School of Pharmaceutical Sciences (Shenzhen), Shenzhen Campus of Sun Yat-sen University, Shenzhen 518107, China
b.
Dongguan Key Laboratory of Pathogenesis and Experimental Diagnosis of Infectious Diseases, School of Medical Technology, Guangdong Medical University, Dongguan 523808, China
c.
Department of Chemistry, National University of Singapore, Singapore 117543, Singapore
Received Date:
25 March 2025 Accepted Date:
06 June 2025 Revised Date:
04 June 2025 Available Online:
15 February 2026
Abstract:
Lysine-targeting reversible covalent inhibitors, particularly salicylaldehyde-based compounds such as the Food and Drug Administration (FDA)-approved drug Voxelotor, exhibit significant therapeutic potential but are limited by challenges including instability and off-target effects. To overcome these limitations in kinase inhibitor A5, we devised a pH-responsive prodrug strategy by masking its reactive aldehyde group with an acid-labile hydrazone linkage and enhancing intracellular delivery through conjugation with FK506. The optimized prodrug demonstrated robust antitumor efficacy in K562 tumor-bearing mice. Furthermore, the incorporation of the photosensitizer chlorin e6 (Ce6) led to the formation of self-assembled nanoparticles (AKNP), which not only improved physiological stability and prolonged tumor retention but also enabled light-triggered release of A5 in conjunction with photodynamic therapy (PDT). Our study thus presents a promising prodrug self-assembly strategy that combines the on-demand release of a novel lysine-targeting, reversible covalent kinase inhibitor with PDT in clinical cancer therapy.
Recent advances in targeted covalent inhibition (TCI) for cancer therapy have underscored the advantages of covalent drugs, including prolonged target engagement, enhanced potency, and selectivity compared to noncovalent agents [1–10]. Although cysteine-targeting strategies currently dominate covalent drug design [5,11–13], their limitations are becoming increasingly apparent. Cysteine residues are relatively scarce in the proteome and exist in unreactive disulfide bonds in the target protein, thereby restricting their applicability. Furthermore, their poor conservation in target proteins promotes rapid resistance mutations under therapeutic pressure [5,14–18]. These challenges necessitate the exploration of alternative strategies targeting non-cysteine residues, such as lysine, methionine, and tyrosine [19,20]. Among these, lysine-targeting covalent inhibitors are particularly promising, especially for proteins lacking accessible cysteines. In protein kinases, a conserved catalytic lysine critical for enzymatic activity is resistant to mutation during drug resistance, making it an ideal target [3,14,15,21,22]. Recent studies have demonstrated diverse lysine-reactive warheads, with aldehydes, such as salicylaldehydes (SA) and o-carbonyl boronic acids, emerging as effective electrophiles for reversible covalent inhibition [23–35]. Our work on benzaldehyde-based kinase inhibitors has revealed that intramolecularly stabilized imine formation via salicylaldehyde enables reversible targeting of the catalytic lysine, achieving high potency and selectivity through prolonged residence times [31]. Unlike irreversible inhibitors, such reversible covalent agents balance potency with reduced off-target risks, offering a refined therapeutic strategy [23,31,33]. This progress highlights lysine-targeting TCI as a complementary approach to overcome the limitations associated with cysteine-centric strategies.
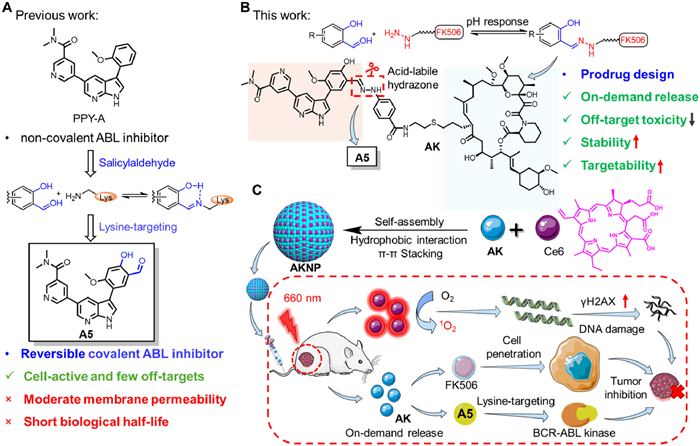
Chronic myeloid leukemia (CML) is caused by a genetic abnormality in the human chromosomes, leading to the formation of a constitutively active breakpoint cluster region-abelson (BCR-ABL) fusion gene kinase [3,31]. First-line therapies, such as imatinib (GleevecTM), target the ATP-binding site non-covalently and effectively reduce tumor burden; however, resistance develops in approximately 20% of patients due to BCR-ABL mutations. The lack of a proximal active-site cysteine in BCR-ABL complicates the development of covalent inhibitors. Our previous research identified A5, a reversible covalent inhibitor based on salicylaldehyde, which forms a hydrogen bond-stabilized imine with the catalytic lysine K271 (Fig. 1A). A5 demonstrated superior cellular selectivity and activity compared to its non-covalent analog PPY-A, particularly against drug-resistant mutants. Although aldehyde-containing drugs, such as VoxelotorTM, exhibit therapeutic potential, the aldehyde moiety of A5 presents certain limitations: its high reactivity increases the risk of off-target interactions, metabolic instability shortens its half-life, and moderate membrane permeability restricts its cellular efficacy [34,35].
Figure 1
Figure 1.
Schematic diagram of pH-responsive on-demand prodrug release strategy of AKNP. (A) Structures of previously reported BCR-ABL inhibitors including PPY-A and A5. (B) Chemical structure of prodrug AK and pH-responsive strategy for on-demand release by using acid-labile hydrazone to bind the aldehyde group in A5. (C) The design strategy of AKNP is based on the combination of prodrug AK and photosensitizer Ce6. After tail vein injection of AKNP, these nanoparticles can effectively accumulate at the tumor site. Upon cellular uptake, AKNP could produce ROS with light irradiation, leading to DNA damage. Meanwhile, the released A5 reversible-covalently inhibits BCR-ABL kinase, further inhibiting tumor growth.
To address A5′s limitations including moderate permeability, metabolic instability, and aldehyde reactivity, we developed a pH-responsive prodrug AK by conjugating A5 with FK506 via acid-labile hydrazone linkage as shown in Fig. 1B [36–40]. This design leverages FK506′s tumor-targeting capacity and hydrazone's pH sensitivity to enable tumor microenvironment (TME) or endolysosomal activation, improving circulatory stability and reducing off-target effects. To overcome the remaining small-molecule prodrug challenges including liver metabolism, rapid renal clearance, and premature activation [41–43], we co-assembled AK with photosensitizer chlorin e6 (Ce6) into AKNP nanoparticles via π-π stacking and hydrophobic interactions (Fig. 1C). The physical encapsulation of small molecules within various organic or inorganic nanocarriers, leading to effective shielding of the reactive sites in the molecules, has recently emerged as an extremely feasible strategy to enhance the absorption, distribution, metabolism, elimination, and toxicity (ADMET) properties of small-molecule (pro)-drugs in cancer therapy [44–48] nano-assembly maintains tumor penetration/accumulation advantages with minimized excipient toxicity [49–58] stimulus-responsive architecture enables tumor-specific drug release through coordinated pH/photodynamic activation [59–65]. In vivo studies demonstrated AKNP's superior antitumor efficacy versus A5/AK in K562 xenografts, attributed to synergistic covalent kinase inhibition by A5 and photodynamic therapy (PDT) Ce6. Structural validation of AK via nuclear magnetic resonance/high-resolution mass spectrometry (NMR/HRMS) confirmed successful hydrazone-functionalized A5 (Hyd-A5) conjugation with FK506-NH2 (Fig. S19 in Supporting information). Our dual-strategy platform combining pH-responsive prodrug chemistry with nano-assembly establishes a generalized approach for optimizing reversible covalent kinase inhibitors' therapeutic potential.
To confirm the effects of FK506 and its conjugate AK on cellular uptake, 4T1 and K562 cells were treated with FK506-Cy5 and Cy5-COOH, respectively, followed by flow cytometry (FACS) analysis (Figs. S1A and S2 in Supporting information). FACS analysis revealed enhanced cellular uptake of FK506-Cy5 compared to Cy5-COOH in both 4T1 and K562 cells (Figs. S1A and S2), consistent with prior reports on FK506-mediated tumor targeting [40]. High-performance liquid chromatography (HPLC) analysis demonstrated that AK exhibited pH-dependent release of native A5: ~50% release occurred within ~6 h at pH 3 and ~12 h at pH 6, while minimal release (< 12%) was observed at pH 7 after 12 h (Figs. S1B and S3 in Supporting information), confirming its TME-specific activation via hydrazone bond cleavage. Pre-treatment assays showed that AK exhibited a 5-fold lower ABL kinase inhibition (half maximal inhibitory concentration (IC50) = 20.66 nmol/L) compared to free A5 (IC50 = 3.452 nmol/L), indicating effective shielding of the aldehyde group in the prodrug form. However, pH 3 pre-treated AK restored inhibition comparable to native A5 (IC50 = 4.851 vs. 3.452 nmol/L) (Fig. S1C in Supporting information), confirming hydrazone bond cleavage under acidic conditions and recovery of kinase inhibitory activity.
Cell viability assay (MTT) was next used to evaluate the inhibition of A5 and the prodrug AK on the proliferation of K562 cancer cells. As shown in Fig. S1D (Supporting information), the growth inhibition value (growth inhibition 50% (GI50)) after 96-h drug treatment in the AK group was 4.5-fold lower than that after 48-h treatment (2.098 µmol/L vs. 9.033 µmol/L, respectively), indicating that inhibition of AK on K562 cell growth increased in a time-dependent manner, probably due to the progressive release of A5 from the prodrug in the cancer cells. Similarly, the inhibitory activity of A5 and AK was measured in BCR-ABL-low-expressing MV4–11 cancer cells, as well as noncancerous HEK293T cells (Figs. S1E and F in Supporting information); even after prolonged drug treatment (96 h), both A5 and AK showed significantly weaker activities in MV4–11 cells (GI50 = 0.389 and 17.62 µmol/L, respectively) when compared to K562 cells, suggesting a high selectivity in their targeting of endogenous BCR-ABL kinase. It is worth noting that, even at the highest concentration (300 µmol/L) with prolonged incubation time (96 h), the cell viability of noncancerous HEK293T cells treated with the prodrug AK was still higher than 60% (Fig. S1F), indicating that AK had a relatively good biocompatibility on normal cells.
To confirm that acid-responsive hydrazone bonds in the AK structure could be cleaved in tumor cells to release functional A5, thereby subsequently inhibiting endogenous BCR-ABL kinase activity and its phosphorylation of downstream targets in K562 cells, we examined AK-treated K562 cells by Western blot (WB) analysis (Fig. S1G in Supporting information); results showed that AK could effectively inhibit the phosphorylation of Crkl and STAT5 (two well-known BCR-ABL kinase targets [31]) in a dose-dependent manner, indicating the successful intracellular release of A5 and its ensuing inhibition of endogenous BCR-ABL kinase activity. To further check whether the reversible covalent lysine-targeting SA moiety in A5 that was released from the prodrug AK was still functional in prolonged inhibition of endogenous BCR-ABL kinase activity from live K562 cells, the expression level of phosphorylation of Crkl and STAT5 and cell viability were measured, under "wash-out" conditions [31]. As shown in Figs. S1H and I (Supporting information), after extensive washing steps, AK-treated cells continued to display a sustained inhibition on both Crkl/STAT5 phosphorylation and K562 cell growth. In contrast, cells treated with PPY-A (a noncovalent BCR-ABL kinase inhibitor) lost almost all of its inhibition effects in both assays after "wash-out". Therefore, our results demonstrated that prodrug AK could successfully release the reversible covalent, functional A5 under the highly acidic intracellular environment of tumor cells, leading to subsequent sustained inhibition of endogenous BCR-ABL kinase in live K562 cells.
Next, we evaluated the antitumor effect of the reversible covalent lysine-targeting BCR-ABL kinase inhibitor prodrug AK in K562 tumor-bearing NSG mice in vivo. The lysine-targeting BCR-ABL kinase inhibitor prodrug AK exhibited superior antitumor efficacy in K562 tumor-bearing NSG mice compared to noncovalent control PPY-A and parent drug A5 (Figs. S7A–C in Supporting information). Specifically, AK achieved the highest tumor inhibition rate (~79%), surpassing the PPY-A control group (~50%) and A5 (< 40%). Notably, this in vivo potency order (AK > PPY-A > A5) contrasted with previous in vitro findings (A5 > PPY-A > AK) [31], likely attributable to AK's acid-responsive prodrug strategy. This strategy shields A5′s reactive aldehyde during circulation while enhancing tumor permeability via conjugated FK506 [40]. Consistent with efficacy outcomes, AK treatment significantly reduced p-STAT5-positive cells (AK > PPY-A > A5, Fig. S7D in Supporting information), and hematoxylin and eosin (H&E) staining of tumors exhibited the lowest pathological degree in the AK-treated group (Fig. S7E in Supporting information). All groups maintained stable body weights (Fig. S7F in Supporting information) and showed no organ toxicity or biochemical abnormalities (Figs. S7G and H in Supporting information), confirming favorable biosafety. These results demonstrate AK's dual optimization of A5 through improved stability and tumor-targeted delivery, establishing its potential as a first-in-class reversible covalent inhibitor with enhanced in vivo anti-tumor effect.
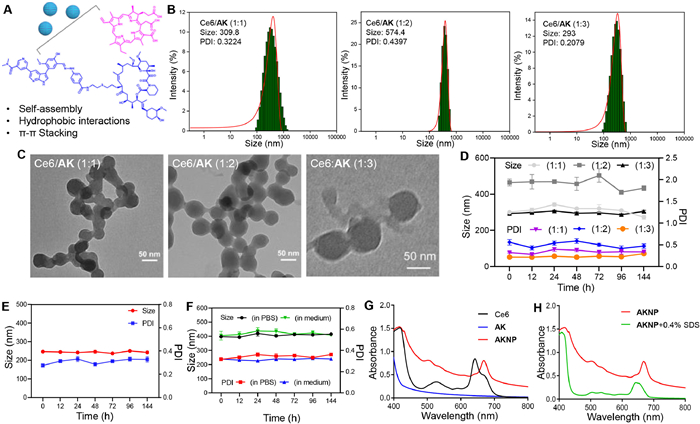
To address these challenges and facilitate clinical transformation, we employed a nanoparticle delivery strategy leveraging the enhanced permeability and retention (EPR) effect [66,67]. Based on Ce6′s well-documented capacity for molecular interactions via hydrophobic forces, π-π stacking, and hydrogen bonding (Fig. 2A), it has been reported that AK's macrocyclic structure with aromatic rings could enable co-assembly with Ce6 into nanoparticles. Through optimized nano-precipitation methods [68–72], we developed spherical nanoparticles (TEM-verified) with variable sizes dependent on Ce6/AK molar ratios (Figs. 2B and C). Systematic screening identified the 1:3 molar ratio formulation as optimal one, demonstrating uniform morphology (~60 nm diameter), stable hydrodynamic size and polydispersity index (PDI) over 4 days in aqueous conditions (Figs. 2D and E), and consistent performance in biological media (cell culture/phosphate-buffered saline (PBS)) (Fig. 2F). This optimized formulation, designated AKNP, exhibited enhanced physicochemical stability compared to other ratios (1:1, 1:2), supporting its selection for subsequent biological evaluation. The strategy synergistically combines molecular self-assembly principles with nanoparticle pharmacokinetic advantages to potentially overcome intrinsic drug limitations.
Figure 2
Figure 2.
Characterizations of AKNP. (A) Schematic illustration of the self-assembly between Ce6 and AK (giving AKNP) through π-π stacking and hydrophobic interactions. (B, C) Particle size histograms and TEM images of nanoparticles prepared at different molar ratios of Ce6 to AK in aqueous solution. (D) The changes of nanoparticles on particle size and PDI in 6 days. Stability test of AKNP incubated in (E) water and (F) PBS or DMEM, for 6 days. (G) Absorbance spectra of Ce6, AK, and AKNP. (H) Absorbance spectra of AKNP with or without treatment with 0.4% SDS. Data are presented as mean ± standard deviation (SD) (n = 3).
To investigate the molecular interactions driving AKNP self-assembly, we analyzed ultraviolet-visible spectroscopy (UV–vis) spectra under different concentrations of NaCl and detergent sodium dodecyl sulfate (SDS) conditions (Figs. 2G and H). AKNP exhibited a red-shifted and broadened Soret band compared to Ce6 alone, confirming π-π interactions and hydrophobic interactions between Ce6′s porphyrin and AK's aromatic groups. SDS exposure induced a spectral blue shift and a notable alteration in the shape of the characteristic absorption peaks in AKNP, further validating hydrophobic forces as critical assembly drivers. Notably, NaCl treatment caused no spectral changes (Fig. S9 in Supporting information), indicating that the electrostatic interactions within AKNP were negligible. These results collectively establish that AKNP self-assembly occurs via synergistic π-π interactions and hydrophobic bonding between Ce6 and AK. The release profiles of AKNP in the microenvironments corresponding to two distinct pH values were characterized using ultraviolet-visible spectroscopy: pH 5.0 (lysosomal environment, 4.5–5.0) and pH 7.4 (physiological pH) (Fig. S22 in Supporting information). At pH 7.4, AKNP exhibited high stability with minimal release (< 20%). In contrast, at an acidic pH of 5.0, AKNP demonstrated a significant release (> 80%). These results indicate that AKNP possesses excellent stability under physiological conditions and exhibits responsive release behavior in acidic environments.
Conventional cancer therapy is often associated with major limitations and severe side effects. A photosensitizer is the key component of PDT that generates cytotoxic reactive oxygen species (ROS) to eradicate cancer cells. Ce6 is a Food and Drug Administration (FDA)-approved second-generation photosensitizer known for its high ROS-generating ability and anticancer potency against many types of cancer [68]. Mechanistically, appropriate light irradiation on Ce6 causes the photosensitizer to generate a large amount of ROS to induce DNA double-strand breaks (DSBs) and initiate cell apoptosis.
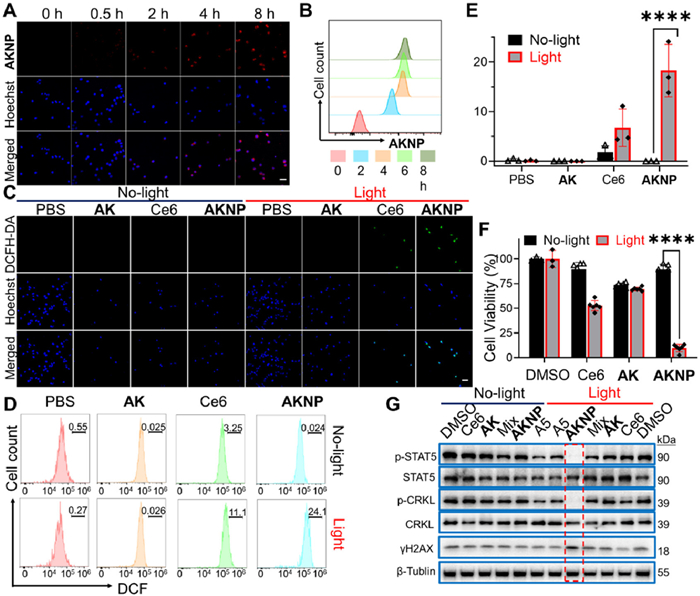
We first assessed the cell uptake behavior of AKNP by subjecting K562 cells to AKNP treatment followed by measurements with confocal laser scanning microscopy (CLSM) and FACS. By using the native fluorescence property of Ce6, we could directly image and quantify the cellular uptake of the nanomedicine (Figs. 3A and B); the red fluorescence intensity of Ce6 in AKNP progressively augmented over time, and after 8-h incubation, the majority of cells exhibited a red fluorescence signal, indicating that the nanomedicine was able to effectively internalized into K562 cells in a time-dependent manner. Congruent results were witnessed by FACS analysis. Our findings thus unequivocally confirmed that AKNP was effectively uptaken in K562 tumor cells. Next, the PDT performance of AKNP-treated K562 cells was evaluated by using 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA), a non-fluorescent probe that is oxidized to fluorescent 2′,7′-dichlorofluorescein (DCF) by ROS. Fluorescence imaging demonstrated minimal background signals in non-irradiated groups (Fig. 3C). Upon exposure to 660 nm light, AKNP-treated cells exhibited significantly stronger DCF fluorescence compared to controls (P < 0.05), indicating enhanced ROS generation. The Ce6 group showed limited ROS production, likely due to its inferior cellular uptake relative to AKNP [68]. FACS analysis (Figs. 3D and E) corroborated these findings, with AKNP displaying the highest DCF fluorescence intensity post-irradiation. These results demonstrate AKNP's superior ROS-generating capacity, which directly correlates with its potential therapeutic efficacy.
Figure 3
Figure 3.
Antitumor activities of AKNP in vitro. (A) CLSM images of AKNP internalization in K562 tumor cells at different time points. Scale bar: 50 µm. (B) FACS analysis of cellular uptake of AKNP in K562 cells. (C) Confocal images of ROS generation in K562 cells after different treatments. Scale bar: 50 µm. (D, E) Quantitative FACS results of ROS generation in K562 cells after different treatments (n = 3). The graph in (E) was plotted from data extrapolated from (D). (F) Cell viability data of Ce6, AK, and AKNP in K562 cells with and without light irradiation (n = 6). (G) WB analysis of AKNP-treated K562 cells, with or without light irradiation. For all light treatments, cells were incubated with the drug for 12 h (for cellular uptake) before 2-min light irradiation (at 660 nm), followed by an additional 12-h cell incubation. ****P < 0.0001. Data are presented as mean ± SD.
Antiproliferative evaluation and mechanism of AKNP in K562 cells MTT assays revealed the antiproliferative effects of AKNP on K562 cells (Fig. 3F and Fig. S10 in Supporting information). Without light irradiation, both Ce6 and AKNP exhibited minimal cytotoxicity (cell viability > 90%), while AK alone caused slight inhibition [12]. Under light exposure, Ce6 induced significant cytotoxicity via photodynamic therapy (PDT), whereas AKNP demonstrated superior tumor growth inhibition (TGI) compared to all controls (Fig. 3F). To elucidate the underlying mechanism, WB analysis showed that light-activated AKNP selectively suppressed the phosphorylation of CRKL and STAT5 (p-CRKL/p-STAT5), downstream targets of BCR-ABL kinase, without altering total CRKL/STAT5 levels (Fig. 3G). This inhibitory effect surpassed that of A5, AK, or Ce6 and AK mixtures under identical conditions. Notably, γH2AX expression—a marker of DNA damage increased markedly in AKNP-treated cells post-irradiation, confirming Ce6-mediated ROS generation and PDT-induced DNA damage (Fig. 3G). These findings indicate that AKNP's antiproliferative activity arises synergistically from three factors: (1) enhanced AK cellular uptake, (2) A5-driven reversible inhibition of BCR-ABL kinase, and (3) Ce6-triggered PDT.
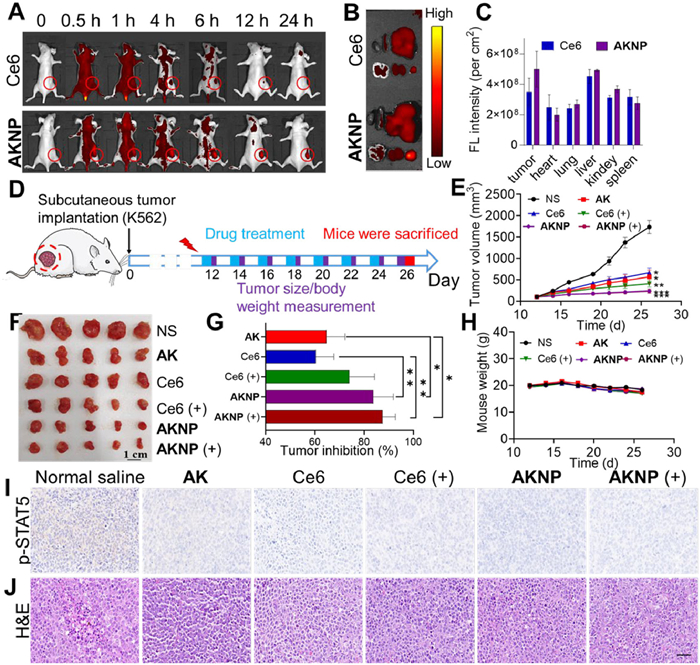
Before an antitumor assessment, we investigated the biodistribution of AKNP in K562 tumor-bearing nude mice using Ce6 as a control (Figs. 4A and B). All the animal studies have been approved by the Animal Experiment Ethics Committee of Guangdong Medical University (approval No. GDMU-2023–000079). Utilizing Ce6′s intrinsic fluorescence for real-time tracking, AKNP exhibited extended tumor retention with sustained fluorescence intensity at 24 h post-injection, contrasting sharply with Ce6-treated mice showing rapid signal decline after 12 h (Fig. 4A and Fig. S11 in Supporting information). Ex vivo imaging at 24 h confirmed superior tumor-specific accumulation of AKNP compared to free Ce6 (Figs. 4B and C). For antitumor evaluation, K562-xenografted NSG mice received treatments with tumor volume and body weight monitored biweekly until sacrifice on day 26 (Fig. 4D).
Figure 4
Figure 4.In vivo property and antitumor effect of AKNP in K562-xenografted mice. (A) Fluorescence imaging and biodistribution of mice treated with Ce6 or AKNP at different time points. (B, C) Fluorescence images of tumor tissues and major organs (1. heart, 2. spleen, 3. liver, 4. brain, 5. kidney, 6. tumor) ex vivo, examined at 24-h post-injection. The graph in C was obtained by quantifying the fluorescence intensity of different tumor tissues/organs from B (n = 3). (D) Schematic diagram of AKNP injection timeline for antitumor experiments (equivalent amount of AK: 6.5 mg/kg). (E) Tumor volumes of K562-bearing mice treated with different formulations (n = 6). (F) Photos of tumors derived from tumor-bearing mice treated with AK, Ce6, and AKNP, with or without light irradiation at the end of antitumor study ex vivo (n = 5). The survival rate of tumor-bearing mice is 100%. (G) Plots of relative TGI values after drug treatments. (H) Body weight changes following drug treatment with or without light irradiation (660 nm) (n = 5). (I, J) p-STAT5 and H&E staining of tumor tissues from drug-treated mice (brown area: p-STAT5-positive cells; blue area: cell nucleus). Scale bar: 50 µm. n.s.: not significant. P < 0.05, **P < 0.01. Data are presented as mean ± SD.
At a dosage of 6.5 mg/kg, the prodrug AK exhibited moderate tumor inhibition (64%), while AKNP achieved superior efficacy (84%) without light irradiation (Figs. 4E–G). The better antitumor efficacy of AKNP (without light) could be attributed to its improved tumor accumulation ability as a nanomedicine. Upon 660 nm light irradiation, AKNP further elevated tumor suppression to 88%, demonstrating synergistic effects of AK-mediated covalent BCR-ABL inhibition and Ce6-driven photodynamic therapy (Figs. 4F and G). Immunohistochemical analysis confirmed significantly reduced p-STAT5 in AKNP light-treated tumors (Fig. 4I), aligning with tumor regression trends. Safety evaluations revealed no significant weight loss (Fig. 4H) or pathological abnormalities in major organs (Fig. 4J and Fig. S12 in Supporting information). Serum biochemistry parameters remained within normal ranges (Fig. S12), confirming minimal systemic toxicity and blood compatibility.
In addition to preventing potential AK depletion in peripheral circulation in vivo, AKNP was shown to improve the physiological stability of AK, prolong its tumor accumulation and retention time, and achieve on-demand release of A5 at the tumor sites. Our study thus presents a promising nanomedicine-based, prodrug self-assembly strategy that combines the on-demand release of a novel lysine-targeting, reversible covalent kinase inhibitor with PDT.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This study was supported by the grants from National Key R & D Program of China (No. 2022YFA1104800), Shenzhen Science and Technology Program (No. JCYJ20210324124214038), National Natural Science Foundation of China (Nos. 52072418, 82300016), Natural Science Foundation of Guangdong Province (No. 2023A1515140072), Shenzhen Key Laboratory of Neural Cell Reprogramming and Drug Research, Social Development Science and Technology Key Project of Dongguan (No. 20231800940512), and the National Medical Research Council (NMRC, No. 23–0740-A0001), the Ministry of Education (MOE, No. T2EP10222–0002) of Singapore.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111423.
[1]
L. Boike, N.J. Henning, D.K. Nomura, Nat. Rev. Drug Discov. 21 (2022) 881–898. doi: 10.1038/s41573-022-00542-z
Figure 1
Schematic diagram of pH-responsive on-demand prodrug release strategy of AKNP. (A) Structures of previously reported BCR-ABL inhibitors including PPY-A and A5. (B) Chemical structure of prodrug AK and pH-responsive strategy for on-demand release by using acid-labile hydrazone to bind the aldehyde group in A5. (C) The design strategy of AKNP is based on the combination of prodrug AK and photosensitizer Ce6. After tail vein injection of AKNP, these nanoparticles can effectively accumulate at the tumor site. Upon cellular uptake, AKNP could produce ROS with light irradiation, leading to DNA damage. Meanwhile, the released A5 reversible-covalently inhibits BCR-ABL kinase, further inhibiting tumor growth.
Figure 2
Characterizations of AKNP. (A) Schematic illustration of the self-assembly between Ce6 and AK (giving AKNP) through π-π stacking and hydrophobic interactions. (B, C) Particle size histograms and TEM images of nanoparticles prepared at different molar ratios of Ce6 to AK in aqueous solution. (D) The changes of nanoparticles on particle size and PDI in 6 days. Stability test of AKNP incubated in (E) water and (F) PBS or DMEM, for 6 days. (G) Absorbance spectra of Ce6, AK, and AKNP. (H) Absorbance spectra of AKNP with or without treatment with 0.4% SDS. Data are presented as mean ± standard deviation (SD) (n = 3).
Figure 3
Antitumor activities of AKNP in vitro. (A) CLSM images of AKNP internalization in K562 tumor cells at different time points. Scale bar: 50 µm. (B) FACS analysis of cellular uptake of AKNP in K562 cells. (C) Confocal images of ROS generation in K562 cells after different treatments. Scale bar: 50 µm. (D, E) Quantitative FACS results of ROS generation in K562 cells after different treatments (n = 3). The graph in (E) was plotted from data extrapolated from (D). (F) Cell viability data of Ce6, AK, and AKNP in K562 cells with and without light irradiation (n = 6). (G) WB analysis of AKNP-treated K562 cells, with or without light irradiation. For all light treatments, cells were incubated with the drug for 12 h (for cellular uptake) before 2-min light irradiation (at 660 nm), followed by an additional 12-h cell incubation. ****P < 0.0001. Data are presented as mean ± SD.
Figure 4In vivo property and antitumor effect of AKNP in K562-xenografted mice. (A) Fluorescence imaging and biodistribution of mice treated with Ce6 or AKNP at different time points. (B, C) Fluorescence images of tumor tissues and major organs (1. heart, 2. spleen, 3. liver, 4. brain, 5. kidney, 6. tumor) ex vivo, examined at 24-h post-injection. The graph in C was obtained by quantifying the fluorescence intensity of different tumor tissues/organs from B (n = 3). (D) Schematic diagram of AKNP injection timeline for antitumor experiments (equivalent amount of AK: 6.5 mg/kg). (E) Tumor volumes of K562-bearing mice treated with different formulations (n = 6). (F) Photos of tumors derived from tumor-bearing mice treated with AK, Ce6, and AKNP, with or without light irradiation at the end of antitumor study ex vivo (n = 5). The survival rate of tumor-bearing mice is 100%. (G) Plots of relative TGI values after drug treatments. (H) Body weight changes following drug treatment with or without light irradiation (660 nm) (n = 5). (I, J) p-STAT5 and H&E staining of tumor tissues from drug-treated mice (brown area: p-STAT5-positive cells; blue area: cell nucleus). Scale bar: 50 µm. n.s.: not significant. P < 0.05, **P < 0.01. Data are presented as mean ± SD.

 DownLoad:
DownLoad:

 下载:
下载:





