Citation:
Xuanbo Zhang, Feng Fang, Na Li, Huicong Zhang, Kaiyuan Wang, Zhiqiang Yu, Jin Sun. From reversible to irreversible: Albumin-hitchhiking gemcitabine prodrugs for enhanced antitumor efficacy and reduced toxicity[J]. Chinese Chemical Letters,
2026, 37(2): 111452.
doi:
10.1016/j.cclet.2025.111452
From reversible to irreversible: Albumin-hitchhiking gemcitabine prodrugs for enhanced antitumor efficacy and reduced toxicity
English
From reversible to irreversible: Albumin-hitchhiking gemcitabine prodrugs for enhanced antitumor efficacy and reduced toxicity
The Tenth Affiliated Hospital, Southern Medical University (Dongguan People's Hospital), Dongguan 523058, China
b.
Department of Pharmaceutics, Wuya College of Innovation, Shenyang Pharmaceutical University, Shenyang 110016, China
c.
Department of Obstetrics and Gynecology, Foshan Sanshui District People's Hospital (Sanshui Hospital, Zhujiang Hospital, Southern Medical University), Foshan 528100, China
d.
CSPC Zhongqi Pharmaceutical Technology (Shijiazhuang) Co., Ltd., Shijiazhuang 050000, China
sunjin@syphu.edu.cn (J. Sun). 1 These authors contributed equally to this work.
Received Date:
18 April 2025 Accepted Date:
12 June 2025 Revised Date:
10 June 2025 Available Online:
15 February 2026
Abstract:
Albumin, owing to its high abundance and excellent biocompatibility, is widely used as a drug carrier to enhance delivery efficiency and reduce systemic toxicity. The Michael addition between albumin thiols and maleimide-functionalized prodrugs is a common in situ macromolecular prodrug strategy. However, the resulting reversible adducts are susceptible to retro-Michael reactions in vivo, leading to premature drug release and off-target effects. To address this limitation, a gemcitabine prodrug (GAB) bearing a chloroacetamide group was designed to form irreversible covalent bonds with albumin via nucleophilic substitution. A maleimide-based prodrug (GAM) was synthesized as a control. Compared to GAM, GAB showed faster and stronger albumin binding in plasma, enhanced blood circulation time, improved tumor accumulation, and superior in vivo antitumor efficacy. Moreover, GAB exhibited a better safety profile, with reduced cytotoxicity in normal tissues and no observable systemic toxicity. These advantages are attributed to the stable albumin-drug conjugate formed by GAB, which improves drug retention and targeted delivery. This study presents an effective and generalizable albumin-hitchhiking strategy for constructing irreversible prodrugs, offering a promising approach to enhance the therapeutic index of chemotherapeutic agents.
Albumin, a 66.5 kDa protein with a plasma half-life of up to 19 days, is the most abundant protein in human plasma and plays a vital role in maintaining colloidal osmotic pressure and transporting a wide range of molecules [1–3]. Its long circulation time, biocompatibility, and low immunogenicity make albumin an attractive drug carrier, particularly for chemotherapeutic agents [4,5]. Albumin-binding drug delivery systems, where drugs are either non-covalently or covalently associated with albumin, have demonstrated improved pharmacokinetics, tumor accumulation, and reduced systemic toxicity compared to traditional nanoparticle formulations [6–8].
Among these strategies, covalent conjugation to albumin's free thiol at cysteine-34 has gained significant interest due to its potential for improved in vivo stability [9,10]. Maleimide-thiol chemistry is widely used in this context, as it enables rapid, selective conjugation under physiological conditions [11]. Prior work, including studies from our group, has shown that maleimide-modified chemotherapeutics, such as gemcitabine (GEM) prodrugs, can form covalent adducts with circulating albumin in situ, thereby prolonging circulation and enhancing tumor delivery [9,12,13].
However, maleimide-thiol conjugation is fundamentally limited by its reversibility [14]. In plasma, maleimide–albumin adducts are prone to hydrolysis and thiol exchange reactions, leading to premature drug release and reduced delivery efficiency [15]. This is particularly problematic for unstable drugs like GEM [16], which can undergo rapid deamination and clearance if prematurely released, reducing its therapeutic effect and increasing the risk of systemic toxicity or drug resistance [17].
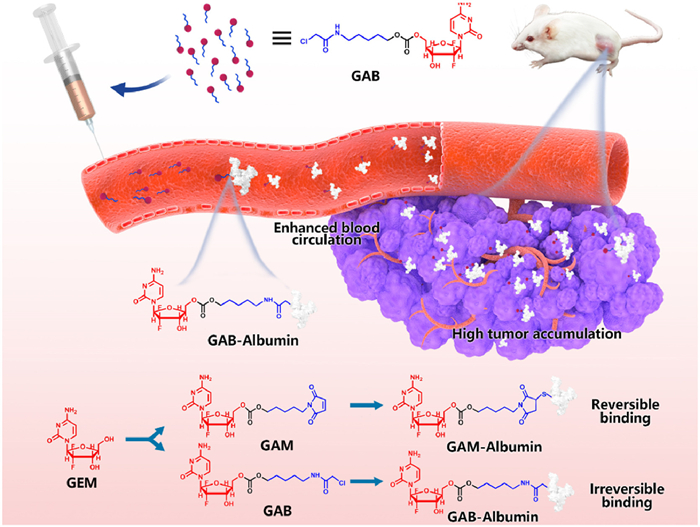
To address these limitations, we developed a GEM prodrug (GAB) functionalized with a chloroacetamide group, a known electrophile capable of undergoing irreversible covalent bonding with thiol groups [18]. In contrast to maleimide-based prodrugs, GAB was designed to stably and irreversibly bind albumin in vivo, improving drug retention and delivery efficiency. We synthesized both GAB and a reference maleimide-based prodrug (GAM) and confirmed their structures by nuclear magnetic resonance (NMR) spectroscopy and mass spectrometry. Binding studies in phosphate-buffered saline (PBS) and rat plasma revealed that while both prodrugs could bind to bovine serum albumin (BSA) rapidly, GAB exhibited significantly stronger and more stable albumin binding in plasma. Importantly, the binding process did not disrupt the structure of albumin. In vitro cytotoxicity assays demonstrated that both prodrugs showed reduced toxicity compared to free GEM, consistent with their prodrug nature. Pharmacokinetic analysis in rats showed that GAB achieved longer circulation times and greater systemic exposure than both GAM and free drug. Biodistribution studies in 4T1 tumor-bearing mice revealed higher tumor accumulation for GAB, likely due to its stable albumin conjugation. Antitumor efficacy studies confirmed that GAB outperformed GAM and free GEM in tumor inhibition while maintaining good systemic safety. These findings demonstrate that irreversible albumin-hitchhiking via chloroacetamide modification offers a promising platform to enhance the delivery and therapeutic index of chemotherapeutic agents such as GEM (Scheme 1).
Scheme 1
Scheme 1.
Schematic illustration of the delivery process of the GEM prodrug (GAB) via albumin hitchhiking. Upon entering the bloodstream, GAB forms irreversible covalent adducts with albumin, in contrast to the reversible binding observed with the maleimide-based prodrug (GAM). The resulting stable GAB-albumin complex enhances circulation time, promotes tumor accumulation, and leads to improved antitumor efficacy.
The GEM prodrug GAB was synthesized using 2–chloro-N-(5-hydroxypentyl)acetamide as the starting material. After esterification with triphosgene, the intermediate was subsequently coupled with GEM to yield the final product, GAB (Fig. S1 in Supporting information). The structure of the compound was confirmed by mass spectrometry and nuclear magnetic resonance spectroscopy (Figs. S2–S7 in Supporting information), verifying successful synthesis. High-performance liquid chromatography (HPLC) analysis showed the purity of the synthesized GAB to be 97.4%. The structure of the synthesized maleimide-based prodrug (GAM) is also shown in Fig. 1A.
Figure 1
Figure 1.
Binding behavior of GEM prodrugs with albumin and cytotoxicity evaluation. (A) Chemical structures of GEM, maleimide-based prodrug (GAM), and chloroacetamide-based prodrug (GAB). (B) Schematic diagram of HPLC-based analysis for prodrug–albumin conjugation. (C) Binding efficiency of GAB and GAM with BSA or BSA blocked by excess EMC at 37 ℃. (D) Time-dependent binding of GAB and GAM with endogenous albumin in fresh whole blood (2, 4, and 6 min). (E) Schematic illustration of GEM metabolism into dFdU. (F) Concentration change of GEM released from albumin–prodrug conjugates during incubation in fresh rat blood. (G) Corresponding concentration change of dFdU as a marker of GEM degradation. (H) Quantification of cellular uptake under different treatment conditions. (I, J) Cytotoxicity of GEM, GAM, and GAB in 4T1 cells after 48 and 72 h of incubation, assessed by MTT assay. Data are presented as mean ± standard deviation (SD) (n = 3). n.s., not significant. P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
The in vitro albumin-binding capabilities of both GEM prodrugs were evaluated in PBS containing BSA and in whole blood. In the BSA incubation experiment, the prodrugs were incubated with BSA in PBS, and the remaining unbound prodrug was quantified via HPLC (Fig. 1B). The reduction in the peak area of the free prodrug was used to assess binding efficiency. As shown in Fig. 1C, significant binding to BSA occurred within 5 min, and the majority of prodrugs had formed albumin complexes within 10 min. After 30 min, over 90% of each prodrug had successfully bound to BSA. To verify the thiol-dependency of this interaction, BSA in the plasma was blocked using excess 6-maleimidocaproic acid (EMC), which quenched free thiol groups [12]. Under this condition, over 90% of the prodrug remained unbound, indicating that the albumin binding is mediated by a thiol-specific reaction between the prodrug and cysteine residues on albumin.
To further simulate physiological conditions, albumin binding was also assessed in freshly collected whole blood from Sprague-Dawley rats. All animal experiments were performed in accordance with the guidelines approved by the Animal Laboratory Ethics Committee of Shenyang Pharmaceutical University and the Tenth Affiliated Hospital of Southern Medical University (approval No. IACUC-AWEC-202503001). As shown in Fig. 1D and Fig. S8 (Supporting information), both GAB and GAM demonstrated enhanced albumin-binding efficiency in whole blood compared to the BSA-PBS system. Notably, after 10 min of incubation, most of the prodrug had already bound to plasma proteins, with GAB displaying significantly stronger and faster binding than GAM. These results collectively suggest that GEM prodrugs can rapidly and efficiently form complexes with albumin under physiological conditions. Moreover, the GAB prodrug, bearing a chloroacetamide moiety, showed superior binding kinetics and efficiency compared to the maleimide-modified prodrug GAM. This indicates that irreversible binding via the chloroacetamide group enables more stable and rapid conjugation with endogenous albumin, which may enhance in vivo delivery efficiency.
In the bloodstream, GEM is rapidly metabolized by cytidine deaminase into 2′,2′-difluorodeoxyuridine (dFdU), which is subsequently excreted in urine [19]. Therefore, the concentration of dFdu in plasma serves as a reliable biomarker for GEM degradation and inactivation (Fig. 1E). As shown in Figs. 1F and G, free GEM exhibited a continuous decline in concentration during incubation with plasma, accompanied by a marked increase in dFdu levels. This indicates significant enzymatic degradation of GEM in circulation. In contrast, both GEM prodrugs (GAB and GAM), after forming complexes with albumin, showed minimal decreases in GEM concentration during plasma incubation, and only slight elevations in dFdu levels. These results suggest that albumin binding provides effective protection against enzymatic degradation of GEM in plasma. Notably, the GAB-albumin complex demonstrated superior stability and protection compared to GAM, likely due to its irreversible covalent binding with albumin. This enhanced protection may contribute to improved pharmacokinetics and antitumor efficacy in vivo.
To investigate cellular uptake, Cy5-labeled BSA and BSA–prodrug conjugates (BSA-GAB and BSA-GAM) were synthesized. The fluorescence spectra and circular dichroism (CD) spectra of BSA and its prodrug conjugates were comparable, indicating that the conjugation process did not significantly alter BSA's structural integrity (Figs. S9 and S10 in Supporting information). 4T1 tumor cells were incubated with Cy5-labeled BSA or BSA-prodrug conjugates at either 4 or 37 ℃ for 2 h, and intracellular fluorescence intensity was quantified. As shown in Fig. 1H and Fig. S11 (Supporting information), uptake was significantly reduced at 4 ℃, suggesting an energy-dependent internalization mechanism for both conjugates and native BSA. To further elucidate the internalization pathway, uptake experiments were performed in the presence of specific endocytosis inhibitors. The presence of chlorpromazine (clathrin-mediated endocytosis inhibitor), indomethacin (caveolae-mediated endocytosis inhibitor), quercetin (non-clathrin/non-caveolae inhibitor), and sodium azide (metabolic inhibitor) all led to reduced cellular uptake of BSA-GAB and BSA-GAM (Fig. 1H and Fig. S11). These findings indicate that the internalization of BSA-prodrug conjugates involves multiple endocytic pathways, including clathrin-mediated, caveolae-mediated, and non-classical endocytosis, all of which are energy-dependent. Moreover, the cellular uptake of BSA-GAB and BSA-GAM conjugates was directly assessed. The results demonstrated that, relative to the free drug, the albumin-bound prodrugs exhibited comparable intracellular accumulation, suggesting that albumin conjugation did not markedly affect cellular uptake (Fig. S12 in Supporting information).
The cytotoxicity of the prodrugs was evaluated using MTT assays in murine breast cancer (4T1) and human lung cancer (A549) cells following 48 and 72 h of incubation. As shown in Figs. 1I and J and Fig. S13 (Supporting information), both GAB and GAM exhibited reduced cytotoxicity compared to free GEM, likely due to protein binding in the culture medium. Given that the prodrugs achieve over 90% conjugation with albumin within 30 min in simulated body fluids and plasma, a similar extent of binding is expected under in vitro conditions, reducing the amount of free active drug. GAM demonstrated slightly higher cytotoxicity than GAB, which may be attributed to its less stable linker, leading to faster drug release. In contrast, the more stable chloroacetamide linkage in GAB results in slower drug release and thus lower cytotoxicity. Interestingly, the difference in cytotoxicity between GAB and GAM diminished over time (Table S1 in Supporting information), likely due to gradual intracellular drug release from both conjugates [20]. These results suggest that although the prodrugs show reduced in vitro cytotoxicity, reflecting their improved systemic safety profile, their actual antitumor efficacy requires further validation in vivo.
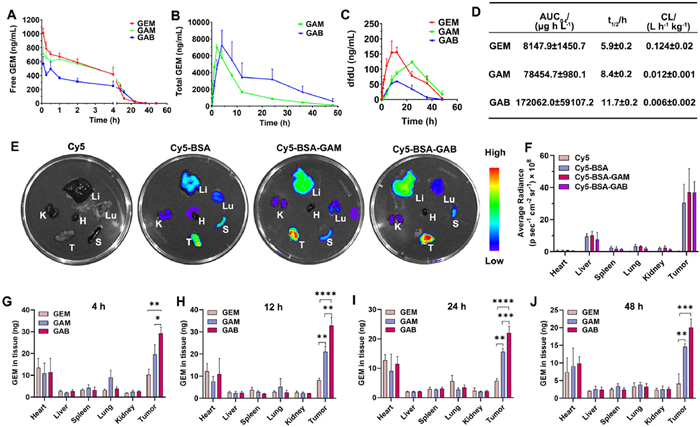
To investigate the pharmacokinetic behavior of the prodrugs, GAB, GAM, and the parent drug GEM were intravenously administered to Sprague-Dawley rats via tail vein injection at 1 equiv. GEM dose of 1 mg/kg. Plasma samples were collected at predetermined time points. The plasma concentration–time profiles are shown in Figs. 2A–C, and the corresponding pharmacokinetic parameters are summarized in Fig. 2D. As expected, the parent drug GEM was rapidly cleared from the bloodstream, with an area under the concentration–time curve (AUC) of 8147.9 ± 1450.7 µg h L−1 and a half-life (t1/2) of 5.9 ± 0.2 h. In contrast, both prodrugs exhibited significantly prolonged circulation time. The total GEM AUC released from GAB and GAM was 21- and 9-fold higher, respectively, than that of the GEM group. These findings demonstrate that the albumin-binding prodrug strategy substantially enhances the systemic retention of GEM and improves its pharmacokinetic profile. Notably, GAB released less free GEM in the bloodstream compared to GAM (Fig. 2A) while achieving a higher total GEM AUC (Fig. 2D), indicating that the covalent linkage between GAB and albumin provides more robust protection against premature degradation. Correspondingly, the level of dFdU, a metabolic degradation product of GEM, was significantly lower in the GAB group than in the GAM group (Fig. 2C). These results confirm that GAB offers superior stabilization and plasma retention of GEM compared to GAM.
Figure 2
Figure 2.
Pharmacokinetics and biodistribution of GEM prodrugs. Plasma concentration–time curves of GEM, GAM, and GAB in rats following intravenous injection at 1 equiv. GEM dose of 8 mg/kg (n = 5): (A) free GEM; (B) total GEM released from prodrugs; (C) GEM metabolite dFdU. (D) Pharmacokinetic parameters of total GEM derived from GEM, GAM, and GAB groups (n = 5). (E) Ex vivo fluorescence imaging of major organs and tumors in 4T1 tumor-bearing mice at 24 h after intravenous injection of Cy5, Cy5-labeled BSA, and the two BSA-prodrug conjugates. (F) Quantitative fluorescence analysis corresponding to (E). Quantification of free GEM levels in tumor tissues at different time points post-injection of GEM, GAM, and GAB (8 mg/kg GEM-equivalent dose): (G) 4 h; (H) 12 h; (I) 24 h; (J) 48 h. Data are presented as mean ± SD (n = 3). P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Given albumin's ability to accumulate in tumor tissues and its clearance primarily via renal and hepatic pathways, we next evaluated the biodistribution of the Cy5-labeled BSA and BSA-prodrug conjugates in 4T1 tumor-bearing mice. As shown in Figs. 2E and F, 24 h after intravenous injection, strong fluorescence was observed predominantly in tumor tissue, indicating excellent tumor-specific accumulation of the macromolecular prodrugs. Additionally, the biodistribution patterns of Cy5-BSA and its prodrug conjugates were similar in intensity and distribution, suggesting that conjugation with small-molecule prodrugs does not compromise albumin's inherent tumor-targeting capacity. To quantify the drug accumulation in tissues, GEM concentrations in homogenized organs were measured via liquid chromatography–tandem mass spectrometry (LC–MS/MS) at 4, 12, 24, and 48 h post-injection. The results are presented in Figs. 2G–J. Following administration, GEM was primarily distributed in tumor tissue. For the free GEM group, the drug peaked in tumors at 4 h and subsequently declined due to rapid systemic clearance. In contrast, GAB and GAM showed a time-dependent increase in tumor accumulation, reaching maximum levels at 12 h, with both groups demonstrating significantly higher GEM concentrations in tumor tissues compared to the free drug group. Notably, GAB achieved greater tumor accumulation than GAM, aligning with the results of in vitro binding and pharmacokinetic studies. These findings suggest that the more stable covalent linkage between GAB and albumin leads to improved pharmacokinetics and enhances tumor-specific delivery, thereby laying a strong foundation for superior antitumor efficacy in vivo.
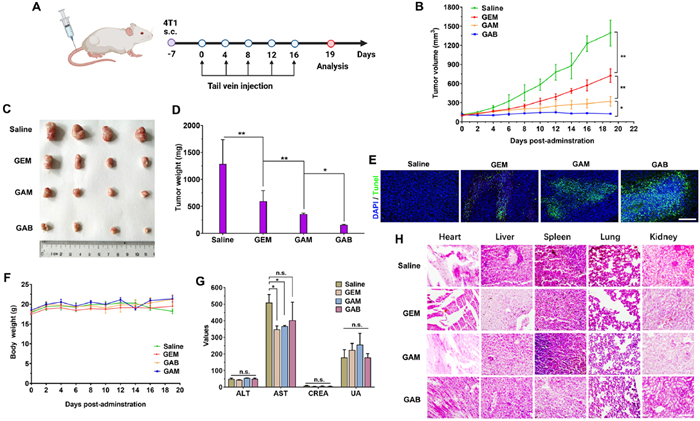
The in vivo antitumor efficacy of the GEM prodrugs was evaluated in a 4T1 murine triple-negative breast cancer (TNBC) xenograft model. Due to the known toxicity of GEM, an optimized dosing regimen was established based on preliminary studies: five intravenous injections were administered at 3-day intervals, each at a GEM-equivalent dose of 8 mg/kg (Fig. 3A). As shown in Figs. 3B–D, tumor growth in the vehicle-treated control group proceeded rapidly. In contrast, mice treated with free GEM initially exhibited a noticeable delay in tumor growth, but the inhibitory effect diminished over time, and tumor progression ultimately could not be effectively controlled. Notably, both GEM prodrugs, GAB and GAM, demonstrated significantly enhanced tumor growth inhibition compared to free GEM, despite their lower in vitro cytotoxicity. This improvement in antitumor efficacy may be attributed to the enhanced in vivo stability and prolonged circulation time conferred by albumin binding. The strong interaction between the prodrugs and endogenous albumin likely facilitated enhanced tumor accumulation via the enhanced permeability and retention (EPR) effect. In particular, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining revealed that GAB induced more extensive apoptosis in tumor tissues than both GEM and GAM (Fig. 3E and Fig. S14 in Supporting information), suggesting superior intratumoral drug activity. The improved therapeutic outcome of the prodrugs, especially GAB, is likely due to their enhanced pharmacokinetic properties and efficient tumor targeting, rather than their intrinsic cytotoxic potency. Additionally, the carbonate linker is likely to undergo more rapid cleavage in the acidic tumor microenvironment (Fig. S15 in Supporting information) and in the presence of tumor-associated enzymes [21,22], thereby facilitating the efficient release of active GEM at the tumor site.
Figure 3
Figure 3.In vivo antitumor efficacy and safety evaluation of GEM, GAM, and GAB in the 4T1 tumor xenograft model. (A) Schematic diagram of the treatment schedule. (B) Tumor growth curves following five intravenous administrations of different formulations (GEM-equivalent dose: 8 mg/kg, administered every 3 days). (C) Representative tumor images collected on day 19. (D) Final tumor weights at the end of treatment. (E) TUNEL staining of tumor tissues to assess apoptosis. Scale bar: 200 µm. (F) Changes in body weight during treatment. (G) Serum biochemical parameters including ALT, AST, CREA, and uric acid (UA). (H) H&E staining of major organs (heart, liver, spleen, lung, kidney) for histopathological assessment. Scale bar: 120 µm. Data are presented as mean ± SD (n = 4). P < 0.05; **P < 0.01.
These results highlight that in vivo antitumor efficacy is not solely determined by in vitro cytotoxicity [23,24]. Prolonged systemic circulation and enhanced tumor accumulation play critical roles in therapeutic outcomes. Among the tested formulations, GAB, bearing a chloroacetamide group that enables more stable albumin conjugation, exhibited the most pronounced antitumor effect, achieving significant tumor growth suppression and even shrinkage following five doses. While no significant changes in body weight or serum biochemical parameters were observed across treatment groups (Figs. 3F and G), histological examination of major organs using hematoxylin and eosin (H&E) staining indicated mild hepatic and renal toxicity in the GEM and GAM groups (Fig. 3H). This underscores the need for further safety evaluations to fully establish the therapeutic window of these prodrugs.
GEM is known to cause side effects involving multiple systems, including hematological, gastrointestinal, hepatic, and renal functions [25]. Common adverse effects include bone marrow suppression and hepatic/renal dysfunction [26]. As shown in Fig. S16 (Supporting information), to evaluate the systemic toxicity of GEM and its prodrugs, healthy mice were treated with escalating doses (dose 1 of 12 mg/kg and dose 2 of 16 mg/kg, GEM-equivalent) of GEM, GAM, and GAB, respectively (Fig. S16A). Following four injections of GEM, mice exhibited significant body weight loss, and the 16 mg/kg dose of free GEM resulted in fatal chemotherapeutic toxicity (Fig. S16B). In contrast, GAB treatment did not cause any noticeable toxicity, even at the highest dose tested.
Serum biochemical and hematological parameters were further assessed to investigate organ toxicity. GAM was associated with hematological toxicity, including marked reductions in platelet and red blood cell counts, whereas GAB showed no such effects even at the maximum dosage tested (Figs. S16C and D). Moreover, GEM treatment at both 12 mg/kg and 16 mg/kg led to significant elevations in serum levels of aspartate aminotransferase (AST), alanine aminotransferase (ALT), and alkaline phosphatase (ALP), indicating liver injury (Figs. S16E–G). Meanwhile, GAM and GAB demonstrated improved safety profiles at the 12 mg/kg dose, with no major abnormalities observed. However, at 16 mg/kg, GAM treatment caused a significant reduction in serum creatinine (CREA), suggesting potential nephrotoxicity possibly due to premature drug release (Fig. S16H). Collectively, these findings indicate that the irreversible albumin-binding prodrug GAB can effectively delay drug release in circulation, thereby significantly improving the in vivo safety profile of GEM.
In summary, this study demonstrates that albumin-binding GEM prodrugs, particularly the chloroacetyl-modified GAB, offer significant advantages over free GEM and its maleimide-based counterpart GAM. GAB exhibited enhanced plasma stability, prolonged circulation time, reduced formation of inactive metabolites, and superior tumor accumulation, resulting in improved pharmacokinetic profiles and enhanced antitumor efficacy in vivo. Despite showing lower cytotoxicity in vitro, both GAB and GAM significantly outperformed free GEM in tumor suppression, highlighting the critical role of pharmacokinetic behavior and tumor-targeted delivery in therapeutic outcomes. Importantly, GAB showed minimal systemic toxicity even at higher doses, with no significant hepatotoxicity, nephrotoxicity, or hematological side effects, underscoring its excellent safety profile. Furthermore, in contrast to pre-conjugated albumin, drug complexes, which are more complex to manufacture and face challenges similar to antibody–drug conjugates, including storage, transportation, high production costs, and potential immunogenicity, this in vivo albumin-binding strategy provides a simpler and more practical alternative. The prodrugs can be handled as conventional small molecules, substantially reducing logistical and manufacturing burdens. Additionally, by utilizing endogenous plasma albumin as a carrier, this approach helps avoid immunogenic risks associated with exogenous proteins. These findings support the potential of stable, albumin-binding prodrug strategies as a promising approach for improving the efficacy and safety of GEM-based chemotherapy.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Xuanbo Zhang: Writing – review & editing, Writing – original draft, Methodology, Investigation, Data curation, Conceptualization. Feng Fang: Methodology, Data curation, Conceptualization. Na Li: Writing – original draft, Methodology, Investigation, Data curation. Huicong Zhang: Supervision, Methodology, Conceptualization. Kaiyuan Wang: Methodology, Investigation, Data curation. Zhiqiang Yu: Writing – review & editing, Funding acquisition, Data curation, Conceptualization. Jin Sun: Writing – review & editing, Visualization, Project administration, Funding acquisition, Conceptualization.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos. 82372115, 52073139 and 82404553), the Postdoctoral Fellowship Program of Chinese Postdoctoral Science Foundation (No. GZC20231085).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111452.
M. Haas, D.T. Waldschmidt, M. Stahl, et al., Eur. J. Cancer 146 (2021) 95–106. doi: 10.1016/j.ejca.2020.12.029
Scheme 1
Schematic illustration of the delivery process of the GEM prodrug (GAB) via albumin hitchhiking. Upon entering the bloodstream, GAB forms irreversible covalent adducts with albumin, in contrast to the reversible binding observed with the maleimide-based prodrug (GAM). The resulting stable GAB-albumin complex enhances circulation time, promotes tumor accumulation, and leads to improved antitumor efficacy.
Figure 1
Binding behavior of GEM prodrugs with albumin and cytotoxicity evaluation. (A) Chemical structures of GEM, maleimide-based prodrug (GAM), and chloroacetamide-based prodrug (GAB). (B) Schematic diagram of HPLC-based analysis for prodrug–albumin conjugation. (C) Binding efficiency of GAB and GAM with BSA or BSA blocked by excess EMC at 37 ℃. (D) Time-dependent binding of GAB and GAM with endogenous albumin in fresh whole blood (2, 4, and 6 min). (E) Schematic illustration of GEM metabolism into dFdU. (F) Concentration change of GEM released from albumin–prodrug conjugates during incubation in fresh rat blood. (G) Corresponding concentration change of dFdU as a marker of GEM degradation. (H) Quantification of cellular uptake under different treatment conditions. (I, J) Cytotoxicity of GEM, GAM, and GAB in 4T1 cells after 48 and 72 h of incubation, assessed by MTT assay. Data are presented as mean ± standard deviation (SD) (n = 3). n.s., not significant. P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Figure 2
Pharmacokinetics and biodistribution of GEM prodrugs. Plasma concentration–time curves of GEM, GAM, and GAB in rats following intravenous injection at 1 equiv. GEM dose of 8 mg/kg (n = 5): (A) free GEM; (B) total GEM released from prodrugs; (C) GEM metabolite dFdU. (D) Pharmacokinetic parameters of total GEM derived from GEM, GAM, and GAB groups (n = 5). (E) Ex vivo fluorescence imaging of major organs and tumors in 4T1 tumor-bearing mice at 24 h after intravenous injection of Cy5, Cy5-labeled BSA, and the two BSA-prodrug conjugates. (F) Quantitative fluorescence analysis corresponding to (E). Quantification of free GEM levels in tumor tissues at different time points post-injection of GEM, GAM, and GAB (8 mg/kg GEM-equivalent dose): (G) 4 h; (H) 12 h; (I) 24 h; (J) 48 h. Data are presented as mean ± SD (n = 3). P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Figure 3In vivo antitumor efficacy and safety evaluation of GEM, GAM, and GAB in the 4T1 tumor xenograft model. (A) Schematic diagram of the treatment schedule. (B) Tumor growth curves following five intravenous administrations of different formulations (GEM-equivalent dose: 8 mg/kg, administered every 3 days). (C) Representative tumor images collected on day 19. (D) Final tumor weights at the end of treatment. (E) TUNEL staining of tumor tissues to assess apoptosis. Scale bar: 200 µm. (F) Changes in body weight during treatment. (G) Serum biochemical parameters including ALT, AST, CREA, and uric acid (UA). (H) H&E staining of major organs (heart, liver, spleen, lung, kidney) for histopathological assessment. Scale bar: 120 µm. Data are presented as mean ± SD (n = 4). P < 0.05; **P < 0.01.

 DownLoad:
DownLoad:


 下载:
下载:





