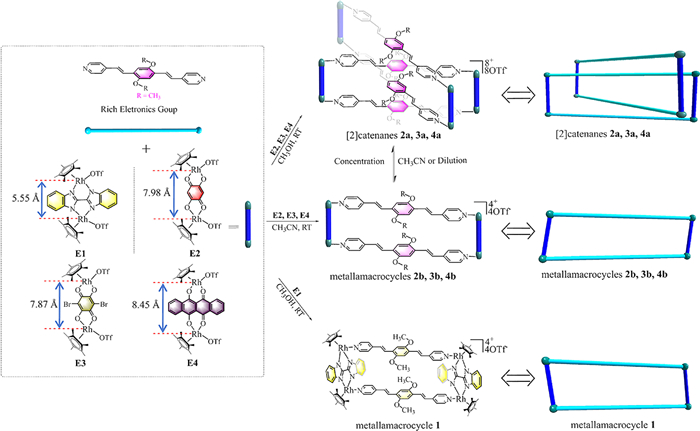
Scheme 1.
Synthesis of organometallic supramolecular compounds and dinuclear building blocks E1-E4 used in this study.
Controllable self-assembly and photothermal conversion of metalla[2]catenanes induced by synergistic effect of free radicals and stacking interactions
Ying Zhao , Yao He , Jian-Xin Yang , Wen-Jie Liu , Dan Tian , Francisco Aznarez , Le-Le Gong , Li-Long Dang , Lu-Fang Ma

The intriguing topological configuration and promising applications of interlocked complexes in molecular machines, photophysical chemistry, or biological simulation, have motivated supramolecular chemists to continuously explore their synthesis [1–9]. Thus, a variety of coordination supramolecular topologies have been isolated either intentionally or accidentally by following different synthetic approaches, including coordination-driven self-assembly or the use of metal-ion templates. While the structures of a number of metallamacrocycles [10–12], catenanes [13–16], Solomon links [17], Borromean rings [18], Russian dolls [19], metallacages [20–24], diverse knots [25–27] and other complicated structures [28,29] have been reported, a reduced amount of studies on their possible application in different fields have been carried out.
In previous works, the host-guest properties of some interlocked structures were investigated by Fujita et al. [30], Jin et al. [31] and Lang et al. [32], and others [33,34]. Thus, a molecular pentafoil knot and triply entwined [2]catenanes were synthesized by Leigh et al., which are suitable to accommodate halide anions in their central cavities through electrostatic and CH···X hydrogen bonding interactions [35]. Lang et al. reported the synthesis of cluster-mediated 3D catenanes by self-assembly of the cluster node [Tp*WS3Cu3Cl]+ and an organic linker (Z)-1,2-diphenyl-1,2-bis(4-(pyridin-4-yl)phenyl)ethene, thereby obtaining astonishing structures with unique cation-in-cation host-guest interactions [36]. Interestingly, the development of interlocked supramolecular compounds based on half-sandwich Rh/Ir fragments has recently experienced a remarkable blossoming [37]. A series of sophisticated topological structures including Solomon links [38], Borromean rings [39], [2–5]catenanes [40–43], molecular knots [44], 4-ravels [45], and others [46–48] were prepared in good yields and have been fully characterized owing to their outstanding solubility in a variety of organic solvent and crystallinity, which enables X-ray diffraction analysis. Studies have proven that due to the free radical effect of half-sandwich building blocks and the stacking effect of interlocked structures, suppressing radiative transitions and promoting non-radiative transitions, these compounds exhibit significant photothermal conversion performance. However, the roles in photothermal conversion performance played by different half-sandwich building blocks have not yet been clarified under the same accumulation effect. Therefore, synthesizing the similar topological structures based on the same ligand and different building blocks is conducive to exploring the role on photothermal conversion played by the half-sandwich building blocks, which is a major challenge as it is necessary to control metal-to-metal distance in half-sandwich building blocks at an appropriate level to ensure the accumulation effect.
Considering that π-π stacking interactions usually lead to the formation of complicated interlocked structures in the solid state, the new ligand (4,4′-((1E, 1′E)-(2,5-dimethoxy-1,4-phenylene) bis(ethene-2,1-diyl))dipyridine) (L1) has been synthesized. Self-assembly of L1 with four different building units (E1-E4), featuring different metal-to-metal distances, led to the formation of metallamacrocycles 1,2b-4b and [2]catenanes 2a-4a (Scheme 1). The structures of these compounds were confirmed by single crystal X-ray diffraction analysis, NMR spectroscopy and ESI-TOF-MS. Moreover, dynamic structural transformation between [2]catenanes and the corresponding metallamacrocycles could be achieved by concentration changes and polar solvent induced effect. In addition, near-infrared photothermal studies with the isolated compounds indicated the existence of a variety of photothermal responses in solution. Thus, while [2]catenane 3a showed an excellent photothermal conversion and its efficiency reached 40.4% at 1.5 W/cm2, the photothermal conversion efficiency of [2]catenanes 2a-4a was in the limit range of 13.3% to 40.4%. Recorded EPR results are in concordance with the measured photothermal conversion data, and suggest that a large number of free radicals produced by the building blocks improve the photothermal efficiency. This study provides a new method to synthesize valuable half-sandwich-based NIR photothermal conversion materials.
([Cp*2Rh2(BiBzlm)]OTf2) E1 was mixed with the ligand precursor L1 in the absence of light, and the obtained mixture was stirred at ambient temperature for 12 h. Then, the mixture was filtered and the filtrate was concentrated under reduced pressure. Thereafter, isopropyl ether was slowly diffused into the indicated concentrated solution, producing yellow crystals suitable for single crystal X-ray diffraction analysis (yield: 84.2% for metallamacrocycle 1; Scheme S1). The structure of metallamacrocycle 1 was also unambiguously confirmed by NMR spectroscopy and ESI-TOF-MS.
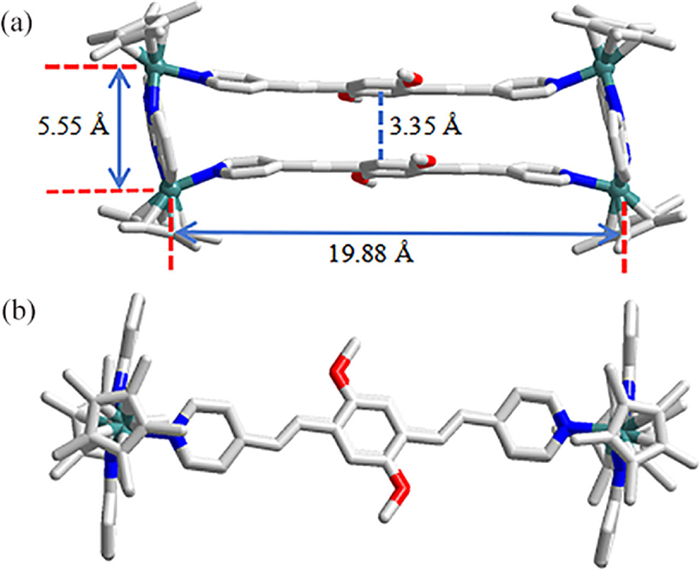
Single-crystal X-ray diffraction analysis of compound 1 indicated that it belongs to the P21/c space group (Figs. 1a and b). Note that two binuclear [Cp*2Rh2(BiBzlm)]2+ cations are coordinated by two dipyridyl ligands L1 in the molecular structure of compound 1, with short Rh−Rh nonbonding distance of 5.55 Å and long Rh−Rh nonbonding distance of 19.88 Å. Previous studies in the same field have shown that a suitable Rh-Rh nonbonding distance leads to the formation of interlocked structures [49]. Compound 1 in the solid state exhibits π-π stacking interaction, and the central distance of the conjugate plane between the two ligands L1 is 3.35 Å.
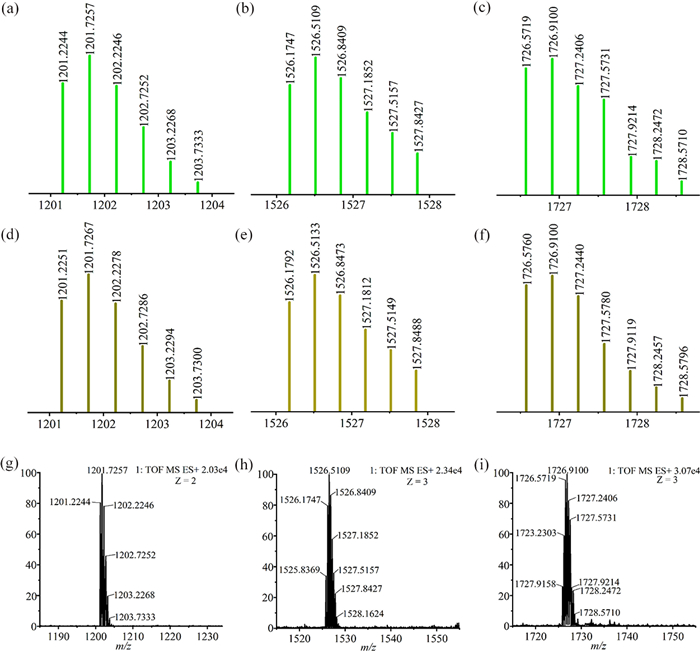
The NMR spectra of compound 1 in CD3OD confirmed the results obtained by single crystal X-ray diffraction analysis. The 1H NMR spectrum of 1 showed two singlets at 6.74 ppm and 3.70 ppm, which have been attributed to the protons of the phenyl and methoxy groups in ligand L1. Furthermore, a resonance corresponding to the protons of the Cp* group appeared at 1.90 ppm. While two doublets at 7.60 ppm and 6.91 ppm were assigned to the pyridinyl protons of L1, the two doublets at 7.29 ppm and 6.74 ppm were attributed to the protons at the olefinic bond of L1. Moreover, the signals corresponding to the protons of the 2,2′-bisbenzimidazole (BiBzlm) group appeared as two multiplets at 8.10–8.09 ppm and 7.57–7.55 ppm (Fig. S4 in Supporting information). The recorded 1H–1H COSY NMR spectrum confirmed the proton-assigned resonances (Fig. S5 in Supporting information). Additionally, the 1H DOSY NMR spectrum of 1 revealed a single diffusion coefficient, thereby indicating the presence of only one compound in solution (D = 3.05 × 10–6 cm2/s) (Fig. S6 in Supporting information). Furthermore, the ESI-TOF-MS showed a strong signal at m/z 1201.73, corresponding to [1–2OTf-]2+ (Figs. 3a and d), which is consistent with the calculated theoretical distributions.
Subsequent self-assembly studies were carried out by using building units E2, E3 and E4, possessing longer longitudinal sizes and different conjugate planes. Mixtures of L1 and E2, E3 or E4 were stirred for 12 h at ambient temperature and in the absence of light. Work-up of the resulting self-assemblies led to the isolation of crystalline compounds in high yields (87.8% for 2a, 88.2% for 3a, 88.6% for 4a) (Scheme 1). The structures of 2a, 3a and 4a were confirmed by single-crystal X-ray diffraction analysis, NMR spectroscopy and ESI-TOF-MS measurements.
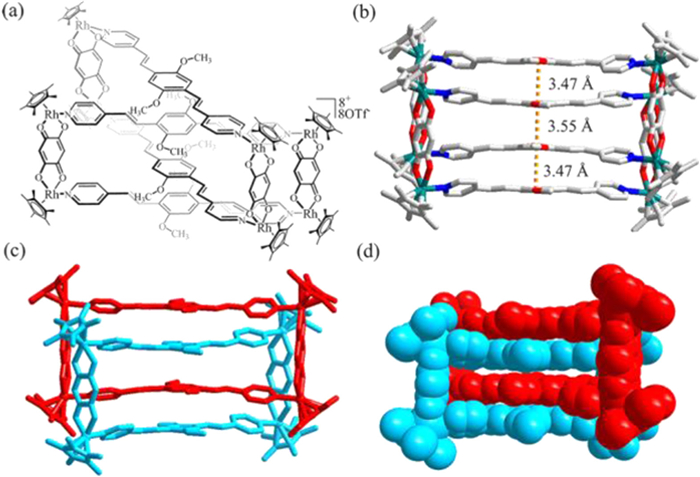
A detailed study of the structure of compound 2a in the solid state indicates that the architecture is stabilized by strong sandwich-type π-π stacking interactions between the central benzene rings of ligands L1 in two different metallamacrocycles 2b. Furthermore, the molecular structure of 2a exhibits the typical topology of a [2]catenane. The distances between the central phenyl planes of L1 ligands are 3.47, 3.55 and 3.47 Å (Fig. 3).
The proposed molecular structure of compound 2a has been confirmed by recorded NMR spectra. The 1H NMR spectrum of 2a in CD3OD showed one singlet at 6.72 ppm, corresponding to the protons of the phenyl groups in ligand L1. One singlet at 3.62 ppm was assigned to protons of the methoxy group of L1. The doublets at 7.45 and 6.92 ppm were attributed to the olefinic protons of L1. Besides, the resonances corresponding to the protons of the Cp* group appeared as two singlets at 1.79 and 1.69 ppm and the doublets at 8.11 and 7.49 ppm were assigned to the pyridiyl protons of L1. Furthermore, the protons of 2,5-dihydroxy-1,4-benzoquinon generated resonances in the NMR spectrum of 2a which appeared as two singlets at 6.11 and 5.83 ppm (Fig. S7 in Supporting information). All 1H NMR signal assignments were supported by recorded 1H–1H COSY NMR data (Fig. S8 in Supporting information). The 1H DOSY NMR spectrum of 2a showed a single diffusion coefficient (D = 2.44 × 10–6 cm2/s), indicating that only one assembled stoichiometry appeared in solution (Fig. S9 in Supporting information). The structure of the compound 2a was also supported by ESI-TOF-MS, exhibiting one strong peak at m/z 1526.51, which corresponds to the species [2a-3OTf-]3+ (Figs. 2b and e), in agreement with the calculated theoretical distribution.
Compound E2 is structurally very similar to E3. Self-assembly of E3 with L1 led to the formation of compound 3a, which has been characterized by single crystal X-ray diffraction analysis. The molecular structure of compound 3a in the solid state belongs to P-1 space group. The structure of compound 3a in the solid state is stabilized by strong sandwich-type π-π stacking interactions. The distances between the central phenyl planes are 3.47, 3.52 and 3.48 Å (Fig. 4).

The recorded NMR spectra of 3a also supported the results of the single crystal X-ray diffraction analysis. The 1H NMR spectrum of the compound 3a showed a triplet and a doublet at 8.22 and 7.58 ppm, which have been attributed to the pyridine protons of ligand L1. Furthermore, the peaks at 1.78 and 1.72 ppm have been assigned to the protons of Cp* groups (Fig. S13 in Supporting information). The 1H–1H COSY spectrum of compound 3a confirmed the proton-assigned resonances (Fig. S14 in Supporting information). In addition, 1H DOSY NMR spectrum of compound 3a (D = 2.76 × 10–6 cm2/s) was used to determine its single diffusion coefficient (Fig. S15 in Supporting information).
Single crystals of 4a suitable for a single crystal X-ray diffraction analysis were subsequently obtained. Similarly to 2a and 3a, compound 4a is also stabilized by strong sandwich-type π-π stacking interactions between the benzene moieties of L1 from two different metallamacrocycles (Fig. 5).

The 1H NMR spectrum of 4a in CD3OD showed a singlet and two doublets at 8.41, 7.37 and 7.32 ppm, which were assigned to the pyridinyl protons of the ligand L1. The signals corresponding to the protons of the Rh(Ⅲ) building block E4 appeared as a doublet at 8.81 ppm, and a multiplet at 8.03 ppm (Fig. S19 in Supporting information). Additionally, the resonances corresponding to the phenyl protons of L1 appeared at 6.58 and 5.66 ppm as two singlets. The 1H–1H COSY spectrum of [2]catenane 4a confirmed the assigned resonances (Fig. S20 in Supporting information) and the 1H DOSY NMR spectrum of compound 4a exhibited a diffusion coefficient of 2.30 × 10–6 cm2/s. This result implies the presence of only one compound in solution (Fig. S21 in Supporting information). Moreover, the ESI-TOF-MS spectrum of 4a showed a peak at m/z 1726.91, corresponding to [4a-3OTf-]3+ (Figs. 2c and f).
Various external stimuli, including the addition of suitable guest molecules, the use of solvents with enhanced polarity or a variation in the concentration can lead to the complete structural transformation of complicated structures into their corresponding metallamacrocycles [50–52]. Previous studies inspired us to explore the behavior of [2]catenanes 2a-4a in solution. Interestingly, a change in the concentration of [2]catenane 2a in methanol led to a significant transformation of its 1H NMR spectra. At low concentrations, the 1H NMR spectrum of 2a showed a set of simple proton signals, while two sets of coupling protons were found in the 1H–1H COSY NMR spectrum. Furthermore, doublets at 8.19 and 7.63 ppm were belonging to the pyridyl protons of L1, and doublets at 7.77, 7.23 ppm were attributed to the olefinic protons of L1. The singlet at 5.69 pm was observed in the 1H NMR spectrum, corresponding to resonances of the protons of the building block E2 (Fig. S10 in Supporting information). A signal generated by the Cp* protons was found at 1.68 ppm. This type of signal pattern indicated the formation of metallamacrocycle 2b Remarkably, the intensity of the initial signals in the 1H NMR spectrum decreased with an increase of the concentration, while new proton signals appeared, suggesting the structural transformation from the simple metallamacrocycle 2b into the more intricate compound 2a (Fig. S7 in Supporting information). Remarkably, when the concentration increased to 10 equiv., the conversion was completed by 66.7%. (Fig. 6). We also observed the transformation of metallamacrocycles 3b and 4b into the corresponding metalla[2]catenanes by changing their concentration (Figs. S26 and S27 in Supporting information). Next, structural changes induced by solvent effects were also observed (Fig. S25 in Supporting information).

Thus, a change in the ratio of methanol to CH3CN induced reversible supramolecular transformations between intricate structures and the corresponding metallamacrocycles [53]. Such structural interconversion might be due to a weakening of the π-π stacking interactions by modifying the concentration or due to the addition of CH3CN molecules. As expected, a dynamic structural transformation between [2]catenane 2a and metallamacrocycle 2b was found when CH3CN-d3 solvent was either added or removed. Gradual addition of CH3CN-d3 to a solution of mixture 2a + 2b in CD3OD generated the apparent transformation of [2]catenane 2a into metallamacrocycle 2b, as revealed by 1H NMR spectroscopy. Upon increasing the proportion of CH3CN-d3, the intensity of the 1H NMR signals generated by compound 2a decreased, while signals corresponding to compound 2b increased steadily. Recorded 1H NMR spectra indicated that the mixture of 2a + 2b were completely converted into 2b at a CD3OD to CH3CN-d3 ratio of 1:6 (Fig. S25 in Supporting information).
Recently, near-infrared photothermal conversion has been extensively used in diverse fields such as photothermal sensing, photodynamic therapy, and photocatalysis [54–60]. The general principle for synthesizing highly efficient near-infrared photothermal materials lies in enhancing near-infrared absorption while minimizing the radiation conversion process.
In the present work, the metallamacrocycle 1 and [2]catenanes 2a-4a are structures that exhibit near-infrared absorption ability. Note that both NIR absorption intensity and π-π stacking interactions play a key role in different processes of photothermal conversion. By controlling the molar ratio of metallamacrocycle 1 to [2]catenanes 2a-4a to 3:1:1:1, the same number of conjugate planes are subject to study. Next, the UV absorption of the compounds at 300–800 nm has been checked, The results indicated that compounds 1, 2a, 3a and 4a have absorbance values at 730 nm (1: A1 = 0.298; 2a: A2 = 0.069; 3a: A3 = 0.530; 4a: A4 = 0.290) (Fig. 7a). Subsequently, the same quantity of discrete structures 1, 2a, 3a and 4a was dissolved in 3 mL of CH3OH for UV–vis absorption experiments. The results revealed that compounds 1, 2a, 3a and 4a have absorbance values at 730 nm (1: A5 = 0.233; 2a: A6 = 0.747, 3a: A7 = 0.264; 4a: A8 = 0.761) (Fig. 7d). Therefore, we next studied the NIR photothermal conversion properties of metallamacrocycle 1, and [2]catenanes 2a, 3a and 4a, featuring half-sandwich fragments of different sizes. We conducted photothermal conversion experiments by using the compounds in solid state as a reference. Experimental results showed that compound 2a has significantly superior photothermal conversion effect than compounds 1, 3a and 4a in the solid state. Remarkably, at an irradiation intensity of 0.6 W/cm2, the temperature of compound 2a (in solid state) increased by 127.7 ℃ (from 24.3 ℃ to 150.0 ℃) (Fig. 7b). In addition, according to the absorption range of the solution, a laser lamp with a wavelength of 730 nm was selected to irradiate the sample, and the corresponding 0.5 mL solution was placed separately in a specific container. Photothermal tests were performed over solutions of the four compounds under the conditions of 1.5, 1.2, 0.9 and 0.6 W/cm2 (Figs. 7c and e), and the photothermal conversion efficiency was calculated by fitting (Figs. S28-S39 in Supporting information).

In this work, metallamacrocycle 1 generated a special near-infrared photothermal conversion curve during the heating progress (1.5 W/cm2, Fig. 7e), and temperature change of metallamacrocycle 1 was not obvious. The [2]catenanes 2a-4a exhibited different temperature change processes (1.5 W/cm2, Fig. 7e). Thus, [2]catenane 3a (from 25.8 ℃ to 50.3 ℃) and [2]catenane 4a (from 24.9 ℃ to 49.1 ℃) showed significant temperature changes (1.5 W/cm2), while a minor change in temperature was observed for [2]catenane 2a (from 25.3 ℃ to 39.3 ℃). After conducting rigorous and thorough fitting calculations for [2]catenane 2a (1.5 W/cm2, conversion efficiency: 13.9%) (Fig. S31 in Supporting information), 3a (1.5 W/cm2, conversion efficiency: 40.4%) (Fig. S35 in Supporting information), and 4a (1.5 W/cm2, conversion efficiency: 22.3%) (Fig. S39 in Supporting information), it has been determined that compound 3a exhibited the highest photothermal conversion efficiency among the three compounds.
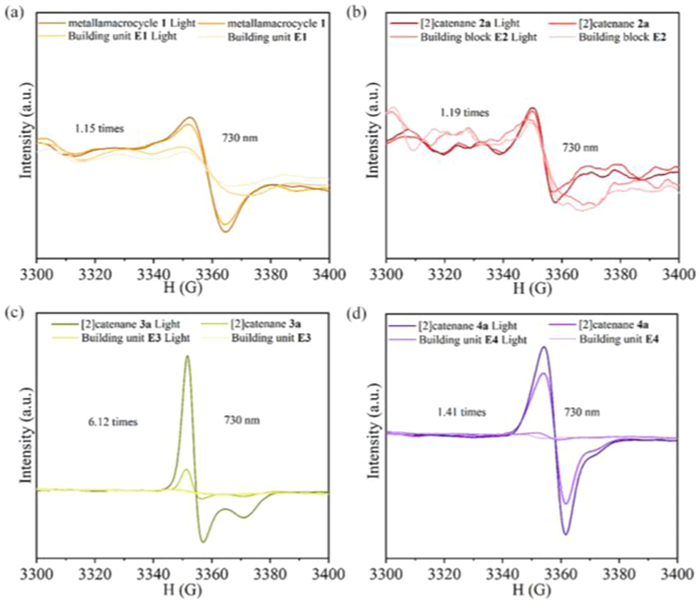
In an effort to shed light on the photothermal conversion mechanism, electron paramagnetic resonance (EPR) spectra of the building blocks E1-E4, metallamacrocycle 1 and [2]catenanes 2a, 3a and 4a were recorded. The strong recorded signal that was detected clearly indicates the existence of unpaired electrons, in concordance with the charge-transfer interactions that were observed in the ground state. Subjecting the metallamacrocycle 1, and [2]catenanes 2a, 3a and 4a to irradiation, results in expansions of their EPR signals by 1.15, 1.19, 6.12 and 1.41 times, respectively (Figs. 8a-d). This phenomenon is most likely due to π-π stacking interactions, which facilitated non-radiative transitions while inhibiting the radiative transitions, thereby leading to an enhancement of the EPR signals. This experimental observation implies that the presence of photothermal effect in the compounds depends on the existence of an EPR signal in the half-sandwich unit. Thus, although multiple π-π stacking interactions exist in the structure, the EPR signal of E1 and E2 changed less in metallamacrocycle 1 and [2]catenane 2a at 730 nm laser, showing a weak photothermal conversion effect. In contrast, the difference in strong EPR signal with 730 nm laser irradiation indicated stronger photothermal conversion for [2]catenane 3a [61–64].
In summary, we have demonstrated that two types of supramolecular topological structures, metalla[2]catenanes and metallamacrocycles, can be obtained by the self-assembly of the well-designed ligand L1 and Cp*Rh-based building blocks E1-E4 possessing different sizes and conjugation effects. Remarkably, dynamic structural transformation between [2]catenanes and the corresponding metallamacrocycles can be observed by changing the concentration or by polar solvent induced effect. Besides, research on photothermal conversion showed that [2]catenane 3a displayed significant temperature changes in solution under a laser irradiation of 1.5 W/cm2 (from 25.8 ℃ to 50.3 ℃), and the photothermal conversion efficiency could reach 40.42%. The energy levels of building units E2-E4 by DFT calculation reflected that the building unit E3 exhibits the smallest bandgap, thus compound 3a has the best photothermal conversion efficiency. This research clarified the mechanism of photothermal conversion, which is of great significance for further exploring the application of half-sandwich photothermal materials.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Ying Zhao: Writing – original draft. Yao He: Validation. Jian-Xin Yang: Data curation. Wen-Jie Liu: Methodology. Dan Tian: Visualization. Francisco Aznarez: Formal analysis. Le-Le Gong: Data curation. Li-Long Dang: Writing – review & editing. Lu-Fang Ma: Supervision.
This work was supported by the National Natural Science Foundation of China (Nos. 22471113 and 22171123), and Natural Science Foundation of Henan Province (Nos. 242300421139, 232300421232), the Science and Technology Innovation Talent Program of University in Henan Province (No. 25HASTIT001).
Supplementary material associated with this article can be found, in the online version, at doi:
A.S. Baluna, M. Dommaschk, B. Groh, et al., J. Am. Chem. Soc. 145 (2023) 27113–27119. doi: 10.1021/jacs.3c11290
Y.T. Wang, Y. Qin, X.L. Zhao, et al., Chin. Chem. Lett. 34 (2023) 107576. doi: 10.1016/j.cclet.2022.05.090
E.G. Percástegui, T.K. Ronson, J.R. Nitschke, Chem. Rev. 120 (2020) 13480–13544. doi: 10.1021/acs.chemrev.0c00672
L.J. Wang, S. Bai, Y.F. Han, J. Am. Chem. Soc. 144 (2022) 16191–16198. doi: 10.1021/jacs.2c07586
K.Y. Wang, M.Z. Zuo, T. Zhang, et al., Chin. Chem. Lett. 34 (2023) 107848. doi: 10.1016/j.cclet.2022.107848
J. Gemen, J. Ahrens, L.J.W. Shimon, et al., J. Am. Chem. Soc. 142 (2020) 17721–17729. doi: 10.1021/jacs.0c08589
W.Q. Lv, Y.Y. Song, X.Y. Lv, et al., Chin. Chem. Lett. 34 (2023) 108179. doi: 10.1016/j.cclet.2023.108179
W. Cullen, A.J. Metherell, A.B. Wragg, et al., J. Am. Chem. Soc. 140 (2018) 2821–2828. doi: 10.1021/jacs.7b11334
Z.L. Jiang, H. Zhao, J. Wang, et al., Chin. Chem. Lett. 34 (2023) 108334. doi: 10.1016/j.cclet.2023.108334
F. Yu, B.Q. Ji, M. Jagodič, et al., Inorg. Chem. 59 (2020) 13524–13532. doi: 10.1021/acs.inorgchem.0c01915
S. Bhowmick, A. Jana, K. Singh, et al., Inorg. Chem. 57 (2018) 3615–3625. doi: 10.1021/acs.inorgchem.7b01561
H. Li, Z.J. Li, J.L. Jiang, et al., Org. Chem. Front. 11 (2024) 6358–6366. doi: 10.1039/d4qo01498g
Y. Zhao, Y.H. Chai, T. Chen, et al., Chin. Chem. Lett. 35 (2024) 109298. doi: 10.1016/j.cclet.2023.109298
S.J. Bao, H.N. Zhang, G.X. Jin, CCS Chem. 6 (2024) 2000–2010. doi: 10.31635/ccschem.024.202303525
Y.W. Zhang, S. Bai, Y.Y. Wang, et al., J. Am. Chem. Soc. 142 (2020) 13614–13621. doi: 10.1021/jacs.0c06470
B. Lan, R.Y. Zhang, J.F. Yan, et al., Chin. J. Struct. Chem. 42 (2023) 100008.
Z. Cui, Y. Lu, X. Gao, et al., J. Am. Chem. Soc. 142 (2020) 13667–13671. doi: 10.1021/jacs.0c05366
H.N. Zhang, G.X. Jin, Nat. Synth. 4 (2025) 488–496. doi: 10.1038/s44160-024-00720-4
T. Chen, Y. Zhao, L.L. Dang, et al., J. Am. Chem. Soc. 145 (2023) 18036–18047. doi: 10.1021/jacs.3c05720
L.L. Dang, J. Zheng, J.Z. Zhang, et al., Angew. Chem. Int. Ed. 63 (2024) e202406552. doi: 10.1002/anie.202406552
Y. Tamura, H. Takezawa, M. Fujita, J. Am. Chem. Soc. 142 (2020) 5504–5508. doi: 10.1021/jacs.0c00459
X.Q. Guo, P.W. Yu, L.P. Zhou, et al., Nat. Synth. 4 (2025) 359–369. doi: 10.1038/s44160-024-00697-0
H. Sepehrpour, W.X. Fu, Y. Sun, et al., J. Am. Chem. Soc. 141 (2019) 14005–14020. doi: 10.1021/jacs.9b06222
Y. Sun, C.Y. Chen, J.B. Liu, et al., J. Am. Chem. Soc. 142 (2020) 17903–17907. doi: 10.1021/jacs.0c08058
H.N. Zhang, H.J. Feng, Y.J. Lin, et al., J. Am. Chem. Soc. 145 (2023) 4746–4756. doi: 10.1021/jacs.2c13416
H.N. Zhang, G.X. Jin, Angew. Chem. Int. Ed. 62 (2023) e202313605. doi: 10.1002/anie.202313605
Z. Cui, Q.S. Mu, X. Gao, et al., J. Am. Chem. Soc. 145 (2023) 725–731. doi: 10.1021/jacs.2c12027
P.P. Hua, J.H. Bai, H.J. Feng, et al., J. Am. Chem. Soc. 146 (2024) 26427–26434. doi: 10.1021/jacs.4c09385
F. Zeng, L.L. Tang, H. Yu, et al., Chin. Chem. Lett. 34 (2023) 108304. doi: 10.1016/j.cclet.2023.108304
K. Iizuka, H. Takezawa, M. Fujita, J. Am. Chem. Soc. 146 (2024) 32311–32316. doi: 10.1021/jacs.4c14509
Y.F. Han, G.X. Jin, Acc. Chem. Res. 47 (2014) 3571–3579. doi: 10.1021/ar500335a
M.F. Wang, Y. Mi, F.L. Hu, et al., J. Am. Chem. Soc. 142 (2020) 700–704. doi: 10.1021/jacs.9b12358
Z.L. Zheng, N. Hanikel, H. Lyu, et al., J. Am. Chem. Soc. 144 (2022) 22669–22675. doi: 10.1021/jacs.2c09756
H. Li, Z.J. Li, C. Lin, et al., Nat. Commun. 15 (2024) 5315. doi: 10.1038/s41467-024-49540-2
H.R. Fu, D.D. Ren, K. Zhang, et al., ACS Mater. Lett. 6 (2024) 2559–2568. doi: 10.1021/acsmaterialslett.4c00619
H.M. Yu, M.H. Du, J. Shu, et al., J. Am. Chem. Soc. 145 (2023) 25103–25108. doi: 10.1021/jacs.3c11398
P.F. Cui, X.R. Liu, Y.J. Lin, et al., J. Am. Chem. Soc. 144 (2022) 6558–6565. doi: 10.1021/jacs.2c01668
H.N. Zhang, W.X. Gao, Y.J. Lin, et al., J. Am. Chem. Soc. 141 (2019) 16057– 16063. doi: 10.1021/jacs.9b08254
H.N. Zhang, Y.J. Lin, G.X. Jin, J. Am. Chem. Soc. 143 (2021) 1119–1125. doi: 10.1021/jacs.0c11925
P.F. Gao, K. Zhang, D.D. Ren, et al., Adv. Funct. Mater. 33 (2023) 2300105. doi: 10.1002/adfm.202300105
T. Feng, X. Li, Y.Y. An, et al., Angew. Chem. Int. Ed. 59 (2020) 13516–13520. doi: 10.1002/anie.202004112
L.L. Dang, J. Zheng, D. Tian, et al., Angew. Chem. Int. Ed. 64 (2025) e202422444. doi: 10.1002/anie.202422444
Y.H. Chai, L.L. Dang, Chin. J. Struct. Chem. 43 (2024) 100322.
D.A. Leigh, L. Pirvu, F. Schaufelberger, J. Am. Chem. Soc. 141 (2019) 6054– 6059. doi: 10.1021/jacs.9b01819
L.L. Dang, T.T. Zhang, T. Chen, et al., Angew. Chem. Int. Ed. 62 (2023) e202301516. doi: 10.1002/anie.202301516
W.L. Shan, H.H. Hou, N. Si, et al., Angew. Chem. Int. Ed. 63 (2024) e202402198. doi: 10.1002/anie.202402198
X.R. Liu, P.F. Cui, Y. García-Rodeja, et al., Chem. Sci. 15 (2024) 9274–9280. doi: 10.1039/d4sc01158a
N.T. Wu, J.K. Shen, X.L. Zhou, et al., Adv. Energy Mater. 15 (2025) 2405729. doi: 10.1002/aenm.202405729
L.J. Wang, X. Li, S. Bai, et al., J. Am. Chem. Soc. 142 (2020) 2524–2531. doi: 10.1021/jacs.9b12309
Y. Zhao, J.X. Yang, Y.H. Chai, et al., Sci. China Chem. 68 (2025) 1383–1391. doi: 10.1007/s11426-024-2502-6
H.J. Feng, W.X. Gao, Y.J. Lin, et al., Chem. Eur. J. 25 (2019) 15687–15693. doi: 10.1002/chem.201904196
H.T. Tang, H.N. Zhang, X. Gao, et al., J. Am. Chem. Soc. 146 (2024) 16020–16027. doi: 10.1021/jacs.4c03019
L.C. Liu, Y.L. Liao, L.X. Li, et al., Inorg. Chem. 64 (2025) 4345–4354. doi: 10.1021/acs.inorgchem.4c04872
P.F. Gao, Y.Y. Jiang, H. Liu, et al., ACS Appl. Mater. Interfaces 14 (2022) 16435–16444. doi: 10.1021/acsami.2c01615
N.T. Wu, W.J. He, S.C. Shi, et al., J. Colloid Interface Sci. 684 (2025) 658–667. doi: 10.1016/j.jcis.2025.01.071
Z. Zhou, X. Wang, H. Zhang, et al., Small 17 (2021) 2007486. doi: 10.1002/smll.202007486
B.L. Xue, X.W. Geng, H.H. Cui, et al., Chin. Chem. Lett. 34 (2023) 108140. doi: 10.1016/j.cclet.2023.108140
Y. Wang, W.G. Zhu, W.N. Du, et al., Angew. Chem. Int Ed. 57 (2018) 3963–3967. doi: 10.1002/anie.201712949
X. Gao, Z. Cui, Y.R. Shen, et al., J. Am. Chem. Soc. 143 (2021) 17833–17842. doi: 10.1021/jacs.1c09333
L.L. Dang, T.T. Li, T.T. Zhang, et al., Chem. Sci. 13 (2022) 5130–5140. doi: 10.1039/d2sc00437b
D.B. Wang, X.N. Kan, C.Y. Wu, et al., Chem. Commun. 56 (2020) 5223–5226. doi: 10.1039/d0cc01834a
Y. Zhang, G.Y. Wu, H. Liu, et al., Mater. Chem. Front. 5 (2021) 6575–6581. doi: 10.1039/d1qm00462j
N.T. Wu, Z.B. Zhao, Y.M. Zhang, et al., J. Colloid Interface Sci. 679 (2025) 990–1000. doi: 10.1016/j.jcis.2024.10.175
Z. Zhou, T. Wang, T. Hu, et al., Adv. Mater. 36 (2024) 2311002. doi: 10.1002/adma.202311002

Scheme 1 Synthesis of organometallic supramolecular compounds and dinuclear building blocks E1-E4 used in this study.

Figure 2 ESI-TOF-MS spectra of metallamacrocycle 1, [2]catenanes 2a and 4a: experimental (jade-green) and calculated (deep yellow) (a, d) 2+ mass peak of 1; (b, e) 3+ mass peak of 2a; (c, f) 3+ mass peak of 4a. Partial ESI-TOF-MS spectra of the complexes (g) 1, (h) 2a and (i) 4a.

Figure 3 (a) Chemical structure of [2]catenane 2a. (b) Solid-state structure of 2a, showing the π-π stacking interactions. (c, d) Two-colors solid-state structure of [2]catenane 2a disordered elements are omitted for clarity (N, blue; O, red; C, gray; and Rh, teal).

Figure 4 (a) Chemical structure of [2]catenane 3a. (b) Solid-state structure of 3a, showing the π-π stacking interactions. Disordered elements are omitted for clarity (N, blue; O, red; C, gray; Br, brown; and Rh, teal).

Figure 5 (a) Chemical structure of [2]catenane 4a. (b) Solid-state structure of 4a, showing the π-π stacking interactions. (c) Side view of [2]catenane 4a (d) Top view of [2]catenane 4a. Disordered elements are omitted for clarity (N, blue; O, red; C, gray; and Rh, teal).

Figure 6 1H NMR (500 MHz, CD3OD, ppm) spectra of showing transformation from metallamacrocyle 2b to [2]catenane 2a up increasing concentration from 1.0 equiv. to 10.0 equiv., the conversion rate is 66.7%.

Figure 7 (a) The UV–vis absorption of compounds 1, 2a, 3a and 4a at 725–800 nm. (b) NIR photothermal conversion curves of compounds 1 and 2a, 3a and 4a at 0.6 W/cm2 in solid state. (c) NIR photothermal conversion curves of compounds 2a-4a at 1.5 W/cm2, 1.2 W/cm2, 0.9 W/cm2 and 0.6 W/cm2 in solution. (d) The UV–vis absorption of compounds 1, 2a, 3a and 4a at 630–800 nm in CH3OH. (e) NIR photothermal conversion curves of 2a-4a at 1.5 W/cm2 in CH3OH. (f) Near infrared thermal imaging of [2]catenane 3a performed in a specific container under 730 nm laser irradiation.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:

