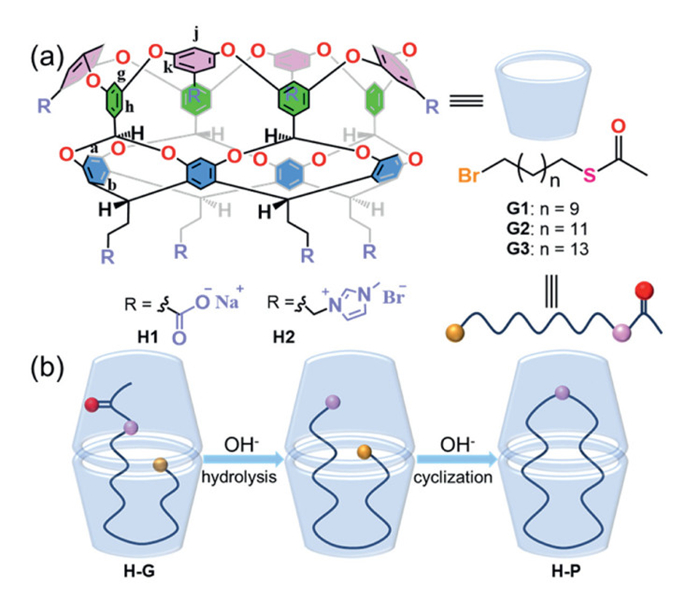
Scheme 1.
(a) The structures of the hosts (H1-H2) and guests (G1-G3). (b) The cascade hydrolysis-cyclization reactions of G1-G3 within the dimeric capsules.
Tailoring cascade hydrolysis and cyclization efficiency in confined spaces via spatial and electrostatic regulation
Qian Liu , Yi Shi , Kaiya Wang , Xiao-Yu Hu

Enzymes, as natural catalysts, exhibit remarkable efficiency and selectivity, playing indispensable roles in biological processes [1]. Inspired by these natural systems, chemists have pursued the design of artificial enzyme mimics, particularly supramolecular macrocycles with confined cavities that effectively emulate enzymatic active sites. A variety of artificial supramolecular macrocyclic hosts have been successfully synthesized, including but not limited to crown ethers [2], cyclodextrins [3], calixarenes [4], cucurbiturils [5], pillararenes [6,7], cavitands [8,9], and metal-organic cages [10]. These systems have demonstrated exceptional catalytic performance in various transformations, such as Diels-Alder reaction, monofunctional reaction, and terpene cyclization [11–18]. The catalytic mechanism relies on host-guest complexation, wherein the preorganization of substrate within the constrained cavity modulates reactivity. Such spatial confinement not only facilitates challenging transformations but also allows for the exploration of novel reaction pathways and the formation of unique products.
The synthesis of macrocycles from linear precursors remains a significant challenge due to the entropic penalty associated with bringing reactive chain termini into proximity, often leading to competing oligomerization. Traditional approaches rely on high-dilution conditions to suppress intermolecular side reactions. However, this approach requires excessive solvent usage and slow addition of reactants, rendering it impractical for large-scale applications. An alternative approach involves preorganizing the substrate within confined nanospaces, thereby positioning reactive termini for efficient intramolecular cyclization. Supramolecular macrocycles, particularly cavitands, have proven highly effective in facilitating such processes due to their unique encapsulation properties. Cavitands, a class of supramolecular host derived from resorcinarenes, were first introduced by Cram and co-workers in the early 1980s [19]. Unlike pillararenes or cucurbiturils, cavitands possess only one open end, allowing guest molecules to enter and egress while imparting them with distinctive properties [20,21]. For instance, long-chain guests bind to cavitands in a folded conformation, contrasting with the more symmetrical pillararenes and cucurbiturils, where guests thread through the cavities to form pseudortaxanes [22–24]. Cavitands serve as exceptional molecular containers and are commonly employed for analogous isolation [25], monofunctionalization [26], and selective chemical catalysis [27]. Particularly, cavitands exhibit unparalleled selectivity in intramolecular cyclization reactions owing to their unidirectional binding characteristics [28–30].
In this study, two water-soluble cavitands H1 and H2 with identical inner space but opposite charges were employed as the nanoreactors for the cascade hydrolysis and intramolecular cyclization of long-chain reactants (Scheme 1a, Figs. S1 and S2 in Supporting information) [31–33]. The eight peripheral carboxyl and methylimidazolyl groups endowed them with water solubility but generated opposite electrostatic potential (EP) fields, respectively. Previous studies demonstrated that H1 could promote the intramolecular cyclization of n-haloalkane-1-thiols [34]. In this work, we expand this system to cascade hydrolysis-cyclization processes involving n-bromoalkane-1-ethanethioates (n = 11, 13, 15). Accordingly, guests G1-G3 (Figs. S3-S5 in Supporting information), bearing terminal halide and ethanethioate groups with varying chain length, were synthesized [35]. In bulk aqueous solution under basic conditions, the ethanethioate moiety of G1-G3 hydrolyzes to generate a thiolate anion, which predominantly undergoes intermolecular reactions with halide termini, yielding linear oligomers (Fig. S6 in Supporting information). However, in the presence of the host molecules, the guests adopt a preorganized conformation driven by hydrophobic effect, bringing the reactive terminals into proximity and potentially altering the reaction pathway. This confinement overrides the inherent preference for oligomerization, redirecting the reaction pathway toward macrocyclic thioethers (Scheme 1b). The reaction of G1-G3 proceeded in two sequential steps, with the host cavity dictating product selectivity. Considering the presence of a negatively charged transition state in the aforementioned reaction, we hypothesized that, in addition to conformational preorganization, the EP field of the hosts also played a crucial role during the reaction. The positively charged H2 could exhibit superior reaction acceleration compared to H1 by stabilizing the negatively charged transition state. Our findings demonstrate that cavitands not only enforce substrate preorganization but also generate tailored EP fields to enhance reaction kinetics.
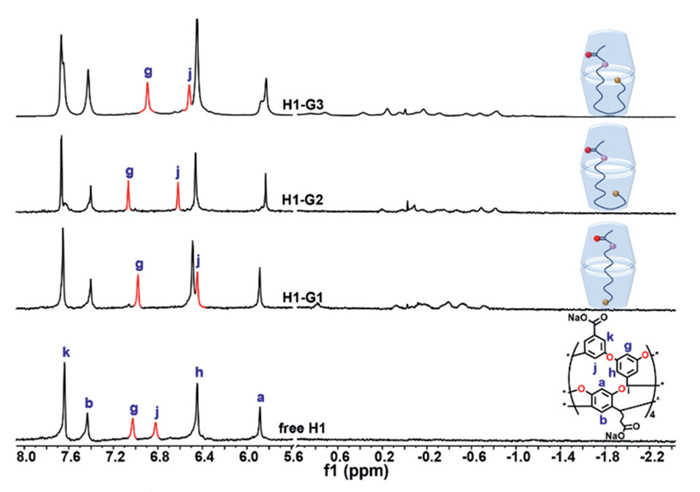
Previous studies have established that H1 could self-assemble into a dimeric capsule via hydrophobic interactions upon binding with long-chain guests [36]. Binding experiments between H1 and G1-G3 were performed using 1H NMR spectroscopy in D2O, revealing the formation of 2:1 complex. Upon binding, protons Hg and Hj of the host exhibited obvious chemical shifts and thus were selected as probes for subsequent reaction monitoring. Moreover, the other four aromatic protons showed minor chemical shift changes. Correspondingly, the guests exhibited substantial upfield shifts and anisotropic effects due to the diamagnetic, shielding walls of the host (Fig. 1). It has been reported that cavitands with same framework but opposite charges exhibited similar binding behaviors [34]. Therefore, H2, possessing an identical cavity as H1, demonstrated comparable binding behaviors (Fig. S8 in Supporting information). According to previous reports, a 15-membered long-chain guest, such as G1, bound to the host in a helical conformation with its two terminals anchored at opposing poles of the capsule. In contrast, a longer 19-membered guest, such as G3, adopted a folded conformation with both ends directed into one cavitand and the turn directed into its partner [37]. The conformation of G2 within the capsule was determined using 1H-1H COSY NMR (Figs. S9 and S10 in Supporting information). Two-dimensional NMR analysis confirmed that G2 bound to the host in a folded conformation. This specific folded conformation facilitated the proximity of the two terminal reactive groups, thereby accelerating the reaction.
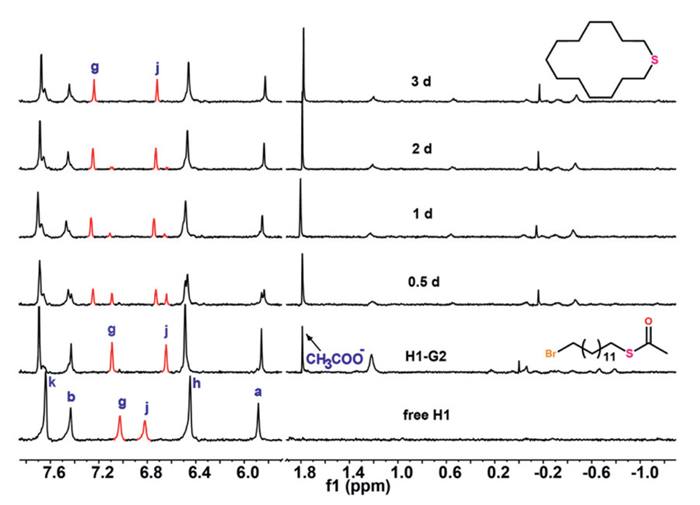
Subsequently, to initiate the reaction, 100 mmol/L NaOH was added to a pre-formed 2:1 host-guest complex, thereby facilitating the subsequent hydrolysis and cyclization process. The ester moiety of the guests underwent hydrolysis to form a thiolate group, which subsequently reacted with the terminal halide moiety on the same alkyl chain to generate a cyclic thioether. The two-step sequential reactions were tracked with 1H NMR. For the H1-G1 complex, as the reaction of G1 progressed, Hg shifted upfield and gradually overlapped with Hj, accompanied by a slight upfield shift of Hk and Ha and a downfield shift of Hb, reflecting the influence of the cyclization product on the host structure. Simultaneously, the number of peaks corresponding to bound G1 gradually decreased, indicating the generation of a symmetrical cyclic product. The complete reaction required approximately 20 days, likely due to the linear conformation where the two terminal residues were anchored at opposing poles of the capsule, thereby reducing their interaction probability (Fig. S11 in Supporting information). In contrast, the H1-G2 complex exhibited significantly faster kinetics, completing within 3 days. During this reaction, both Hg and Hj moved downfield, confirming the progression of cyclization (Fig. 2). Notably, the peak corresponding to hydrolyzed product (CH3COO−, 1.8 ppm) gradually emerged and was consistently observed throughout each reaction, providing clear evidence of ongoing hydrolysis processes within the system. The appropriate detection interval for G2 enabled us to monitor its transformation process. Concurrently, the number of peaks corresponding to G2 significantly decreased, strongly suggesting the formation of the cyclization product. Collectively, these observations confirm the occurrence of cascade reaction mechanisms. The rapid kinetics are attributed to G2's folded conformation, which brings reactive groups into closer proximity. Subsequently, the reaction of G3 was also monitored. As anticipated, G3 exhibited the fastest reaction rate, completing the process in less than 12 h. This is attributable to its longer chain length, which promotes the optimal preorganization of reactive termini within the capsule (Fig. S13 in Supporting information). The observed trend of G3 > G2 > G1 in reaction rates is directly correlated with the guests' propensity to adopt folded conformations that facilitate intramolecular ring closure.
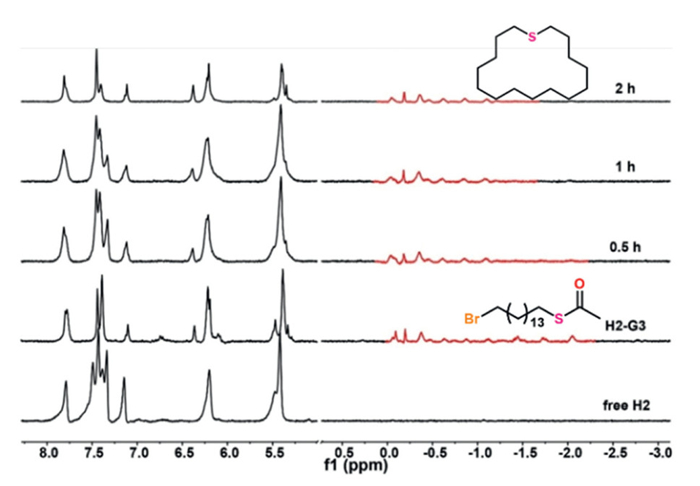
To elucidate the influence of EP fields on reaction kinetics, we conducted comparative studies using the positively charged host H2. The distribution of signal peaks in the aromatic region of H2 is more intricate than that of H1, making it challenging to accurately track the reaction process based solely on the chemical shift changes of Hg and Hj. Consequently, the reactions within the H2 capsule were monitored by observing the signal peak changes of the guests. The results showed that the intramolecular cyclization products were obtained with excellent selectivity, and the reaction times for all three reactions were significantly reduced in the presence of H2 compared to H1. Specifically, the reaction time of G1 was shortened 20-fold, completing in just one day (Fig. S14 in Supporting information). The reaction of G2 took three hours, representing a 24-fold reduction in reaction time (Fig. S15 in Supporting information). For G3, the reaction completed within two hours after initiation, which represents a 6-fold reduction in reaction time (Fig. 3). As the reaction progressed, the number of signal peaks corresponding to the guests decreased significantly, indicating the generation of symmetrical cyclic products. The products were subsequently extracted and characterized by 1H NMR and HR-ESI-MS (Figs. S17-S20 in Supporting information). This dramatic rate enhancement can be attributed to H2's positive EP field, which stabilizes the anionic transition state more effectively than H1's negative EP field. However, as the chain length increases progressively, the influence of the EP field on reaction time diminishes gradually, suggesting that optimal preorganization in longer chains can partially overcome electrostatic disadvantages (Table S2 in Supporting information). Additionally, competition experiments were conducted between the guests and their cyclic products to confirm the existence of catalytic cycles. The results revealed that the guests showed negligible competitive binding, indicating strong product inhibition (Fig. S21 in Supporting information).
We then performed density functional theory (DFT) calculations to gain a deeper mechanistic understanding of the EP field effects and guest conformational control on reaction kinetics. First, the molecular EP surfaces were calculated for the optimized structures of the hosts. The results revealed that the two hosts exhibited opposite charge distributions on their surfaces, with the charges being concentrated near the carboxyl and methylimidazole groups. This aligns with the observation that the host capsules provide opposing EP field environments for intramolecular cyclization reactions (Fig. 4a). Later, to further explore the binding conformations of the guests, the host-guest interaction energies of the complexes were calculated based on their optimized geometries. The optimal conformations of the host-guest complexes are shown in Fig. 4b. Compared to the optimal conformations, the alternative conformations displayed less favorable interaction energies (Fig. S22 in Supporting information). For G1, the two conformations exhibited minor differences, likely due to its short chain length, which enabled G1 sufficient flexibility to transition between the two conformations within the capsule. Conversely, for G2 and G3, the folded conformation demonstrated significantly lower interaction energies compared to their linear counterparts, indicating a preference for the folded conformation within the capsule. It is worth noting that G2 exhibited similar folded conformations in both H1 and H2, while G3 adopted markedly different conformations in the two hosts: Specifically, G3 adopted a more compact conformation in the negatively charged H1 compared to the positively charged H2. If G3 were to assume a similar conformation in H1 as in H2, its extended chain would increase the repulsion between the thioester group and the negatively charged cavity of H1, thereby destabilizing the complex. Moreover, all of the abovementioned optimal binding conformations were consistent with the 1H NMR results and facilitated the intramolecular cyclization reactions. Simultaneously, to clarify the phenomenon of product inhibition, the interaction energies of the complexes formed between the cyclic products and the capsules were calculated. The results revealed that these complexes exhibited comparable interaction energies to those of host-guest complexes, providing a theoretical explanation for the failure to establish an efficient catalytic cycle (Fig. S23 in Supporting information).

In summary, this study demonstrates that water-soluble cavitands form well-defined capsular assemblies capable of directing selective cascade hydrolysis-cyclization reactions through synergistic confinement effects and the influence of an EP field. The shorter guest G1 exhibited a linear conformation within the capsule, whereas the longer guests G2 and G3 adopted folded conformations. 1H NMR tracking experiments revealed that cascade hydrolysis and cyclization reactions occurred with distinct reaction rates. In confined spaces, longer chains were found to be more conducive to intramolecular cyclization. Additionally, the EP field significantly influenced the reaction rates, with the positively charged host H2 accelerating the reaction rates more efficiently by stabilizing the negatively charged transition state. These findings offer a strategic approach for designing enzyme-mimetic nanoreactors leveraging well-defined nanoconfinement. Future research could explore the application of these hosts in more challenging reactions, such as catalytic processes with low catalyst loadings or enantioselective transformations by introducing chiral functional groups to achieve excellent enantiomeric excess values.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Qian Liu: Writing – original draft, Validation, Methodology, Investigation, Formal analysis, Data curation. Yi Shi: Methodology, Investigation, Formal analysis, Data curation. Kaiya Wang: Writing – review & editing, Supervision, Project administration, Methodology, Investigation, Funding acquisition. Xiao-Yu Hu: Writing – review & editing, Supervision, Project administration, Investigation, Funding acquisition.
This research was funded by the National Natural Science Foundation of China (Nos. 22271154, M-0411), the China Postdoctoral Science Foundation (No. 2023M731658), the Science Fund for Distinguished Young Scholars of Jiangsu Province (No. BK20240078), the Innovation Support Program of Jiangsu Province (No. BZ2023055), and the High-Performance Computing Platform of Peking Uniersity. We also acknowledge the Center for Microscopy and Analysis at Nanjing University of Aeronautics and Astronautics for HR-MS measurements.
Supplementary material associated with this article can be found, in the online version, at doi:
B. Hauer, ACS Catal. 10 (2020) 8418–8427. doi: 10.1021/acscatal.0c01708
G.W. Gokel, W.M. Leevy, M.E. Weber, Chem. Rev. 104 (2004) 2723–2750.
Y.M. Zhang, Y.H. Liu, Y. Liu, Adv. Mater. 32 (2020) 1806158.
Y. Razuvayeva, R. Kashapov, L. Zakharova, Supramol. Chem. 32 (2020) 178–206. doi: 10.1080/10610278.2020.1725515
R.C. Mutihac, A.A. Bunaciu, H.J. Buschmann, et al., J. Incl. Phenom. Macrocycl. 98 (2020) 137–148. doi: 10.1007/s10847-020-01019-5
T. Ogoshi, S. Kanai, S. Fujinami, et al., J. Am. Chem. Soc. 130 (2008) 5022–5023. doi: 10.1021/ja711260m
K. Wang, J.H. Jordan, V. Krishnasamy, et al., Angew. Chem. Int. Ed. 60 (2020) 9205–9214.
S. Mosca, Y. Yu, J. Rebek Jr., Nat. Protoc. 11 (2016) 1371–1387. doi: 10.1038/nprot.2016.078
J. Murray, K. Kim, T. Ogoshi, et al., Chem. Soc. Rev. 46 (2017) 2479–2496.
H. Takezawa, K. Iizuka, M. Fujita, Angew. Chem. Int. Ed. 63 (2024) e202319140.
Q. Zhang, K. Tiefenbacher, Nat. Chem. 7 (2015) 197–202. doi: 10.1038/nchem.2181
K. Wang, J.H. Jordan, X.Y. Hu, et al., Angew. Chem. Int. Ed. 59 (2020) 13712–13721. doi: 10.1002/anie.202000045
M. Morimoto, S.M. Bierschenk, K.T. Xia, et al., Nat. Catal. 3 (2020) 969–984. doi: 10.1038/s41929-020-00528-3
K. Wang, X. Tian, J.H. Jordan, et al., Chin. Chem. Lett. 33 (2022) 89–96.
Q. Liu, M. Zuo, K. Wang, et al., Chem. Commun. 59 (2023) 13707–13710. doi: 10.1039/d3cc04040b
K. Wang, Y. Shen, P. Jeyakkumar, et al., Curr. Opin. Green Sustain. Chem. 41 (2023) 100823.
K. Wang, M. Zuo, T. Zhang, et al., Chin. Chem. Lett. 34 (2023) 107848.
C. Wang, L. Xu, Z. Jia, et al., Chin. Chem. Lett. 35 (2024) 109075.
J.R. Moran, S. Karbach, D.J. Cram, J. Am. Chem. Soc. 104 (1982) 5826–5828. doi: 10.1021/ja00385a064
K. Wang, Q. Liu, L. Zhou, et al., Chin. Chem. Lett. 34 (2023) 108559.
K. Wang, K. Yan, Q. Liu, et al., Molecules 29 (2024) 5854–5871. doi: 10.3390/molecules29245854
R. Eelkema, K. Maeda, B. Odell, et al., J. Am. Chem. Soc. 129 (2007) 12384–12385. doi: 10.1021/ja074997i
Y. Gao, X. Tian, X. Xiong, et al., Int. J. Biol. Macromol. 265 (2024) 130680–130689.
X.Q. Xu, X.Q. Wang, W. Wang, Chin. Chem. Lett. 34 (2023) 107665.
F.U. Rahman, J.M. Yang, Y.H. Wan, et al., Chem. Commun. 56 (2020) 6945–6948. doi: 10.1039/d0cc02778b
N.W. Wu, J. Rebek Jr., J. Am. Chem. Soc. 138 (2016) 7512–7515. doi: 10.1021/jacs.6b04278
I. Martin-Torres, G. Ogalla, J.M. Yang, et al., Angew. Chem. Int. Ed. 60 (2021) 9339–9344. doi: 10.1002/anie.202017035
Q. Shi, D. Masseroni, J. Rebek Jr., J. Am. Chem. Soc. 138 (2016) 10846–10848. doi: 10.1021/jacs.6b06950
J.M. Yang, Y. Yu, J. Rebek Jr., J. Am. Chem. Soc. 143 (2021) 2190–2193. doi: 10.1021/jacs.0c12302
W. Yao, K. Wang, Y.A. Ismaiel, et al., J. Phys. Chem. B 125 (2021) 9333–9340. doi: 10.1021/acs.jpcb.1c05238
M.B. Hillyer, C.L.D. Gibb, P. Sokkalingam, et al., Org. Lett. 18 (2016) 4048–4051. doi: 10.1021/acs.orglett.6b01903
J. Kiruthika, S. Srividhya, M. Arunachalam, Org. Lett. 22 (2020) 7831–7836. doi: 10.1021/acs.orglett.0c02710
H. Sun, S. Li, Q. Liu, et al., Chin. Chem. Lett. 36 (2025) 109999.
K. Wang, X. Cai, W. Yao, et al., J. Am. Chem. Soc. 141 (2019) 6740–6747. doi: 10.1021/jacs.9b02287
M. Zarska, F. Novotny, F. Havel, et al., Bioconjugate Chem. 27 (2016) 2558–2574. doi: 10.1021/acs.bioconjchem.6b00491
S. Liu, D.H. Russell, N.F. Zinnel, et al., J. Am. Chem. Soc. 135 (2013) 4314–4324. doi: 10.1021/ja310741q
J.W. Barnett, B.C. Gibb, H.S. Ashbaugh, J. Phys. Chem. B 120 (2016) 10394–10402. doi: 10.1021/acs.jpcb.6b06496

Scheme 1 (a) The structures of the hosts (H1-H2) and guests (G1-G3). (b) The cascade hydrolysis-cyclization reactions of G1-G3 within the dimeric capsules.

Figure 1 Partial 1H NMR spectra of H1⊃G1-G3 in D2O ([H1] = 1.0 mmol/L, [G] = 0.5 mmol/L).

Figure 2 Stacked 1H NMR spectra of the cyclization of G2 within the dimeric capsule formed by H1.

Figure 3 Stacked 1H NMR spectra of the cyclization of G3 within the dimeric capsule formed by H2.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
