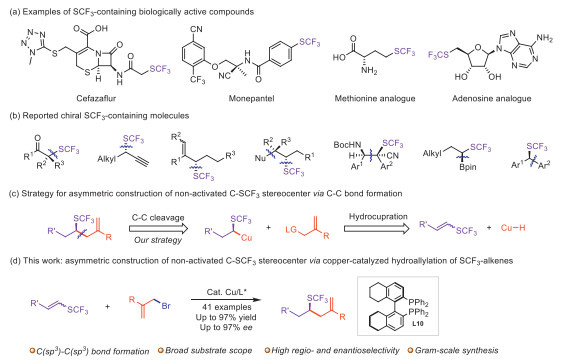
Scheme 1.
SCF3-containing biologically compounds and stoichiometric or catalytic asymmetric synthesis of stereocenters bearing the SCF3 group.
Asymmetric construction of non-activated C-SCF3 stereocenter via copper-catalyzed hydroallylation of SCF3-alkenes
Wei-Cheng Zhao , Yan He , Chen-Hui Jiang , Peng Liu , Qian Gao , Duo-Duo Hu , Xi-Sheng Wang

Chirality is an intrinsic characteristic of nature and pervades the biological systems [1-3]. Most organic molecules that comprise living organisms are chiral. Living systems have a strong chiral recognition ability, contributing to distinct physiological properties of different enantiomeric forms of biologically active molecules. Accordingly, the idea of incorporating chiral motifs in target compound has profoundly affected the modern pharmaceutical industry, and represents a major theme in drugs design and discovery [4-7]. Meanwhile, the adoption of fluorine or fluorinated motifs into drug design has been continuing to expand, facilitated by an ever-increasing understanding of ‘fluorine effects’ on physicochemical properties and efforts to efficient syntheses of novel fluorinated molecules [8-25]. However, most fluorine-containing molecules used in drugs so far are normally achiral compounds, with majority found as fluorinated or fluorine-containing functionalized arenes. The drug-like molecules installed with fluorinated stereocenters are still relatively rare, possibly due to the lack of highly efficient and enantioselective synthetic methods for preparation of single enantiomers to meet the requirement of FDA’s policy. The past several decades have witnessed the rapid development of novel synthetic methodologies for enantioselective construction of fluorinated or fluorine-containing functionalized stereocenters to make chiral fluorine-containing bioactive molecules [26-35]. However, most protocols provide only settlements to construct chiral centers at activated sites, such as benzylic, allylic, propargyl positions, or α-position of carbonyl groups and heteroatoms, and the rational construction of non-activated stereocenters featuring fluorine or fluorinated functional groups remains extremly challenging.
The trifluoromethylthio group (SCF3), characterized by its strong lipophilic and electron-withdrawing nature, could remarkedly improve the metabolic stability and transmembrane permeability of drug molecules [36-38], making it an attractive functional group for drug design and development. For instance, Cefazaflur is a parenteral cephalosporin antibiotic administered via injection [39-49], and Monepantel is specifically marketed for veterinary applications (Scheme 1a). While most reported methods focused on the synthesis of trifluoromethylthioated arenes [50], the asymmetric construction of C(sp3)-SCF3 stereocenter were less explored with known approaches predominantly centered on activated sites [51-81], including α-position adjacent to carbonyl groups or heteroatoms, benzylic, allylic and propargyl position (Scheme 1b). To resolve these issues in asymmetric synthesis of SCF3-containing molecules and deliver the construction of C(sp3)-SCF3 stereocenter at non-activated carbon centers in enantioconvergent synthesis of diverse trifluoromethylthiolated compounds, we reason a conceptually divergent route might address these challenges. Considering the presence of sulfur atom in a SCF3 group of SCF3-alkene could potentially controll the regioselectivity of CuH-catalyzed hydrocupration process, we posited that this in-situ generated alkylcopper intermediate could further react with a alkyl coupling partner for construction of trifluoromethylthiolated carbon center at a non-activated site (Scheme 1c). Notably, employing chiral ligated copper catalyst might contribute to a ‘chiral pocket’ for subsequent C(sp3)-C(sp3) coupling [82-85], constructing C-SCF3 stereocenter at non-activated site via copper-catalyzed three-component hydrofunctionalization of SCF3-alkenes
Taking this hypothesis driven design into practice (Scheme 1d), we report copper-catalyzed asymmetric hydroallylation of 1-trifluoromethylthioalkenes for enantioselective synthesis of trifluoromethylthiolated carbon center at non-activated site. Excellent regioselective and enantioselective control, good functional group tolerance, and mild reaction conditions are demonstrated in this protocol, providing facile syntheses of optically pure allyl SCF3-containing compounds. Given that trifluoromethylthiolated chiral centers could serve as key structural motifs, this method offers a convenient approach to SCF3-containing drug-like molecules and agrochemicals.
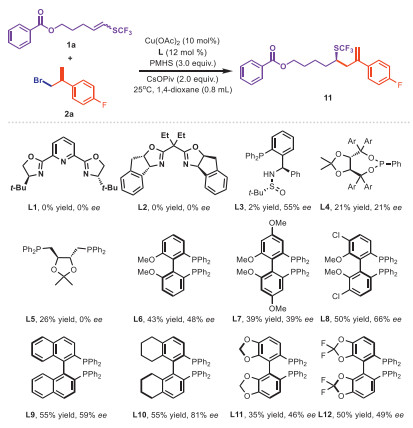
We commenced our study by using trifluoromethylthiolated alkene 1a and exo-alkene 2a as starting materials, with PMHS as the hydrogen source, CsOPiv as additive, and Cu(OAc)2 (10 mol%) as the catalyst in 1, 4-dioxane at 25 ℃ (Fig. 1). Given the critical role of chiral ligands in controlling enantioselectivity and catalytic activity, we first optimized various types of chiral ligand, including nitrogen-based ligands, N, P-based ligands, mono and disphosphine (L1-L6) where we observed relatively better reactivity when using biphenyl disphosphine ligands (L6), giving moderate yield and enantioselectivity. Further optimization of chiral biphenyl disphosphine ligands indicated that H8-BINAP (L10) could provide expected product in 55% yield with 81% ee. Lowering reaction temperature offered improved enantioselectivity with comparable yield (Table 1, entry 1). Considering the low freezing point of 1, 4-dioxane, we sought to test alternative solvents. Screening revealed that aromatic and ether solvents performed well, with diethyl ether yielding the product with 87% ee at −10 ℃ (Table 1, entry 3). Continuing optimization of other parameters showed that using CuCN could achieve higher yields and ee values and lowering the temperature furnish the product in higher enantioselectivity but decreased yield. To address this issue, extending the reaction time emerged as an effective strategy. Indeed, we conducted the reaction with CuCN at −20 ℃ for 72 h to access the expected product in a yield of 85% and an ee values of 92% (Table 1, entry 7) while other additives or hydrogen sources did not lead to further improvements (Table 1, entries 8–11).
 DownLoad:
CSV
DownLoad:
CSV
 |
|||||
| Entry | Cu cat. | Temp (℃) | Solvent | Yield (%)b | ee (%)c |
| 1 | Cu(OAc)2 | 10 | 1, 4-Dioxane | 45 | 82 |
| 2 | Cu(OAc)2 | −10 | Benzene | 45 | 79 |
| 3 | Cu(OAc)2 | −10 | Ether | 49 | 87 |
| 4 | Cu(OAc)2 | −10 | Diglyme | 27 | 84 |
| 5 | CuBr2 | −10 | Ether | 70 | 89 |
| 6 | CuCN | −10 | Ether | 72 | 89 |
| 7d | CuCN | −20 | Ether | 85 | 92 |
| 8d | CuCN | −30 | Ether | 75 | 92 |
| 9d,e | CuCN | −20 | Ether | 0 | – |
| 10d,f | CuCN | −20 | Ether | 45 | 79 |
| 11d,g | CuCN | −20 | Ether | 84 | 92 |
| a Reaction conditions: 1a (0.1 mmol, 1.0 equiv.), a 2a (0.2 mmol, 2.0 equiv.), [Cu] (10 mol%), Ligand (12 mol%), PMHS (3.0 equiv.), CsOPiv (2.0 equiv.), 1, 4-dioxane (0.8 mL), Ar. b Yield was determined by 19F NMR spectroscopy using PhCF 3 as an internal standard; numbers in parentheses were yields of isolated products. c The ee values were determined by HPLC on a chiral stationary phase. d The reaction was carried out for 72 h. e LiOPiv was used as the additive. f Cs2CO3 was used as the additive. g Trimethoxysilane was used as the hydrogen source. | |||||
With the optimized conditions in hand, we sought to investigate substrate generality of our protocol (Scheme 2). First, a series of aryl-substituted allylic bromides were found to be well compatible with this catalytic system. When the aryl para-position was substituted with bulky groups such as phenyl (4, 5), cyclohexyl (7), and tert‑butyl (8), the reactions maintained at least 90% ee values, with product 5 having an impressive 97% ee. It is worth noting that the gram-scale synthesis under standard conditions successfully produced the desired product without apparent loss of enantioselectivity, thereby demonstrating the practicality of this method. The reaction was unaffected by electron-donating (9) or electron-withdrawing groups (10). Similarly, when the aryl meta-position was substituted with electron-donating (11, 12) or electron-withdrawing groups (13, 14), high yields and excellent enantioselectivities were showcased. Additionally, polycyclic aryl group (15, 16) did not interfere with the reaction. Having explored aryl substituents, we turned our attention to alkyl chain-substituted allylic bromides. Satisfyingly, both small (17, 18) and bulky alkyl substituents (19, 20) were well tolerated by the reaction system.
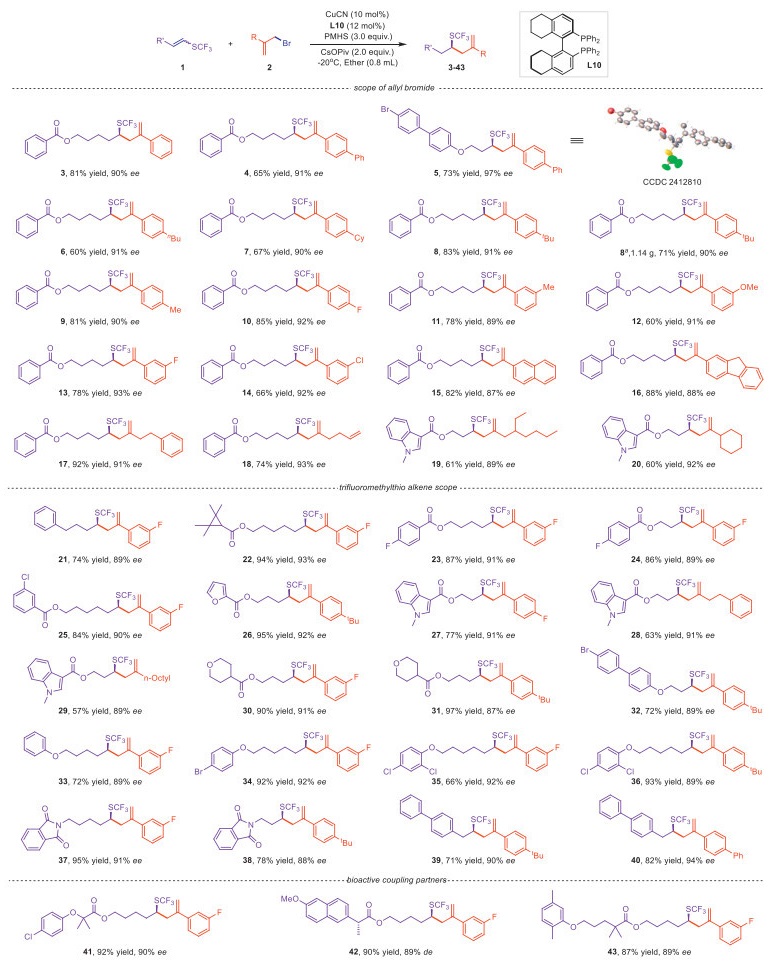
Next, we explored the functional group tolerance of 1-trifluoromethylthiolated alkenes. As shown in Scheme 2, various types of 1-trifluoromethylthiolated alkenes underwent hydroallylation smoothly, yielding the desired products (21–40) with excellent enantioselectivities, including aliphatic chain (21), stained ring system (22), substituted arenes (23–25) and alkyl chains with ether (36) and amide (37, 38) functionalities. Notably, the reactions proceeded smoothly regardless of the alkyl chain length (23, 24 and 37, 38) and the presence of heteroaromatic groups (26–29), or halogen substituents (32, 34, 35, 36) indicated excellent compatibility. Of particular importance is that 1-trifluoromethylthioalkenes bearing aryl conjugated systems exhibited exceptional regioselectivity, furnishing products in high yields and excellent enantioselectivities without detectable isomers (39, 40). This phenomenon may be attributed to the electron-donating effect of the sulfur atom, in favor of Cu-H insertion positioning copper adjacent to trifluoromethylthiolated carbon.
Encouraged by these exciting results, we proceeded to perform late-stage asymmetric modifications on biologically active molecules, such as clofibric acid (41), naproxen (42), and gemfibrozil (Lopid, 43 ). All modifications resulted in excellent yields and good enantioselectivities, highlighting the substantial potential of this method to efficiently construct chiral trifluoromethylthiolated derivatives of pharmaceuticals or drug candidates.
To gain deeper insights into the reaction mechanism of this system, we subsequently carried out a series of mechanistic experiments. Firstly, using Ph2SiD2 for asymmetric catalysis successfully synthesized deuterium-labeled chiral product 39′ (Scheme 3a). The result indicates that the copper hydride species A is formed from hydrosilane, ultimately transferring hydride to 1-SCF3 substituted alkene to form an alkyl copper intermediate. Next, we investigated the hydroallylation step (the transition from D and 2 to A and product in Scheme 3e). Under non-diastereoselective conditions, we tested regioisomeric allylic bromides 3‑bromo‑but-1-ene (2c) and (E)-1-bromobut-2-ene (2d) using the Xantphos ligand (Scheme 3b). The branched allylic bromide 2c reacted with 1a to yield linear products 44 and 44′ in 74% yield, along with 4% of the branched product 45.
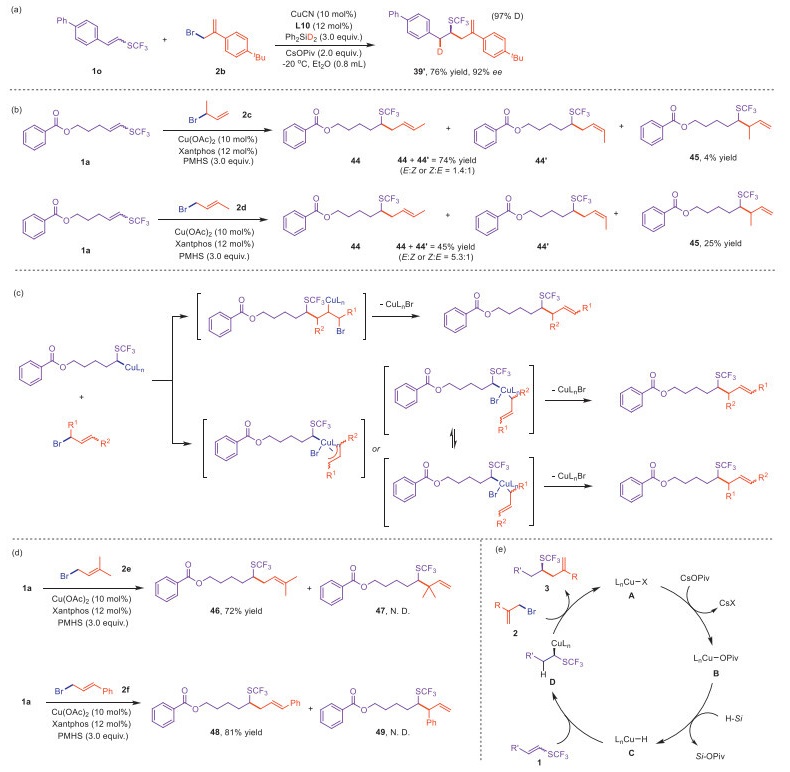
Products 44 and 44′ could be generated either through an SN2′-type addition-elimination mechanism or via the formation of π-allyl or π-en-σ-yl copper intermediates, whereas 45 can only be produced through the latter pathway. On the other hand, the linear allylic bromide 2d reacted with 1a to yield linear products 44 and 44′ in 45% yield, which can only be formed via π-allyl or π-en-σ-yl copper intermediates. Additionally, rapid E: Z (syn/anti) isomerization resulted in the formation of Z-isomers. Although the mechanism for the 25% branched product 45 remains unclear, it is evident that the reaction primarily proceeds through the formation of π-allyl or π-en-σ-yl copper intermediates (Scheme 3c). Furthermore, when steric hindrance increased, such as allylic bromides 2e and 2f, the reaction exclusively proceeded through the formation of π-allyl or π-en-σ-yl copper intermediates, followed by reductive elimination at more accessible spatial positions, yielding the corresponding linear hydroallylated products 46 and 48 (Scheme 3d). Notably, the choice of allylic substrate has a significant impact on the reaction efficiency. When the leaving group is a carbonate, acetate, or phosphate, the hydroallylation product is not observed at all. Allylic chlorides can yield the desired product but with lower yields compared to bromides. In fact, due to high reactivity of allylic bromide and β-elimination followed by Cu-H inserstion to allylic bromide side reactivity, allylic bromides are rarely used in known hydroallylation reactions. Benefited from strong electron-withdrawing effect of SCF3 on alkene components, the nucleophilic copper-hydride is prone to inserting to the alkenes that are more electron deficient, precluding unproductive insertion to allylic bromide, contributing to the excellent regioselectivity of Cu–H insertion into substrate 1a in followed by the participation of allylic bromide 2a to form π-allyl or π-en-σ-yl copper species. Given that allylic bromides are more reactive in forming π-allyl or π-en-σ-yl copper species compared to chlorides and the unproductive metal hydride insersion would be circumvented, higher reactivities with allylic bromide could also correlate to these the mechanistic studies, indicating that the formation of π-allyl or π-en-σ-yl copper species is the predominant pathway (Scheme 3c).
Based on the collective experimental results, we propose a possible reaction mechanism as shown in Scheme 3e. Initially, the in situ formed ligated copper complex LnCu-X (A) undergoes salt metathesis with the base CsOPiv to form the LnCu-OPiv species (B). Next, copper hydride C was furnished by σ-bond metathesis with the hydrosilane. Under the influence of the strong electron-withdrawing nature of the SCF3 group, the double bond of 1-trifluoromethylthioalkene 1 inserts into the Cu–H bond with high regio- and enantioselectivity, yielding the α-SCF3 alkylcopper intermediate D, followed by coupling with allylic bromide 2 via a possible π-allyl or π-en-σ-yl copper intermediate, to give homoallylic trifluoromethylthiolated products and ligated copper.
In conclusion, we have successfully developed an asymmetric construction of homoallylic trifluoromethylthiolated alkanes featuring C-SCF3 stereocenter at non-activated site via copper-catalyzed hydroallylation of SCF3-substituted alkenes. The asymmetric Cu catalysis effectively controls the chirality at the α-position adjacent to the SCF3 group, without the proximal directing heteroatoms. Comprehensive experimental studies also indicate that the reaction involves with the formation of π-allyl or π-en-σ-yl copper intermediates for subsequent coupling process. This transformation provides a robust and efficient method for synthesizing SCF3-containing chiral molecules with potential for pharmaceutical synthesis.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Wei-Cheng Zhao: Writing – original draft, Validation, Methodology, Investigation, Data curation. Yan He: Writing – original draft, Validation. Chen-Hui Jiang: Validation. Peng Liu: Validation. Qian Gao: Validation. Duo-Duo Hu: Writing – review & editing, Writing – original draft, Supervision, Methodology. Xi-Sheng Wang: Writing – review & editing, Writing – original draft, Validation, Supervision, Resources, Investigation, Funding acquisition, Data curation.
We gratefully acknowledge the National Key R & D Program of China (No. 2021YFF0701700) and the National Natural Science Foundation of China (Nos. 22271264 and 21971228) for financial support.
Supplementary material associated with this article can be found, in the online version, at doi:
J. Patocka, A. Dvorak, J. Appl. Med. 2 (2004) 95–100. doi: 10.32725/jab.2004.011
L.A. Nguyen, H. He, C. Pham-Huy, Int. J. Biomed. Sci. 2 (2006) 85–100. doi: 10.1002/mrdd.20103
N. Chhabra, M.L. Aseri, D. Padmanabhan, Int. J. Appl. Basic Med. Res. 3 (2013) 16–18. doi: 10.4103/2229-516X.112233
M. Eichelbaum, A.S. Gross, Adv. Drug Res. 28 (1996) 1–64.
S.W. Smith, Toxicol. Sci. 110 (2009) 4–30. doi: 10.1093/toxsci/kfp097
N. Chhabra, M.L. Aseri, D. Padmanabhan, Int. J. Appl. Basic Med. Res. 3 (2013) 16–18. doi: 10.4103/2229-516X.112233
H. Alkadi, R. Jbeily, Infect. Disord. Drug Targets 18 (2018) 88–95. doi: 10.2174/1871526517666170329123845
K. Müller, C. Faeh, F. Diederich, Science 317 (2007) 1881–1886. doi: 10.1126/science.1131943
S. Purser, P.R. Moore, S. Swallowb, V. Gouverneur, Chem. Soc. Rev. 37 (2008) 320–330. doi: 10.1039/B610213C
J. Wang, M. Sánchez-Roselló, J.L. Aceña, et al., Chem. Rev. 114 (2014) 2432–2506. doi: 10.1021/cr4002879
E.P. Gillis, K.J. Eastman, M.D. Hill, D.J. Donnelly, N.A. Meanwell, J. Med. Chem. 58 (2015) 8315–8359. doi: 10.1021/acs.jmedchem.5b00258
N.A. Meanwell, J. Med. Chem. 61 (2018) 5822–5880. doi: 10.1021/acs.jmedchem.7b01788
H. Mei, J. Han, S. Fustero, et al., Chem. Eur J. 25 (2019) 11797–11819. doi: 10.1002/chem.201901840
M. Inoue, Y. Sumii, N. Shibata, ACS Omega 5 (2020) 10633–10640. doi: 10.1021/acsomega.0c00830
B.M. Johnson, Y.Z. Shu, X. Zhuo, N.A. Meanwell, J. Med. Chem. 63 (2020) 6315–6386. doi: 10.1021/acs.jmedchem.9b01877
Y. Yu, A. Liu, G. Dhawan, et al., Chin. Chem. Lett. 32 (2021) 3342–3354. doi: 10.1016/j.cclet.2021.05.042
J. Li, W. Xi, S. Liu, et al., Chin. Chem. Lett. 33 (2022) 3007–3011. doi: 10.1016/j.cclet.2021.12.002
N. Meng, Y. Lv, X. Zhao, W. Wei, Chin. Chem. Lett. 32 (2021) 258–262. doi: 10.1016/j.cclet.2020.11.034
R. Wang, J. Xu, J.X. Li, et al., Chin. Chem. Lett. 34 (2023) 108490. doi: 10.1016/j.cclet.2023.108490
D.S. Deng, S.Q. Tang, Y.T. Yuan, et al., Chin. Chem. Lett. 35 (2024) 109417.
L. Yang, Y. Li, M. Jiang, et al., Chin. Chem. Lett. 35 (2024) 109512. doi: 10.1016/j.cclet.2024.109512
C.C. Wu, B.L. Wang, J.B. Liu, et al., Chin. Chem. Lett. 28 (2017) 1248–1251.
X.J. Song, Y. Shao, X.G. Dong, Chin. Chem. Lett. 22 (2011) 1036–1038. doi: 10.1016/j.cclet.2011.05.012
X. Chen, Z. Zhu, S. Liu, Y.H. Chen, X. Shen, Chin. Chem. Lett. 33 (2022) 2391–2396. doi: 10.1016/j.cclet.2021.10.083
H. Wang, Y. Xie, Y. Zhou, N. Cen, W. Chen, Chin. Chem. Lett. 33 (2022) 221–224.
T.D. Beeson, D.W.C. MacMillan, J. Am. Chem. Soc. 127 (2005) 8826–8828. doi: 10.1021/ja051805f
M. Marigo, D. Fielenbach, A. Braunton, A. Kjærsgaard, K.A. Jørgensen, Angew. Chem. Int. Ed. 44 (2005) 3703–3706. doi: 10.1002/anie.200500395
D. Cahard, X. Xu, S. Couve-Bonnaire, X. Pannecoucke, Chem. Soc. Rev. 39 (2010) 558–568. doi: 10.1039/B909566G
S. Lectard, Y. Hamashima, M. Sodeoka, Adv. Synth. Catal. 352 (2010) 2708–2732. doi: 10.1002/adsc.201000624
C.Y. Huang, A.G. Doyle, J. Am. Chem. Soc. 134 (2012) 9541–9544. doi: 10.1021/ja3013825
R.J. Phipps, F.D. Toste, J. Am. Chem. Soc. 135 (2013) 1268–1271. doi: 10.1021/ja311798q
N.A. Cochrane, H. Nguyen, M.R. Gagne, J. Am. Chem. Soc. 135 (2013) 628–631. doi: 10.1021/ja3116795
X. Yang, R.J. Phipps, F.D. Toste, J. Am. Chem. Soc. 136 (2014) 5225–5228. doi: 10.1021/ja500882x
S.Y. Lee, S. Neufeind, G.C. Fu, J. Am. Chem. Soc. 136 (2014) 8899–8902. doi: 10.1021/ja5044209
X. Jiang, M. Gandelman, J. Am. Chem. Soc. 137 (2015) 2542–2547. doi: 10.1021/jacs.5b00473
A. Leo, C. Hansch, D. Elkins, Chem. Rev. 71 (1971) 525–616. doi: 10.1021/cr60274a001
C. Hansch, S.H. Unger, A.B. Forsythe, J. Med. Chem. 16 (1973) 1217–1222. doi: 10.1021/jm00269a004
C. Hansch, A. Leo, R.W. Taft, Chem. Rev. 91 (1991) 165–195. doi: 10.1021/cr00002a004
G.W. Counts, D. Gregory, D. Zeleznik, M. Turck, Antimicrob. Agents Chemother. 11 (1977) 708–711. doi: 10.1128/AAC.11.4.708
N. Aswapokee, H.C. Neu, Antimicrob. Agents Chemother. 15 (1979) 444–446. doi: 10.1128/AAC.15.3.444
N.J.W. Straathof, B.J.P. Tegelbeckers, V, Hessel, X. Wang, T. Noël, Chem. Sci. 5 (2014) 4768–4773. doi: 10.1039/C4SC01982B
K. Jouvin, C. Matheis, L.J. Goossen, Chem. Eur. J. 21 (2015) 14324–14327. doi: 10.1002/chem.201502914
S. Alazet, T. Billard, Synlett 26 (2015) 76–78. doi: 10.1055/s-0034-1380555
X. -H. Xu, K. Matsuzaki, N. Shibata, Chem. Rev. 115 (2015) 731–764. doi: 10.1021/cr500193b
S. Barata-Vallejo, S. Bonesi, A. Postigo, Org. Biomol. Chem. 14 (2016) 7150–7182.
M. Zhao, X. Zhao, P. Zheng, Y. Tian, J. Fluorine Chem. 194 (2017) 73–79. doi: 10.1016/j.jfluchem.2017.01.007
A. -L. Barthelemy, E. Magnier, G. Dagousset, Synthesis (Mass) 50 (2018) 4765–4776. doi: 10.1055/s-0037-1611278
C. Ghiazza, T. Billard, A. Tlili, Chem. Eur. J. 25 (2019) 6482–6495. doi: 10.1002/chem.201806234
Y. Huang, M. Zhang, Q. Lin, Z. Weng, Synlett. 32 (2021) 109–118. doi: 10.1055/s-0040-1707211
L. Liao, X. Zhao, Acc. Chem. Res. 55 (2022) 2439–2453. doi: 10.1021/acs.accounts.2c00201
X. Wang, T. Yang, X. Cheng, Q. Shen, Angew. Chem. Int. Ed. 52 (2013) 12860–12864. doi: 10.1002/anie.201305075
T. Bootwicha, X. Liu, R. Pluta, I. Atodiresei, M. Rueping, Angew. Chem. Int. Ed. 52 (2013) 12856–12859. doi: 10.1002/anie.201304957
M. Rueping, X. Liu, T. Bootwicha, R. Pluta, C. Merkens, Chem. Commun. 50 (2014) 2508–2511. doi: 10.1039/c3cc49877h
X.L. Zhu, J.H. Xu, D.J. Cheng, et al., Org. Lett. 16 (2014) 2192–2195. doi: 10.1021/ol5006888
Q.H. Deng, C. Rettenmeier, H. Wadepohl, L.H. Gade, Chem. Eur. J. 20 (2014) 93–97. doi: 10.1002/chem.201303641
B.L. Zhao, D.M. Du, Org. Lett. 19 (2017) 1036–1039. doi: 10.1021/acs.orglett.6b03846
M. Li, X.S. Xue, J.P. Cheng, ACS Catal. 7 (2017) 7977–7986. doi: 10.1021/acscatal.7b03007
H. Zhang, X. Leng, X. Wan, Q. Shen, Org. Chem. Front. 4 (2017) 1051–1057. doi: 10.1039/C7QO00042A
M.Y. Jin, J. Li, R. Huang, et al., Chem. Commun. 54 (2018) 4581–4584. doi: 10.1039/c8cc02097c
F. Gelat, T. Poisson, A.T. Biju, X. Pannecoucke, T. Besset, Eur. J. Org. Chem. 2018 (2018) 3693–3696. doi: 10.1002/ejoc.201800418
M.A. Hardy, H. Chachignon, D. Cahard, Asian J. Org. Chem. 8 (2019) 591–609. doi: 10.1002/ajoc.201900004
V. Capaccio, M. Sicignano, R.I. Rodriguez, G. Della Sala, J. Aleman, Org. Lett. 22 (2020) 219–223. doi: 10.1021/acs.orglett.9b04195
M. Sicignano, R.I. Rodriguez, V. Capaccio, et al., Org. Biomol. Chem. 18 (2020) 2914–2920. doi: 10.1039/d0ob00476f
A. Eitzinger, J.F. Briere, D. Cahard, M. Waser, Org. Biomol. Chem. 18 (2020) 405–408. doi: 10.1039/c9ob02666e
A. Eitzinger, J. Otevrel, V. Haider, et al., Adv. Synth. Catal. 363 (2021) 1955–1962. doi: 10.1002/adsc.202100029
X. Liu, R. An, X. Zhang, J. Luo, X. Zhao, Angew. Chem. Int. Ed. 55 (2016) 5846–5850. doi: 10.1002/anie.201601713
J. Luo, Y. Liu, X. Zhao, Org. Lett. 19 (2017) 3434–3437. doi: 10.1021/acs.orglett.7b01392
J. Luo, X. Liu, X. Zhao, Synlett 28 (2017) 397–401. doi: 10.1055/s-0036-1588926
X. Liu, Y. Lang, J. Ji, J. Luo, X. Zhao, J. Am. Chem. Soc. 140 (2018) 4782–4786. doi: 10.1021/jacs.8b01513
J. Luo, Q. Cao, X. Cao, X. Zhao, Nat. Commun. 9 (2018) 527. doi: 10.1038/s41467-018-02955-0
J. Xu, Y. Zhang, T. Qin, X. Zhao, Org. Lett. 20 (2018) 6384–6388. doi: 10.1021/acs.orglett.8b02672
T. Qin, Q. Jiang, J. Ji, J. Luo, X. Zhao, Org. Biomol. Chem. 17 (2019) 1763–1766. doi: 10.1039/c8ob02575d
Q. Jiang, X. Zhao, Chin. J. Org. Chem. 41 (2021) 443–454. doi: 10.6023/cjoc202010005
X. Gao, Y.L. Xiao, S. Zhang, J. Wu, X. Zhang, CCS Chem. 3 (2021) 1463–1471. doi: 10.31635/ccschem.020.202000353
Q. Wang, T. Nilsson, L. Eriksson, K.J. Szabo, Angew. Chem. Int. Ed. 61 (2022) e202210509. doi: 10.1002/anie.202210509
J. Zhou, Q.F. Zhang, W.H. Zhao, G.F. Jiang, Org. Biomol. Chem. 14 (2016) 6937–6941. doi: 10.1039/C6OB01176D
L. Xu, H. Wang, C. Zheng, G. Zhao, Adv. Synth. Catal. 359 (2017) 2942–2948. doi: 10.1002/adsc.201700321
Z. Zhang, Z. Sheng, W. Yu, et al., Nature Chem. 9 (2017) 970–976. doi: 10.1038/nchem.2789
H. Kondo, M. Maeno, K. Sasaki, et al., Org. Lett. 20 (2018) 7044–7048. doi: 10.1021/acs.orglett.8b02998
D.D. Hu, T.M. Nie, X. Xiao, et al., Angew. Chem. Int. Ed. 63 (2024) e202400308. doi: 10.1002/anie.202400308
Y. Kojima, Y. Nishii, K. Hirano, Angew. Chem. Int. Ed. 63 (2024) e202403337.
Y.M. Wang, S.L. Buchwald, J. Am. Chem. Soc. 138 (2016) 5024–5027. doi: 10.1021/jacs.6b02527
J.T. Han, W.J. Jang, N. Kim, J. Yun, J. Am. Chem. Soc. 138 (2016) 15146–15149. doi: 10.1021/jacs.6b11229
J. Lee, S. Torker, A.H. Hoveyda, Angew. Chem. Int. Ed. 56 (2017) 821–826. doi: 10.1002/anie.201611444
Y. Kojima, M. Miura, K. Hirano, ACS Catal. 11 (2021) 11663–11670. doi: 10.1021/acscatal.1c02947

Scheme 1 SCF3-containing biologically compounds and stoichiometric or catalytic asymmetric synthesis of stereocenters bearing the SCF3 group.

Scheme 2 Copper-catalyzed regio- and enantioselective hydroallylation of 1-trifluoromethylthioalkenes. Isolated yields were given. a Gram-scale synthesis.
Table 1. Copper-catalyzed hydroallylation of SCF3-alkenes: optimization of conditions.a
|
|
|||||
| Entry | Cu cat. | Temp (℃) | Solvent | Yield (%)b | ee (%)c |
| 1 | Cu(OAc)2 | 10 | 1, 4-Dioxane | 45 | 82 |
| 2 | Cu(OAc)2 | −10 | Benzene | 45 | 79 |
| 3 | Cu(OAc)2 | −10 | Ether | 49 | 87 |
| 4 | Cu(OAc)2 | −10 | Diglyme | 27 | 84 |
| 5 | CuBr2 | −10 | Ether | 70 | 89 |
| 6 | CuCN | −10 | Ether | 72 | 89 |
| 7d | CuCN | −20 | Ether | 85 | 92 |
| 8d | CuCN | −30 | Ether | 75 | 92 |
| 9d,e | CuCN | −20 | Ether | 0 | – |
| 10d,f | CuCN | −20 | Ether | 45 | 79 |
| 11d,g | CuCN | −20 | Ether | 84 | 92 |
| a Reaction conditions: 1a (0.1 mmol, 1.0 equiv.), a 2a (0.2 mmol, 2.0 equiv.), [Cu] (10 mol%), Ligand (12 mol%), PMHS (3.0 equiv.), CsOPiv (2.0 equiv.), 1, 4-dioxane (0.8 mL), Ar. b Yield was determined by 19F NMR spectroscopy using PhCF 3 as an internal standard; numbers in parentheses were yields of isolated products. c The ee values were determined by HPLC on a chiral stationary phase. d The reaction was carried out for 72 h. e LiOPiv was used as the additive. f Cs2CO3 was used as the additive. g Trimethoxysilane was used as the hydrogen source. | |||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

