alkene 25, leading to the formation of an alkenyl radical cation species 4c. This species then undergoes nucleophilic addition with aryl-sulfonylacetamide 26, generating the alkyl radical intermediate 4d. Subsequently, a regioselective cyclization event occurs, wherein the alkyl radical interacts with the ipso-position of the aromatic ring to yield intermediate 4b. A rapid radical desulfonylation of 4b follows, furnishing the N-centered radical intermediate 4e and facilitating catalyst turnover.
Alternatively, homolytic fragmentation of the CAr–S bond in 4b, accompanied by desulfonylation, proceeds via a single-electron transfer (SET) process with the catalyst, ultimately leading to the formation of the final product 27. In 2022, the same group [40] disclosed a visible light-induced intramolecular aminoarylation of alkenes with arylsulfonamides via a radical Smiles-Truce rearrangement process to produce β-aryl lactams (Scheme 4d). A variety of simple and unactivated alkenes were tolerated, including some sterically encumbered alkenes, such as 1,2-disubstituted olefins. The authors propose a catalytic cycle consistent with aforementioned catalytic mode, in which arylsulfonamide moiety undergoes a deprotonation to form an N-centered anion 4f in the presence of base. Next, a single-electron oxidation event occurs between the photoexcited iridium catalyst IrⅢ* and 4f to form N-centered radical 4g, which proceeds through subsequent 5-exo-trig cyclization to deliver cyclic alkyl radical intermediate 4h Then, 4h undergoes a radical Smiles rearrangement (including radical cyclization and desulfonylation) and followed by SET with IrⅡ, ultimately yielding the final cyclic product 29.
More recently, Stephenson et al. [41] developed a visible-light-induced aminoarylation of unactivated alkenes 31 and sulfinamides 30 via a radical Smiles-Truce rearrangement to yield arylethylamines 32 (Scheme 4e). The authors suggest that the reaction commences with deprotonation of sulfinamides 30 with the base (K3PO4) to form N-centered anion, then oxidation of this anion by the photocatalysis giving rise to sulfinamidyl radical, which adds to alkenes 31 to generate radical species 4j. Next, a radical Smiles-Truce rearrangement follows from radical species 4j, maybe through a dearomatized spirocyclic transient state such as 4k. Rearomatization and C-S bond homolysis of 4k gives N-sulfinyl radical 4l. Subsequently, the species 4l likely undergoes direct homolysis or reductive cleavage by the reduced photocatalyst to form the amidyl radical or anion, which ultimately undergoes a protonation yields to corresponding products 32.
Almost simultaneously, visible light-mediated asymmetric aminoarylation of alkenes using chiral arylsulfinylamides or sulfoximines are demonstrated by Nevado [42] and Zhu [43], respectively (Scheme 4f). Compared to Stephenson's work, Nevado and co-workers employ a chiral multifunctional arylsulfinylamide reagent bearing a traceless chiral auxiliary group to forge adjacent Csp3-Csp2 and Csp3-N bonds across the π-system in a stereocontrolled manner. Akin to Nevado's work, Zhu and colleagues capitalize on a chiral all-in-one sulfoximine reagent containing a chiral auxiliary to give β-(het)arylethylamine. Mechanistically, the N-centered radical (NCR) intermediate is initially formed under visible-light irradiation with the help of base, followed by addition of NCR intermediate to alkene gives a benzylic radical species 4m, which undergoes intramolecular radical cyclization to deliver spirocyclic transition state 4n. The resultant 1,4-aryl (or hetaryl) migration leads to SO-centered radical species 4o, which may undergo direct SO-extrusion and ultimately hydrogen atom abstraction will complete the sequence and yield to corresponding β-(het)arylethylamine 35.
In 2022, a simple and efficient method for the synthesis of sterically hindered α-quaternary amides via a N-centered radical transient state and 1,4-aryl migration pathway was reported by Studer and co-workers (Scheme 4g) [44]. As authors pointed out, all kinds of N-alkyl or N-arylsulfonamides 36 are reacted with disubstituted ketenes 37, and consecutively underwent SET oxidation, 1,4-aryl migration, desulfonylation, and HAT process to deliver the corresponding amide 38 in good to excellent yields with excellent functional group tolerance.
Similar to previously discussed N-centered radical (NCR) added to alkenes and then induced the radical Smiles rearrangement, C-centered radical (CCR) can also accomplish this rearrangement process through photocatalysis or transition-metal catalysis (Scheme 5). A key mechanistic feature is the generation of an alkyl radical (5a or 5b) upon SET oxidation between catalyst and special substrates (39 or 40) in the presence of base. A series of radical addition and Smiles rearrangement reactions lead to difunctionalization products of alkenes. From the general formula of the reactions depicted in Scheme 5a, it is evident that the structural specificities of the substrates play a crucial role in determining whether the reaction can proceed smoothly through the subsequent rearrangement. For instance, the substrate requires a stabilizing group at the alpha-position to support the radical, or the formed C-centered radical itself must possess a certain degree of stability and reactivity. Under photo-induced (or transition metal catalyzed) reaction conditions, the radical intermediates (5c or 5d) generated from the substrates can rapidly undergo radical addition to the alkenes 41. The resulting Csp3-centered alkyl radical can in turn attack the aromatic hydrocarbons connected with special groups (such as sulfonyl or amino), forming the spirocyclic intermediates (5e or 5f), which can further facilitate the cleavage of the C-Y (Y=S, N) bond, thereby enabling the migration of the (Het)aryl. Among them, it is noteworthy that the 1,4-aryl migration process occurs alongside the cleavage of C-S or C—N bonds. Of note, the model of C—N bond cleavage has greatly enriched the types of Smiles rearrangement reactions. This has not only offered researchers a novel perspective but also provided substantial assistance and inspiration for the design of new substrates in subsequent analogous reactions. We will herein delineate photochemical or transition-metal-catalyzed Truce-Smiles rearrangement involving radical reactions. For the methods demonstrated in this section, whatever radical precursors are, all proceed through a common Csp3-centered radical species, and thus we will discuss these functionalities in parallel.

The seminal report on radical Smiles rearrangement was published by Zhu et al. [45] in 2021 and disclosed a Cu-catalyzed radical addition, functional group migration, intramolecular cyclization, and isomerization to provide multi-functionalized N-fused heteroarenes in high product diversity and broad functional group compatibilities (Scheme 5b). On the basis of control studies, the authors propose that the reaction commences with Single electron transfer between CuⅠ and the C-Br bond of 43 gives the intermediate 5g, which radical addition onto alkene 42 to deliver intermediate 5h. Next, a sequence of ipso-heteroaryl migration and SO2 extrusion generates the radical 5j, which might abstract a Cl or Br atom from the solvent DCM or 43. The ultimate deprotonation through isomerization and HF elimination produces the final product 44.
Similarly, the same group in 2022 developed a new and efficient photoinduced intermolecular fluoroalkylative olefination of unactivated alkenes (Scheme 5c) [46]. The reaction proceeded through a radical Smiles rearrangement-type cascade, where a capacity of smartly designed difunctionalization alkenylated reagents was employed to provide vinylated products with exclusive E-configuration. To gain deeper insight into the mechanism, a series of control experiments were conducted, and a plausible mechanism is depicted in Scheme 5c. Initially, the excited state of IrⅢ*, which formed from visible-light irradiation of IrⅢ catalyst, is oxidatively quenched by the substrates 45, giving rise to the intermediate 5k and IrⅣ species. Radical addition of 5k to alkene 46 produces the alkyl radical 5l, which easily undergoes a 5-exo-trig process to deliver the cyclic intermediate 5m. Subsequently, a sequence of ring-opening homolysis and SO2 extrusion, gives rise to the intermediate 5n. Ultimately, a SET process occurs between 5n and TBAB to furnish the final product 47 and regenerates IrⅢ species. Alternatively, the alkyl radical 5n might directly abstract the Br atom from 45 to give the product.
Ethylene, as the simplest alkene and the most abundant petrochemical product, is mainly used to prepare commodity chemicals, such as polyethylene, polyvinylchloride, ethylene oxide, styrene [47]. Although ethylene is extensively utilized in numerous classical reactions (such as Heck-type reaction [48], Wacker oxidation [49], hydrovinylations [50]) and difunctionalization of ethylene, its application in radical reactions remains scarce. This is due to the fact that when a radical adds to ethylene, the resulting primary alkyl radicals exhibit exceptionally high reactivity and instability. This leads to rapid self-polymerization, making functionalization with other molecules challenging. As a result, this simplest C2 feedstock is often overlooked by chemists.
In an impressive display of the rearrangement ability of heterocyclic sulfone to react with alkenes, Zhu and Wu et al. [51] reported photoinduced three-component selective ethylene difunctionalization through a metal-free radical Smiles rearrangement (Scheme 5d). This methodology is characterized by its mild reaction conditions, simple operation, good functional group tolerance and excellent product diversity. Mechanistically, the authors suggested that the reaction commenced with homolytic cleavage of 48 to form electrophilic difluoromethyl radical 5o under the visible-light irradiation. Addition of 5o to ethylene generates the primary alkyl radical 5p, which undergoes a radical Smiles rearrangement to afford alkyl radical species 5q. After the Smiles rearrangement, the nucleophilic radical intermediate 5q is provided and subjected to a Minisci-type addition with quinoxalinone 49 to deliver final product 51.
In 2024, the group of Nacsa [52] published visible-light mediated alkyl–(hetero)arylation of olefins to afford (het)arylated alkyl ester with excellent yield (up to 99% yield) and broad functional group compatibility (Scheme 5e). Akin to aforementioned mechanism, a series of radical addition and Smiles rearrangement leads to difunctionalized products of olefins.
Simultaneously, the seminal report on intermolecular carbamoylarylation of alkenes was disclosed by Sekine and Kuninobu et al. in 2024 and discovered a visible-light-catalyzed radical addition of carbamoyl radicals to alkenes to produce arylpropanamides in high yields and chemoselectivities (Scheme 5f) [53]. A tentative mechanism is shown in Scheme 5f, N-aryl oxamic acid derivatives 55 rapidly generates carbamoyl radicals 5t in the presence of base and under the visible-light irradiation. Then, the radical addition of 5t to olefins 54 gives alkyl radicals 5u, which easily undergoes ortho-radical addition to an aryl amide moiety giving rise to spiral intermediate 5v. Finally, C(aryl)−N bond cleavage and 1,4-aryl migration leads to carbamoylarylated product 56. It is noteworthy that if the rearrangement product is to be obtained, the introduction of a bulky group onto the N-atom of N-aryl oxamic acids to facilitate the transformation is essential. Consequently, this significantly restricts the scope of substrates. Although some preliminary mechanistic studies have been conducted, the authors still believe that the reaction mechanism on the 1,4-aryl migration seemingly more complex than initially presumed.
In another report showcasing the glamour of the radical Smiles rearrangement, Wang and co-workers [54] in 2022 described a novel photoinduced successive 1,2-boron migrations and Smiles-type rearrangement between allylboronic esters and sodium arylsulfonate (Scheme 5g). A series of terminally functionalized alkyl boronates with broad scope of substrates were obtained. A possible mechanism was proposed as shown in Scheme 5g. Initially, the excited-state photocatalyst is generated from its ground state under visible light irradiation. It then undergoes a single-electron transfer (SET) with arylsulfonates 57, forming the sulfone radical 5w. The radical addition of 5w onto allylboronates 58 produces the secondary alkyl radical 5x, which rapidly undergoes the first 1,2-boron shift, generating the tertiary radical species 5y. Subsequently, a 5-exo-trig cyclization occurs, yielding intermediate 5z. This intermediate then undergoes sequential transformations, including SO2 extrusion, a second 1,2-boron shift, and a hydrogen atom transfer (HAT), ultimately furnishing the final product 59.
When both migratory arenes and alkenes coexist within the same molecule, the alkene moiety serves as the initial acceptor site for external radicals, leading to the formation of a Csp3 radical. This intermediate then triggers an intramolecular 1,4-aryl migration, ultimately yielding the Smiles rearranged product. As illustrated in the general reaction formula (Scheme 6a), radical-active intermediates (R') generated from various radical precursors undergo addition to the alkene moiety of substrates 60, forming the alkyl radical intermediate 6a. Subsequently, this Csp3 radical attacks an aromatic ring bearing a leaving group (such as SO2 or SO), leading to the formation of a five-membered spirocyclic radical transition state 6b. Finally, cleavage of the C–Z bond facilitates the 1,4-aryl migration, yielding the final product 61. Alkenyl sulfonamide compounds 62 are well established as typical substrates for the Smiles rearrangement and are frequently employed to investigate the chemoselectivity of various radical precursors during the rearrangement process. In recent years, under photocatalytic, electrocatalytic, and transition-metal catalytic conditions, alkenyl sulfonamides have undergone rearrangement reactions with diverse radical precursors, leading to the synthesis of a wide array of functionalized amides (Scheme 6b). As depicted in Scheme 6c, in the field of photocatalysis, numerous groups (e.g., Greaney, Ji, Liang, Masson) have developed diverse radical precursors 64 for the radical Smiles rearrangement reactions with alkenyl sulfonamides 63 to afford a variety of functionalized amides 65 (65A-65G, 65I) [55-63]. Additionally, the area of electro catalysis has offered a fertile breeding ground for the development of radical Smiles rearrangement reactions. For example, Pan et al. [64] published a novel electrochemical method for preparing the β-difluoromethylamide compounds 65H using N-benzenesulfonylacrylamide with difluorine reagents as raw material. Furthermore, it is well-established that many radical precursors can readily form reactive radical intermediates in the presence of transition metals (such as copper and silver). These radical species subsequently undergo radical addition to alkenes, thereby initiating follow-up migration processes. Several research groups have made outstanding contributions in this field. For instance, the teams of Liu, Liang and Zheng have reported Cu- or Ag-catalyzed strategies involving radical initiation, radical addition, and aryl migration processes to synthesize corresponding substituted amides (65I-65l) [65-68]. With the continuous in-depth research on radical Smiles rearrangement reactions, it has been discovered that Smiles rearrangements are no longer limited to the cleavage of S-N bonds in sulfonamide but can also involve C-S and S-O bonds. Additionally, the resulting rearrangement products are no longer confined to amides (67, e.g., alcohols, amines) [69-72].

While the aforementioned examples have enabled access to numerous rearranged products, a distinct pattern of substrate engineering tailored to the radical Truce-Smiles rearrangement is evident. As a result, the reactivity scope of transition-metal-, photo-, or electro-catalyzed intramolecular 1,4-aryl migration remains inherently limited. Ongoing efforts toward the development of asymmetric methods employing simpler chiral substrates are encouraged. As shown in Scheme 7a, the enantioenriched N-arylsulfinyl acrylamides 68, which are readily prepared from commercially available cheap chiral starting materials, are employed as radical acceptors for a variety of radicals produced in situ under mild photoredox conditions. Subsequently, a key chiral five-membered spirocyclic intermediate 7a rapidly forms. Notably, during the formation of the new C–C bond, the preexisting sulfur chirality in the molecule induces steric hindrance, restricting the bond formation to a single configuration. This may be thermodynamically favoured, thereby determining the establishment of quaternary carbon stereocenters upon 1,4-aryl migration. Namely, this process effectively achieves the transfer of chirality from sulfur to carbon.

The asymmetric construction of all-carbon quaternary centers in acyclic systems has long posed a significant congestion surrounding the stereogenic carbon center, which is even more pronounced in acyclic settings and must be overcome to achieve efficient synthesis [73,74]. For example, the Nevado group [75] in 2021 reported visible light-induced asymmetric radical sulfinyl-Smiles rearrangement to access quaternary carbon stereocenters (Scheme 7b). As privileged structural units, Csp3-hybridized atoms, in particular chiral quaternary C stereocenters, are universal in varied bioactive natural products, agrochemicals and pharmaceutical molecules whose function could span from Dicorantil, CCR5 antagonist to antiarrhythmic and anti-HIV activities (Scheme 7c) [76-78]. Several control experiments were conducted to provide mechanistic insight, and the authors propose a catalytic cycle consistent with Scheme 7ⅲ, in which R-X undergoes a SET with the activation of the IrⅢ photocatalyst to form the initiating radical R· 7b (and IrⅣ photocatalyst species), which proceeds through subsequent radical addition onto the enantioenriched N-arylsulfinyl acrylamides 70 to give the alkyl radical intermediate 7c. Although one of strongest arguments that experimental evidence has been obtained for the participation of Meisenheimer complex in radical Smile rearrangement reactions [79,80], all tries of the authors to gain the radical intermediate 7c met with failure. Instead, the authors suggested that the 7c would undergo a spirocyclic transition state 7d and 1,4-aryl migration to deliver the SO-centered radical 7e by DFT calculations. Oxidation of the SO-centered radical 7e by the IrⅣ and followed by a protonolysis reaction with two molecules of H2O ultimately yielding the corresponding product 72 and regenerating the IrⅢ catalyst (Scheme 7d).
Similar contributions to substrate-induced stereoselectivities were made by Wu et al., who documented the first asymmetric synthesis of β-chiral sulfones containing all-carbon stereocenters through photo-mediated radical SO2-insertion and radical Smiles rearrangement (Scheme 7e) [81]. A striking feature of this reaction is that, unlike the aforementioned reactions, it proceeds as a three-component process. While diverse radical precursors (74 or 76) generate aryl or alkyl radicals under light irradiation, these radicals can in situ combine with sulfur dioxide (generate from Na2S2O4 or DABCO·(SO2)2) to afford a variety of sulfonyl radical intermediates 7b Consequently, the reaction exhibits a broad substrate scope, and the resulting rearranged products (75 or 77) significantly enrich the library of β-chiral sulfones bearing quaternary carbon stereocenters. A key mechanistic feature is the formation of a spirocyclic transition state 7j upon radical addition onto alkyl radical 7i. Notably, when different radical precursors are used, the choice of photocatalyst also varies, and the corresponding mechanisms differ slightly-specifically in the process of generating sulfonyl radicals. However, under both sets of reaction conditions, the transformations undergo a same radical Smiles rearrangement pathway, ultimately affording products with analogous core structures. Importantly, quantum yields (2.43 and 3.28) experiments of these two catalytic systems support the existence of a radical chain process (Scheme 7f).
In 2024, Nevado and Merino et al. delineated asymmetric remote arylation of Csp3-H bonds via a sulfinyl-Smiles rearrangement and HAT process to produce structurally diverse α-arylated amides with excellent er up to > 99:1 under mild conditions (Scheme 7g) [82]. Mechanistic studies, including computations and extensive control experimental investigations, reveal that the reaction pathway proceeds via radical addition, 1, n-HAT, and 1,4-aryl migration, leading to the formation of the desired α-aryl-substituted amides 79 in moderate to good yields (Scheme 7h). Additionally, the authors have shown the robustness of this protocol with various types of radical precursors (e.g., perfluoroalkyl radicals, phosphonate-containing radical, fluorinated secondary radicals, primary alkyl radicals), albeit with reduced yields.
In 2020, Clayden et al. reported an intramolecular photocatalyzed difunctionalization of vinyl ureas via a radical Smiles rearrangement cascade, proceeding through a spirocyclic-type intermediate (Scheme 8a) [83]. The authors propose that radical intermediates 81, generated from CnFmSO2Na or HP(O)Ar2 via a photocatalyst-mediated single-electron transfer (SET) process, undergoes radical addition to vinyl urea substrates 80, forming radical species 8a. Subsequently, 8a undergoes single-electron reduction, yielding the anionic species 8b The rearrangement of 8b then furnishes the final products 82.
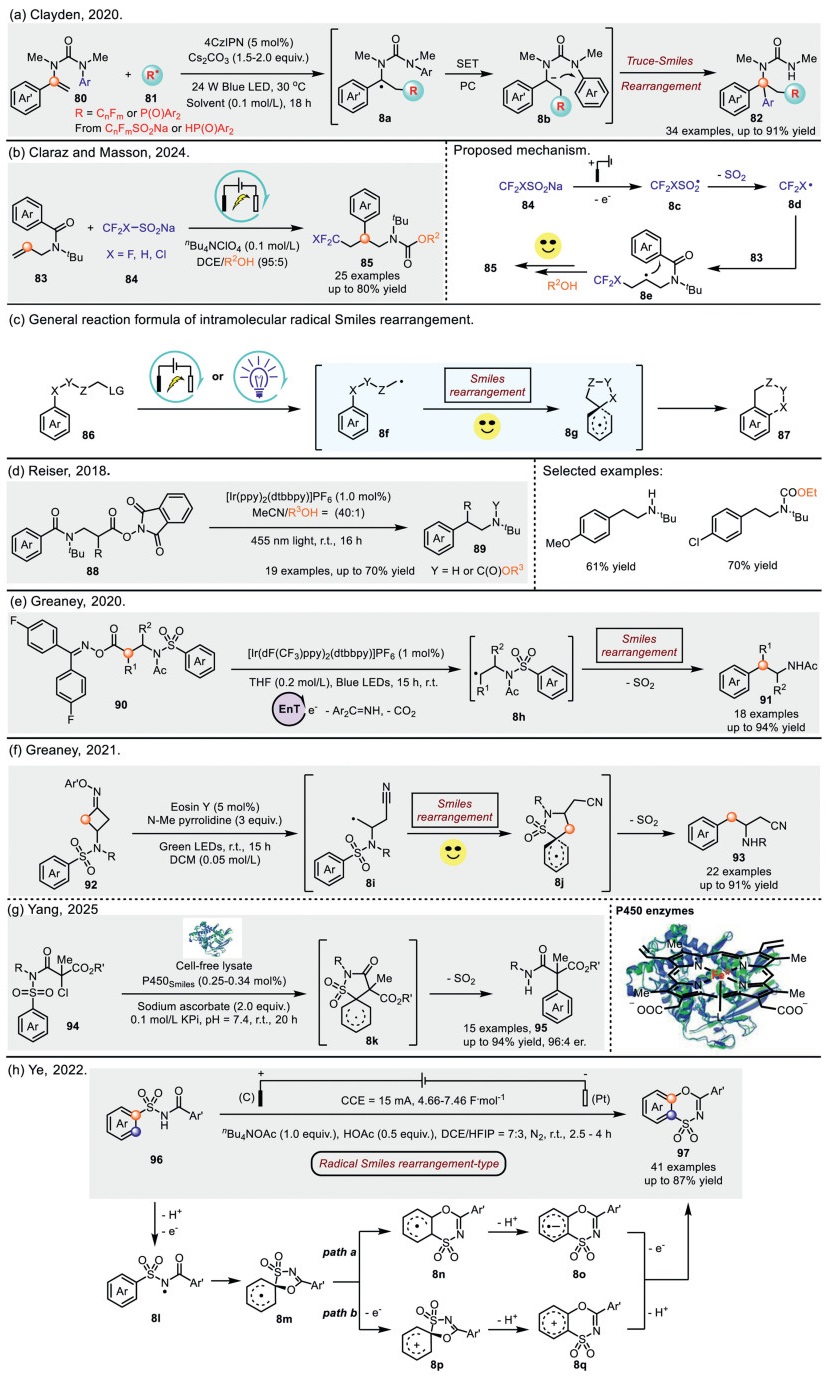
In another study highlighting the potential of electrocatalysis in facilitating radical Smiles rearrangement transformations, Claraz and Masson et al. recently introduced an electro-driven methodology for radical fluoromethylation of N-allylbenzamides, enabling the construction of functionalized β-arylethylamines (Scheme 8b) [84]. The authors' protocol is based on the design of a sterically hindered group tethered to the nitrogen atom of N-allylbenzamides 83. This bulky steric group prevents competitive 6-endo cyclization, thereby promoting more efficient formation of the aryl migration product 85. Computational studies of the rearrangement mechanism in different protecting group systems (t-Bu vs. Ts) indicate that the t-Bu group is essential for the transformation. Mechanistically, the reaction is proposed to initiate with the anodic oxidation of 84, accompanied by SO2 extrusion, yielding the CF3 radical 8d. This radical undergoes addition to N-allylbenzamides 83, generating the alkyl radical intermediate 8e. Finally, 8e undergoes a radical Smiles rearrangement, leading to the formation of the desired products 85 (Scheme 8b, right).
Additionally, a unique type of intramolecular Smiles rearrangement reaction exists, as illustrated in Scheme 8c. This transformation demands exceptional creativity from synthetic chemists, requiring the precise design of substrates 86 with distinct structural features. Through photocatalysis, electrocatalysis, or other activation methods, an in situ-generated radical 8f forms within the molecule. This radical then attacks another segment of the same molecule, initially producing an endocyclic intermediate 8g, which rapidly undergoes bond cleavage and ring-opening, ultimately facilitating aryl migration. The driving force behind this migration-induced ring-opening is the intrinsic ring strain of the resulting spirocyclic intermediate, which promotes the cleavage of existing bonds and the formation of new ones.
On the basis of this concept, Reiser and co-workers realized an intramolecular decarboxylation and Smiles-type rearrangement of ω-aryl-N-(acyloxy)phthalimides by visible-light-promoted (Scheme 8d) [85]. Using [Ir(dtbbpy)(ppy)2]PF6 as a photocatalyst and leveraging the unique structure of the starting material, the oxime ester moiety of the substrate undergoes conversion to the corresponding alkyl radical. This radical then undergoes energy transfer (EnT) with the Ir(Ⅲ) catalyst, followed by an intramolecular electron transfer (IET). Depending on the length of the alkyl chain, this process facilitates the formation of a key spirocyclic intermediate, which subsequently undergoes 1,4-aryl migration to yield the rearranged product 89 in moderate yields.
Smilarly, the Greaney group in 2020 developed a visible light catalyzed decarboxylative and desulfonylative Smiles rearrangement for synthesis of arylethylamine 91 employing activated-ester undergoes an EnT process (Scheme 8e) [86]. In 2021, the same research group reported a photocatalyzed strain-release-triggered radical Truce-Smiles rearrangement reaction for the synthesis of valuable 1,3-diamines and unnatural β-amino acids (Scheme 8f) [87]. Their method demonstrated broad substrate tolerance, accommodating both simple and poly-substituted aromatics. A key mechanistic distinction between this work and their previously reported system lies in the initiation step. In the earlier case, the reaction was driven by an energy transfer (EnT) process between the photocatalyst and the substrate, leading to homolytic cleavage of the N–O bond, followed by decarboxylation to generate the alkyl radical 8h. In contrast, the present reaction proceeds via a single-electron reduction of the substrate by the photocatalyst, forming an iminyl radical intermediate. This intermediate subsequently undergoes ring-opening, driven by ring strain, to afford the alkyl radical 8i. Although both pathways involve N–O bond cleavage, their mechanistic underpinnings differ significantly. The authors conducted extensive mechanistic studies to validate these distinctions.
In recent years, photobiocatalytic radical reactions have witnessed burgeoning development [88-90]. For instance, the Yang and Liu et al. [91] published a directed evolution-derived P450 aryl migration enzyme (P450Smiles) capable of catalyzing asymmetric Smiles rearrangement with high chemoselectivity and enantioselectivity, constructing a series of acyclic amide products bearing all-carbon quaternary stereocenters (Scheme 8g). Key highlights of this work include: (a) Compared to reported Smiles rearrangements, this strategy does not rely on substrate control to achieve chemoselectivity, demonstrating broad substrate compatibility. (b) DFT calculations reveal that the dearomatized spirocyclic radical intermediate 8k is more stable, suggesting that this cyclization step is irreversible and likely the key factor governing stereoselectivity. (c) Classical molecular dynamics (MD) simulations indicate that: (ⅰ) P450Smiles suppresses the inherent cyclization tendency of substrate intermediates through conformational constraints and hydrogen bonding with key residues (e.g., R267), significantly enhancing selectivity toward acyclic amides; (ⅱ) Compared to the P450rad3 variant, P450Smiles features a more compact active site with stronger substrate confinement, leading to superior chemoselectivity and stereoselectivity.
Benzoxathiazine dioxide is a widely used diazoxide bioisostere in clinical practice, exhibiting intriguing biological activities. However, there have been relatively few literature reports on efficient synthetic methods for such compounds [92]. The Ye and Zhu group in 2022 developed an electrochemical migratory cyclization strategy of N-acylsulfonamides to synthesize a series of benzoxathiazine dioxides via radical Smiles rearrangement (Scheme 8h) [93]. Moreover, this approach features a broad substrate scope, excellent functional group compatibility, and mild reaction conditions. These findings highlight the unique characteristics of electrochemistry in exploring novel reactivity and will inspire the discovery of more electrochemically mediated new chemical transformations. To further elucidate the reaction mechanism, the authors conducted cyclic voltammetry (CV) studies. The results revealed that N-Ts-benzamide 96 exhibits a significantly higher oxidation potential than nBu4NOAc. Additionally, NMR studies revealed the presence of an intermolecular hydrogen bond between the N–H group in 96 and the acetate anion. On the basis of CV and NMR studies, the authors proposed a plausible reaction mechanism. The reaction is initiated by the generation of N-centered radical 8l from substrate 96 via proton-coupled electron transfer (PCET), followed by two possible pathways. DFT calculations revealed that radical 8l undergoes facile radical spirocyclization to form radical 8m. In path a, the mechanism involves radical-type sulfonyl migration (8m→8n) followed by base-promoted homolytic aromatic substitution 8o. In path b, radical intermediate 8m may undergo single-electron oxidation to form the corresponding carbocation 8p, which subsequently proceeds via cationic sulfonyl migration (8p→8q) and base-mediated deprotonation to afford the migratory cyclization product 97. Notably, computational studies indicate that both radical-type and cationic sulfonyl migrations represent viable pathways for the formation of the final benzoxathiazine dioxide product.
As was introduced in the previous section, whether in ionic or radical-type Smiles rearrangements, the critical 1,4-aryl migration process (forming the key spirocyclic intermediate) typically requires cleavage of the CAr–S bond to proceed smoothly. However, a review of classical literature clearly reveals that in ionic-type Smiles rearrangements, the heteroatom involved in aryl migration can also be oxygen, that is, CAr–O bond cleavage similarly facilitates efficient and controlled 1,4-aryl migration, enabling the ionic Smiles rearrangement to occur. Interestingly, under modern catalytic systems (e.g., photocatalysis or electrocatalysis), radical-type Smiles rearrangements can also proceed via analogous intramolecular CAr–O bond cleavage and aryl migration processes. As outlined in Scheme 9a, ortho-acyl aryl ethers 98 have been designed that, under photo- or electro-induced conditions, generate radical intermediates 9a (or 9b). These radical intermediates subsequently undergo six- or five-membered spirocyclization to form 9c (or 9d), respectively. Finally, CAr–O bond cleavage facilitates 1,4-aryl migration, yielding the rearranged products 99 (or 100). As such, photoredox-catalyzed radical Smilesrearrangement of 2-aryloxybenzoic acids to afford aryl salicylates was successively demonstrated by Gonzalez-Gomez [94], Yatham [95] and Shen [96] (Schemes 9b, e and f). A key commonality among these three studies is that, under the combined action of a base and a photocatalyst, the carboxylate anion of the substrate (101, 107, 109) first generates a carboxyl radical. This radical then forms a six-membered spirocyclic radical intermediate with the aryl moiety of the aryl ether, ultimately leading to the final product (102, 108, 110) via CAr–O bond cleavage. In 2019, Chen and co-workers reported photoredox-induced acyl radical Smiles rearrangement to convert biaryl ethers 103 to hydroxybenzophenones 104 by using the dual catalysis of hypervalent iodine reagents (BI-OAc) and organophotocatalysts (Acr-Mes+ClO4–) (Scheme 9c) [97]. Moreover, Ye et al. in 2020 developed NHC/photo-co-catalyzed radical rearrangement to synthesize aryl salicylates from O-aryl salicylaldehydes (Scheme 9d) [98]. Accordingly, a possible mechanism is shown in Scheme 9d, the addition of NHC-cat. to 105 produces intermediate 9e, which is oxidized by O2 in the presence of photocatalyst (PC) and NaI as the cocatalysts via a SET process to give the radical cation 9f. Deprotonation of 9f furnishes zwitterionic radical species 9g, which was oxidized by the in situ generated HO2· to deliver acyl azolium 9h. O-Aryl salicylic acid 9i is generated from acyl azolium 9h via hydrolysis and regenerate NHC-cat. enter next catalytic cycle. Next, 9i is oxidized by the excited-state Acr-Mes+ to give the carboxyl radical 9j. Finally, intermediate 9j undergoes a radical Smiles rearrangement to afford 106.

The seminal report on electro-catalyzed radical Smiles rearrangement was reported by Guo et al. in 2019 and describes an amidyl radical generated from the cleavage of the N-O bond of hydroxylamines 111 to yield amides 112 in high yields (up to 86%) (Scheme 9g) [99]. Additionally, Lawson and Murphy in 2020 published visible-light-induced intramolecular radical Smiles rearrangements and annulations of non-activated aromatics at ambient temperature and in the presence of an acridinium catalyst (Scheme 9h) [100]. The authors suggest that the catalytic cycle commences with visible light excitation of the ground-state acridinium salt (Acr-Mes+) to its excited state (Acr-Mes+*) enables the oxidation of phenoxyalkylamine 113 to an arene radical cation 9k. The tethered primary amine subsequently adds onto the ipso-position gives the key spirocyclic species 9l via a radical Smiles rearrangement process. H atom transfer and reduction by the acridinyl radical Acr-Mes· deliver rearranged product 114 and concurrently regenerate the ground state catalyst.
The past decade has witnessed a remarkable surge in radical Smiles rearrangement reactions. The rapid advancements in this area, driven by diverse catalytic strategies, including photocatalysis, electrocatalysis, and transition-metal catalysis, highlight the immense synthetic potential of this methodology, ensuring its continued relevance in the years to come. The primary objective of this paper is to identify gaps in the current understanding and stimulate critical thinking among readers regarding alternative approaches. By highlighting these areas, we aim to inspire further research and innovation in this evolving field.
Particularly, asymmetric radical Smiles rearrangements remain a significant challenge, despite their promising reactivity. In this regard, a broad range of examples presented throughout this review highlights an evident gap in approaches describing asymmetric intermolecular radical-type Smiles rearrangement. In fact, only one reported method has demonstrated this pattern. Further advancements in expanding the reactivity and diversity of chiral substrates, as well as developing intermolecular systems, are highly warranted. Our group's recent work on photo-catalyzed asymmetric radical Smiles rearrangement has opened new avenues for such transformations, and continued progress in this area is anticipated.
Despite astounding progress in achieving radical Smiles rearrangement by photocatalysis, electro- or transition-metal-catalyzed asymmetric radical Smiles rearrangements remain elusive. In general, for radical-type Smiles rearrangements, the use of photoredox or electrocatalytic strategies offers milder reaction conditions compared to traditional transition metal catalysis. Although conventional transition metal catalysis typically requires harsh conditions (e.g., high temperatures), it often exhibits greater robustness. For instance, in scale-up reactions, transition metal catalysis remains less affected, whereas photoredox or electrochemical methods tend to encounter more pronounced challenges with increasing scale. Consequently, transition metal-catalyzed processes are often more readily adopted in industrial production. Looking ahead, we anticipate that in the near future, radical Smiles rearrangement reactions will extend beyond the pre-design and synthesis of corresponding chiral starting materials. Instead, a wider range of substrates will undergo Smiles rearrangement via asymmetric catalytic strategies. For instance, the integration of new-to-nature enzymatic approaches with transition-metal-catalyzed rearrangement has already provided significant inspiration for chemists, paving the way for further advancements in this field.
Moreover, the radical Smiles rearrangement's unique capacity for diverse Csp2-Y (Y = C, O, N, S, CO, etc.) bonds formation and construction of sterically demanding quaternary carbons make this strategy particularly valuable for: (1) Synthesizing challenging chiral quaternary carbon-containing pharmaceuticals, and (2) Enabling novel developments in materials science and agrochemical design.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Nianhua Luo: Investigation. Jiayi Jiang: Investigation. Muhammad Suleman: Investigation. Zhaowen Liu: Investigation. Shuping Huang: Investigation. Wei Xiao: Investigation. Jie Wu: Writing – review & editing. Jiapian Huang: Writing – original draft.
Financial support from the Fundamental Research Funds for Gannan Medical University (No. QD202429), National Natural Science Foundation of China (No. 22171206), Natural Science Foundation of Zhejiang Province (No. LZ23B020001), Zhejiang Provincial Ten Thousand Talent Program (No. 2023R5244) and Open Research Fund of School of Chemistry and Chemical Engineering, Henan Normal University (No. 2020ZD04) is gratefully acknowledged.
L.M. Harwood, Polar Rearrangements, Oxford Chemistry Primers, Oxford University Press, Oxford, UK, 1992.
W.E. Truce, E.M. Kreider, W.W. Brand. Org. React. 18 (1970) 99–215.
J.J. Li, Smiles rearrangement. Name Reactions: A Collection On Detailed Reaction Mechanisms, 3rd Ed, Springer, Berlin, Heidelberg, 2006, pp. 549–554.
T.J. Snape, Chem. Soc. Rev. 37 (2008) 2452–2458. doi: 10.1039/b808960d
A.A. Levy, H.C. Rains, S. Smiles, J. Chem. Soc. (1931) 3264–3269.
R. Loven, W.N. Speckamp, Tetrahedron Lett. 13 (1972) 1567–1570. doi: 10.1016/S0040-4039(01)84687-6
J.J. Koehler, W.N. Speckamp, Tetrahedron Lett. 18 (1977) 631–634. doi: 10.1016/S0040-4039(01)92711-X
W.B. Motherwell, A.M.K. Pennell, J. Chem. Soc. Chem. Commun. (1991) 877–879.
Y. Zhang, J.J. Chen, H.M. Huang, Angew. Chem. Int. Ed. 61 (2022) e202205671. doi: 10.1002/anie.202205671
A.J. Perkowski, C.L. Cruz, D.A. Nicewicz, J. Am. Chem. Soc. 137 (2015) 15684–15687. doi: 10.1021/jacs.5b11800
T. McCallum, Green Synth. Catal. 4 (2023) 10–19.
J.W. Tucker, C.R.J. Stephenson, Org. Lett. 13 (2011) 5468–5471. doi: 10.1021/ol202178t
E. Brachet, L. Marzo, M. Selkti, B. König, P. Belmont, Chem. Sci. 7 (2016) 5002–5006. doi: 10.1039/C6SC01095D
J. Douglas, H. Albright, M. Sevrin, K. Cole, C.R.J. Stephenson, Angew. Chem. Int. Ed. 54 (2015) 14898–14902. doi: 10.1002/anie.201507369
I. Allart-Simon, S. Gérard, J. Sapi, Molecules 21 (2016) 878–890. doi: 10.3390/molecules21070878
I.V. Alabugin, B. Gold, J. Org. Chem. 78 (2013) 7777–7784. doi: 10.1021/jo401091w
N.P. Tsvetkov, E. Gonzalez-Rodriguez, A. Hughes, et al., Angew. Chem. Int. Ed. 57 (2018) 3651–3655. doi: 10.1002/anie.201712783
G. Stork, N.H. Baine, J. Am. Chem. Soc. 104 (1982) 2321–2323. doi: 10.1021/ja00372a042
H.S. Ibrahim, W.M. Eldehna, H.A. Abdel-Aziz, M.M. Elaasser, M.M. Abdel-Aziz, Eur. J. Med. Chem. 85 (2014) 480–486.
E.N. Scott, G. Meinhardt, C. Jacques, D. Laurent, A.L. Thomas, Expert Opin. Invest. Drugs 16 (2007) 367–379. doi: 10.1517/13543784.16.3.367
E. Brachet, L. Marzo, M. Selkti, B. Konig, P. Belmont. Chem. Sci. 7 (2016) 5002–5006. doi: 10.1039/C6SC01095D
M.D. Abreu, P. Belmont, E. Brachet, J. Org. Chem. 86 (2021) 3758–3767. doi: 10.1021/acs.joc.0c02540
Z.S. Wang, Y.B. Chen, H.W. Zhang, et al., J. Am. Chem. Soc. 142 (2020) 3636–3644. doi: 10.1021/jacs.9b13975
J. Yan, H.W. Cheo, W.K. Teo, et al., J. Am. Chem. Soc. 142 (2020) 11357–11362. doi: 10.1021/jacs.0c02052
O. Ito, M. Matsuda, J. Am. Chem. Soc. 101 (1979) 1815–1819. doi: 10.1021/ja00501a031
X. Su, H. Huang, W. Hong, et al., Chem. Commun. 53 (2017) 13324–13327. doi: 10.1039/C7CC08362A
S. Wei, S. Le, Z. Lei, et al., Org. Biomol. Chem. 20 (2022) 2064–2068. doi: 10.1039/d2ob00186a
L. Zhou, X. Liu, H. Lu, et al., Org. Chem. Front. 8 (2021) 5092–5097. doi: 10.1039/d1qo00703c
S. Li, Y. Wang, Z. Wu, et al., Org. Lett. 23 (2021) 7209–7214. doi: 10.1021/acs.orglett.1c02519
S. Pujals, J. Fernández-Carneado, M.J. Kogan, et al., J. Am. Chem. Soc. 128 (2006) 8479–8483. doi: 10.1021/ja060036c
J. Friedrich, S. Dorrich, A. Berkefeld, P. Kraft, R. Tacke, Organometallics 33 (2014) 796–803. doi: 10.1021/om401181h
S.E. Denmark, R.F. Sweis, Acc. Chem. Res. 35 (2002) 835–846. doi: 10.1021/ar020001r
G. Simons, M.E. Zandler, E.R. Talaty, J. Am. Chem. Soc. 98 (1976) 7869–7870. doi: 10.1021/ja00440a093
T.A. Blumenkopf, L.E. Overman, Chem. Rev. 86 (1986) 857–873. doi: 10.1021/cr00075a009
F. Chen, Y. Shao, M. Li, et al., Nat. Commun. 12 (2021) 3304. doi: 10.1038/s41467-021-23326-2
J.A. Joule, K. Mills, Heterocyclic Chemistry, 5th edn, Wiley, 2010.
G. Hollopeter, H.M. Jantzen, D. Vincent, et al., Nature 409 (2001) 202–207. doi: 10.1038/35051599
S.Z. Zard, Chem. Soc. Rev. 37 (2008) 1603–1618. doi: 10.1039/b613443m
T.M. Monos, R.C. McAtee, C.R.J. Stephenson, Science 361 (2018) 1369–1373. doi: 10.1126/science.aat2117
E.A. Noten, R.C. McAtee, C.R.J. Stephenson, Chem. Sci. 13 (2022) 6942–6949. doi: 10.1039/d2sc01228f
E.A. Noten, C.H. Ng, R.M. Wolesensky, C.R.J. Stephenson, Nat. Chem. 16 (2024) 599–606. doi: 10.1038/s41557-023-01404-w
C. Hervieu, M.S. Kirillova, Y. Hu, et al., Nat. Chem. 16 (2024) 607–614. doi: 10.1038/s41557-023-01414-8
Z. Cao, Y. Sun, Y. Chen, C. Zhu, Angew. Chem. Int. Ed. 63 (2024) e202408177. doi: 10.1002/anie.202408177
N. Radhoff, A. Studer, Nat. Commun. 13 (2022) 3083. doi: 10.1038/s41467-022-30817-3
H. Zhang, M. Wang, X. Wu, C. Zhu, Angew. Chem. Int. Ed. 60 (2021) 3714–3719. doi: 10.1002/anie.202013089
J. Yu, H. Zhang, X. Wu, C. Zhu, CCS Chem. 4 (2022) 1190–1198. doi: 10.31635/ccschem.021.202100944
Independent Commodity Intelligence Services. (2022).
R. Matsubara, A.C. Gutierrez, T.F. Jamison, J. Am. Chem. Soc. 133 (2011) 19020–19023. doi: 10.1021/ja209235d
J. Smidt, W. Hafner, R. Jira, et al., Angew. Chem. Int. Ed. 1 (1962) 80–88. doi: 10.1002/anie.196200801
N. Chatani, T. Asaumi, T. Ikeda, et al., J. Am. Chem. Soc. 122 (2000) 12882–12883. doi: 10.1021/ja002561w
J. Yu, X. Zhang, X. Wu, et al., Chem 9 (2023) 472–482.
D.J. Babcock, A.J. Wolfram, J.L. Barney, et al., Chem. Sci. 15 (2024) 4031–4040. doi: 10.1039/d3sc06476j
A. Shiozuka, D. Wu, K. Kawashima, et al., ACS Catal. 14 (2024) 5972–5977. doi: 10.1021/acscatal.4c00523
X. Tao, S. Ni, L. Kong, Y. Wang, Y. Pan, Chem. Sci. 13 (2022) 1946–1950. doi: 10.1039/d1sc06760e
D.M. Whalley, H.A. Duong, M.F. Greaney, Chem. Eur. J. 25 (2019) 1927–1930. doi: 10.1002/chem.201805712
X. Wu, X. Zhang, X. Ji, G.J. Deng, H. Huang, Org. Lett. 25 (2023) 5162–5167. doi: 10.1021/acs.orglett.3c01933
R.Q. Jiao, Y.N. Ding, M. Li, et al., Org. Lett. 25 (2023) 6099–6104. doi: 10.1021/acs.orglett.3c01988
W.Y. Ma, M. Leone, E. Derat, et al., Angew. Chem. Int. Ed. 63 (2024) e202408154. doi: 10.1002/anie.202408154
S.W. Tian, Z.T. Luo, B.Q. Xiong, et al., Green Chem. 26 (2024) 6774–6778. doi: 10.1039/d4gc00186a
H. Zhou, D. Lunic, N. Sanosa, et al., Angew. Chem. Int. Ed. 64 (2025) e202418869. doi: 10.1002/anie.202418869
J.H. Zhang, H.J. Miao, H. Xin, et al., Chem. Commun. 60 (2024) 5334–5337. doi: 10.1039/d4cc01324g
K. Mondal, M. Baidya, Adv. Synth. Catal. 366 (2024) 148–153. doi: 10.1002/adsc.202301180
S.P. Zhang, J.X. Lan, M.L. Yang, et al., Org. Lett. 26 (2024) 9990–9995. doi: 10.1021/acs.orglett.4c03830
Z.L. Lei, Z.C. Ding, S.H. Li, et al., Chem. Commun. 60 (2024) 7614–7617. doi: 10.1039/d4cc02543a
K. Liu, L.C. Sui, Q. Jin, D.Y. Li, P.N. Liu, Org. Chem. Front. 4 (2017) 1606–1610. doi: 10.1039/C7QO00209B
X. Chen Q. Wang, Z. Zhang, et al., Org. Lett. 24 (2022) 4338–4343. doi: 10.1021/acs.orglett.2c01427
J.L. Wang, M.L. Liu, J.Y. Zou, W.H. Sun, X.Y. Liu, Org. Lett. 24 (2022) 309–313. doi: 10.1021/acs.orglett.1c03973
J. Liu, B. Zhang, J. Hu, et al., Eur. J. Org. Chem. 26 (2023) e202201378. doi: 10.1002/ejoc.202201378
C. He, K. Zhang, D.N. Wang, et al., Org. Lett. 24 (2022) 2767–2771. doi: 10.1021/acs.orglett.2c00875
B. Wang, Z. Hu, L. Huang, et al., Chem. Commun. 59 (2023) 7247–7250. doi: 10.1039/d3cc01994b
J. Lan, K. Lin, X. Zhang, T. Zhu, Green Chem. 24 (2022) 6138–6144. doi: 10.1039/d2gc00960a
X. Wang, J. Liu, Z. Yu, et al., Org. Lett. 20 (2018) 6516–6519. doi: 10.1021/acs.orglett.8b02840
I. Marek, Y. Minko, M. Pasco, et al., J. Am. Chem. Soc. 136 (2014) 2682–2694. doi: 10.1021/ja410424g
J. Feng, M. Holmes, M.J. Krische, Chem. Rev. 117 (2017) 12564–12580. doi: 10.1021/acs.chemrev.7b00385
C. Hervieu, M.S. Kirillova, T. Suárez, et al., Nat. Chem. 13 (2021) 327–334. doi: 10.1038/s41557-021-00668-4
F. Lovering, J. Bikker, C. Humblet, J. Med. Chem. 52 (2009) 6752–6756. doi: 10.1021/jm901241e
F. Lovering, MedChemCommun 4 (2013) 515–519. doi: 10.1039/c2md20347b
T. Ling, F. Rivas, Tetrahedron 43 (2016) 6729–6777.
R.L. Sheng, K. Okada, S. Sekiguchi, J. Org. Chem. 43 (1978) 441–447. doi: 10.1021/jo00397a014
A.C. Knipe, N. Sridhar, J. Chem. Soc., Chem. Commun. 1979 (1979) 791–792.
J. Huang, F. Liu, L.H. Zeng, et al., Nat. Commun. 13 (2022) 7081. doi: 10.1038/s41467-022-34836-y
Y. Hu, C. Hervieu, E. Merino, C. Nevado, Angew. Chem. Int. Ed. 63 (2024) e202319158. doi: 10.1002/anie.202319158
R. Abrams, J. Clayden, Angew. Chem. Int. Ed. 59 (2020) 11600–11606. doi: 10.1002/anie.202003632
E. Derat, G. Masson, A. Claraz, Angew. Chem. Int. Ed. 63 (2024) e202406017. doi: 10.1002/anie.202406017
C. Faderl, S. Budde, G. Kachkovskyi, D. Rackl, O. Reiser, J. Org. Chem. 83 (2018) 12192–12206. doi: 10.1021/acs.joc.8b01538
D.M. Whalley, H.A. Duong, M.F. Greaney, Chem. Commun. 56 (2020) 11493–11496. doi: 10.1039/d0cc05049k
D.M. Whalley, J. Seayad, M.F. Greaney, Angew. Chem. Int. Ed. 60 (2021) 22219–22223. doi: 10.1002/anie.202108240
H. Fu, T.K. Hyster, Acc. Chem. Res. 57 (2024) 1446–1457. doi: 10.1021/acs.accounts.4c00129
W. Harrison, X. Huang, H. Zhao, Acc. Chem. Res. 55 (2022) 1087–1096. doi: 10.1021/acs.accounts.1c00719
Q. Zhou, J. Jiang, N. Luo, J. Huang, Acta Chim. Sinica 83 (2025) 274–286. doi: 10.6023/a25010015
W. Fu, K. Murcek, J. Chen, et al., J. Am. Chem. Soc. 147 (2025) 12197–12205. doi: 10.1021/jacs.5c01179
W.A. Coetzee, Pharmacol Ther. 140 (2013) 167–175. doi: 10.1016/j.pharmthera.2013.06.007
Z. Shi, Y. Li, N. Li, et al., Angew. Chem. Int. Ed. 61 (2022) e202206058. doi: 10.1002/anie.202206058
J.C. Gonzalez-Gomez, N.P. Ramirez, T. Lana-Villarreal, P. Bonete, Org. Biomol. Chem. 15 (2017) 9680–9684. doi: 10.1039/C7OB02579C
A.R. Tripathy, G.S. Yedase, V.R. Yatham, RSC Adv. 11 (2021) 25207–25210. doi: 10.1039/d1ra04130d
D. Shen, L. Li, T. Ren, et al., J. Org. Chem. 89 (2024) 2691–2702. doi: 10.1021/acs.joc.3c02762
J. Li, Z. Liu, S. Wu, Y. Chen, Org. Lett. 21 (2019) 2077–2080. doi: 10.1021/acs.orglett.9b00353
Z.H. Xia, L. Dai, Z.H. Gao, S. Ye, Chem. Commun. 56 (2020) 1525–1528. doi: 10.1039/c9cc09272b
X. Chang, Q. Zhang, C. Guo, Org. Lett. 21 (2019) 10–13. doi: 10.1021/acs.orglett.8b03178
C.A. Lawson, A.P. Dominey, G.D. Williams, J.A. Murphy, Chem. Commun. 56 (2020) 11445–11448. doi: 10.1039/d0cc04666c

Scheme 4 N-centered radical addition to unsaturated double bonds followed by radical Smiles rearrangement.

Scheme 6 The radical Smiles rearrangement initiated by the addition of exogenous radicals to alkenyl sulfonamides.

Scheme 7 Photoinduced asymmetric radical sulfinyl-Smiles rearrangement to access all-C quaternary stereocenters.

Scheme 8 Several other types of intramolecular radical Smiles rearrangement reactions.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:



