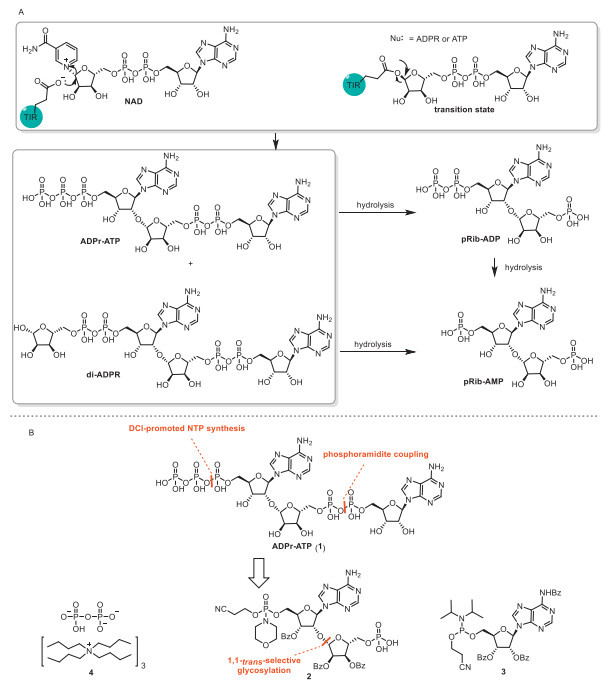
Figure 1.
(A) TIR-catalyzed synthesis and hydrolysis, four important immune molecules. (B) Retrosynthetic analysis of ADPr-ATP.

Nucleoside(tide) is an important class of biomolecule composed of base and sugar, which has wide applications in various fields such as medicine, agriculture, and scientific research [1]. Nucleoside modification is a common practice in the development of antiviral and anti-tumor drug [2]. Appropriate structural changes may generate promising nucleosides for the discovery of new drugs. In addition, nucleotides could promote plant growth, improve crop disease and stress resistance, thereby increasing crop yield and quality. With the continuous deepening of research on nucleoside, the scope of their applications is steadily expanding. In the foreseeable future, nucleosides are anticipated to confer great advantages to human health and significantly contribute to the social development [3].
ADPr-ATP/di-ADPR are universal natural immune activating molecules to resist plant diseases [4]. As nucleotide second messenger molecules, they are produced from NAD+ or NAD+ with ATP mediated by activated TIRs and TNLs in response to cell death, and identified through Cryo-Electron Microscopy analysis of the complex of EDS1-SAG101 and ADPr-ATP in 2022 (Fig. 1A). The pRib-ADP/AMP nucleoside small molecules arise from hydrolysis of ADPr-ATP or di-ADPR and could trigger the ADR1-mediated immune signaling pathway [5,6] via binding and activating EDS1-PAD4. The small molecule pRib-AMP is capable of mobilizing multiple proteins to form immune complexes in rice, thereby activating the immune system and improving the broad-spectrum disease resistance of crops [7,8]. Therefore, the synthesis of ADPr-ATP could provide a basis for analyzing the EDS1-SAG101-NRG1 signaling pathway and serve as a template for developing novel biological disease resistant small molecules.
Up to now, the synthesis of monophosphate [9], diphosphate [10], triphosphate [11], and ADP-ribose and their polymers [12,13] has been established. However, the separation of these compounds is challenging and commonly employs the techniques of multiple reverse chromatography and ion exchange purifications, resulting in low isolated yield [14]. Although the significant application prospects have been envisioned, there are no reports on the chemical or enzymatic synthesis of ADPr-ATP, probably due to the formidable purification resulting from chemical instability, including hydrolysis of diphosphate and triphosphate. As part of our ongoing efforts to design and synthesize nucleotide small molecules [4,5,11], we have been focusing on synthesizing ADPr-ATP, containing diphosphate diester internucleotide linker and triphosphate. Here, we report the first chemical total synthesis of ADPr-ATP in 14 steps with 6.4% overall yield. The structures of all separated compounds were confirmed by NMR and HRMS.
For the structural novelty of ADPr-ATP compared to reported ADP-ribose dimer, several difficulties have to be resolved prior to its total synthesis. (1) There is the issue of β-linked 2′-O-ribosyladenosine. Selective glycosylation and gram scale synthesis are significant challenges due to the abundance of hydroxyl groups [15,16]. (2) The construction of adenosine 5′-hydroxytriphosphate and ribose 5′′–hydroxyl pyrophosphates is challenging in the synthesis of nucleosides. While synthetic methods for triphosphate [17-19] and pyrophosphate [20,21] are still in wide use, the lack of two functional groups related to their orthogonality and efficiency has motivated us to develop a suitable route. We conducted retrosynthetic analysis on ADPr-ATP (1), which involves a phosphoramidite coupling of compound 2 with compound (3), then subsequent 4, 5-dicyanoimidazole (DCI)-promoted NTP synthesis in the absence of tris(tetrabutylammonium) hydrogen pyrophosphate (4) respectively (Fig. 1B) [22].
The natural immune small molecule pRib-AMP, as the hydrolysis product of ADPr-ATP with the simplest structure, plays a crucial role in plant immune regulation. Various natural compounds of nucleotides participate in the regulation of various physiological functions in vivo of animals, for instance, the nucleotide secondary messenger cAMP was involved in metabolism, antiviral activity, gene transcription, and other processes [23-26]. We performed protide modification strategy on pRib-AMP to mask polar groups, reduce molecular polarity, and increase membrane permeability, aiming to test the anti-influenza activity of the modified compound.
To construct β-linked 2′-O-ribosyladenosine (9), selective glycosylation is a crucial step. In order to achieve specific reaction at the 2′-position of N-benzoyladenosine (5), the 3′–hydroxy and 5′–hydroxy groups were selectively protected with 1, 3-dichlorotetraisopropyldisiloxane, giving rise to a single compound (6). Vorbrüggen glycosylation of 3′, 5′-protected adenosine derivative (6) and 1-O-acetyl-2, 3, 5-tri-O-benzoyl-β-D-ribofuranose (7) was carried out in the presence of SnCl4, followed by deprotection to give target compound (9) in 57% overall yield. 5′′-Hydroxy and 2′′, 3′′-dihydroxy were protected with DMTr and Bz in sequential order, and then DMTr-group was removed easily with p-toluenesulfonic acid, affordingcompound (11) in 81% yield. The phosphoramidite coupling with (i-Pr)2NP(OBn)2 in the DCI-activated condition and oxidation resulted in compound (13) in 96% yield (Scheme 1 and Scheme 2).
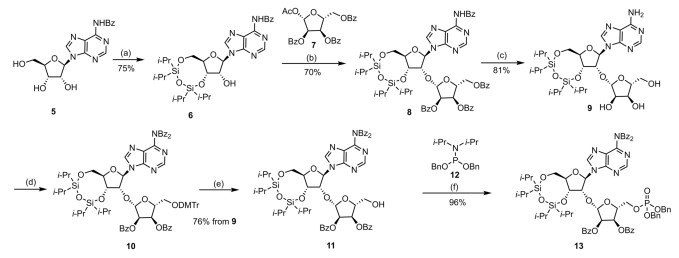
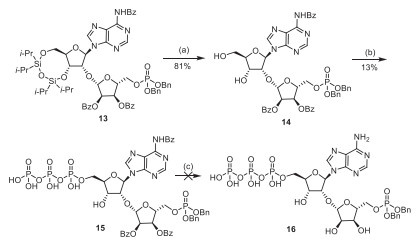
To introduce triphosphate at the desired 5′-position, we initiated a model reaction. After removing the 3′, 5′-silyl groups to obtain compound (14), triphosphate was successfully introduced using common triphosphate synthesis method [11]. The synthesis of triphosphates (15) consists of two steps, with an overall yield of merely 13%. During the first-step reaction for the formation of dichlorophosphate, the generation of N-benzoyladenosine (5) was detected. We hypothesize that the employment of phosphorus oxychloride might cleave the glycosidic bond between the two sugars. Due to the unstable chemical properties of triphosphate, decomposition was observed during removing of Bz group. We also attempted the DCI-promoted P(V)−N activation strategy, which split the synthesis of triphosphate into two steps. It is found that the protected adenosine 5′-phosphoromorpholidate (19) formed in the first step hardly affect the synthesis of pyrophosphate upon the removal of Bz protecting groups.
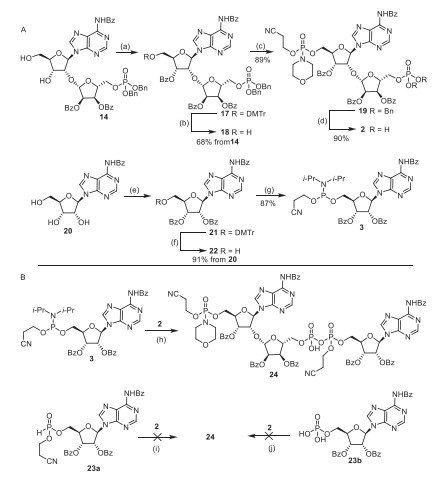
The alcohols (18 and 22) were synthesized with similar procedure to the case of compound (11). Three consecutive steps from compound (18) and 2-cyanoethyl-N, N-diisopropylamidochlorophosphite can furnish intermediate (19), which can be directly used without further separation and purification, thus resulting in excellent yields evenscaled up to gram scale. Removing benzyl protecting group can give pyrophosphate receptor (2). H-phosphonate coupling and the ZnCl2-catalyzed two monophosphate condensation methods reported were unsuccessful, leading us to synthesize N, N-diisopropylchlorophosphoramidite (3) as the building block in 80% overall yield (Scheme 3A). Phosphate (2) and amidite (3) were employed for the synthesis of pyrophosphate (24) (Scheme 3B).
The treatment of compound (24) with saturated ammoniated methanol solution overnight can successfully remove the Bz group as well as the 2-cyanoethyl group, affording compound (25) in 60% yield. Subsequently, 6 equiv. of DCI and 2 equiv. of tris(tetra-n-butylammonium)hydrogen pyrophosphate (4) were added, converting the phosphate propionate intermediate into the target product (1) in 79% yield within 6 h at 20 ℃ (Scheme 4A).
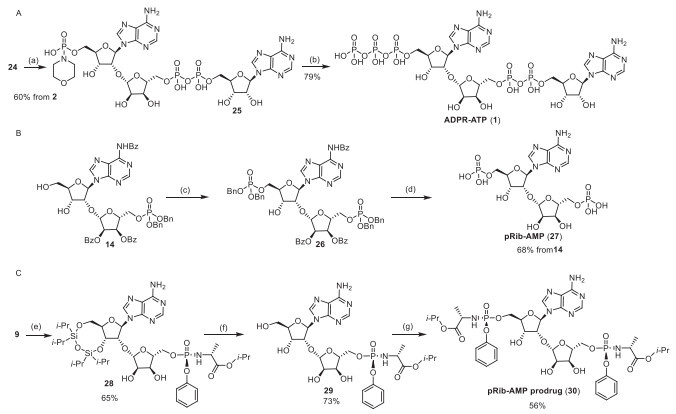
By using intermediate (14) as the starting material, compound (27) could be obtained through introduction of phosphoric acid into 5′–hydroxy and removal of all protective groups. Although the synthesis of pRib-AMP (27) has been reported [5], no NMR data is available for this compound (Scheme 4B). Consequently, we performed full characterization of pRib-AMP (27), including NMR and HRMS.
Due to the high polarity of pRib-AMP, it is difficult to cross cell membranes, resulting in poor bioavailability. To address the issue of druggability, the approach of masking polar groups is employed to reduce molecular polarity and enhance membrane permeability. Once the pRib-AMP prodrug is absorbed, it is hydrolyzed by specific enzymes, releasing the parent drug, pRib-AMP.
A chiral phosphorus amide esterification reagent was used to react with compound (9) to give compound (28), which underwent a stereoselective nucleophilic substitution reaction. The single diastereomer was efficiently prepared in gram scale. After removing 3′, 5′-protecting group, the target compound pRib-AMP prodrug (30) was obtained by introducing phosphoramide ester into 5′-OH again using the above method (Scheme 4C).
We assessed the cytotoxicity of compound (30) to determine optimal dosing regimens for follow-up in vitro testing. Due to the fact that the main target cells of influenza virus are the lungs, considering the feasibility of the experiment, we firstly employed lung cancer cells A549 for cytotoxicity testing. Initial screening revealed that the compound exhibited low cytotoxicity (IC₅₀ > 4000 µmol/L) in A549 cells under the evaluated dose range (0–4000 µmol/L) (Fig. 2A and Table S1 in Supporting information), and the maximum safe concentration could reach 500 µmol/L. Subsequent dose-response experiments therefore employed graded concentrations (1, 25, 50, 100, 200, 400 µmol/L), revealing no significant antiproliferative effects up to 400 µmol/L exposure (Fig. 2B). The results showed that there was no significant difference in the survival rate of A549 cells at our administration concentration (400 µmol/L), compared to the blank control (P > 0.05, Fig. 2B and Table S2 in Supporting information), which strongly indicates the low toxicity of compound 30 to cells, revealing no significant antiproliferative effects up to 400 µmol/L exposure (Fig. 2B). We further tested the cytotoxicity of 30 on renal epithelial cells MDCK. The results showed that at a concentration of 500 µmol/L of 30 (slightly higher than our administration concentration of 400 µmol/L), the cell survival rate remained above 85%, further demonstrating the low toxicity of 30 (Fig. 2C and Table S3 in Supporting information).
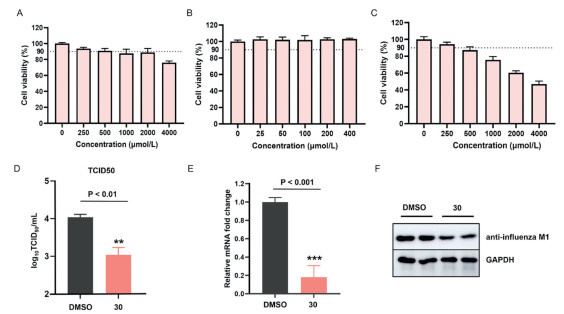
In the in vitro antiviral assay against influenza A virus, pRib-AMP prodrug (30) exhibited significant inhibition of viral infectivity. Quantitative analysis revealed a 10-fold reduction in viral titers compared to DMSO-treated controls, as measured in culture supernatants at 48 h post-infection. The Log10TCDI50/mL of 30 was 3.042 ± 0.156, whichindicates the TCID50 value of the culture medium supernatant was on average about 10-fold lower than that of the DMSO control group (Fig. 2D and Table S4 in Supporting information).
M1 protein is the most abundant protein located under the viral envelope in influenza virus. It plays a crucial role in connecting the viral envelope and nucleocapsid, and can affect the morphology and configuration of the virus. We have also determined qPCR experiments for quantitative analysis of viral RNA replication. The qPCR results showed that compound (30) could inhibit the replication of viral RNA by over 70% (Fig. 2E and Table S5 in Supporting information), further demonstrating its potent antiviral activity. Western blot analysis revealed a marked decrease in M1 protein expression levels in the compound-treated group (Fig. 2F), indicating that 30 effectively inhibits the replication of influenza A virus through its antiviral activity.
In conclusion, we have developed the first chemical total synthesis of natural ADPr-ATP (1), and conducted structural determination through full characterization. This work discloses an alternative pathway to obtain ADPr-ATP besides enzymatic synthesis in vivo, and provides a foundation for the synthesis of similar compounds. Remarkably, we applied selective glycosylation to construct β-linked 2′-O-ribosyladenosine skeleton (8), phosphine(Ⅲ) reagent to construct pyrophosphate, and two-step strategy to construct triphosphate, respectively. In addition, this route also utilizes selective modification methods for all positions (1′-, 2′-, 3′-, 5′-) on the sugar ring, which provides a paradigm for subsequent nucleoside synthesis. Natural nucleoside small molecules have immunomodulatory and antiviral effects in animals and plants. The modified pRib-AMP prodrug molecule (30) was used for preliminary activity study against influenza virus, and inhibitory effect on influenza virus activity was observed with low cytotoxicity. We envision that this compound will find potential inhibitors for a new generation of influenza virus.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Tiantian Zhang: Data curation. Hanbo Liu: Data curation. Junbiao Chang: Supervision, Funding acquisition, Conceptualization. Yonggang Meng: Writing – review & editing, Writing – original draft, Funding acquisition, Data curation.
This work was supported by the National Natural Science Foundation of China (Nos. 82204209 and 82130103), Natural Science Foundation of Henna Province (No. 242300421084). We are grateful to Xianfeng Hui and Hui Wang from Xinxiang Medical University for evaluation of the antiviral activity of pRib-AMP prodrug.
Supplementary material associated with this article can be found, in the online version, at doi:
H. Yao, J. Qin, Y. Wang, et al., Chin. Chem. Lett. 35 (2024) 109154. doi: 10.1016/j.cclet.2023.109154
M. Meanwell, S.M. Silverman, J. Lehmann, et al., Science 369 (2020) 725–730. doi: 10.1126/science.abb3231
L. Liu, R. Chen, G. Xue, et al., Chin. Chem. Lett. 35 (2024) 108455. doi: 10.1016/j.cclet.2023.108455
A. Jia, S. Huang, W. Song, et al., Science 377 (2022) eabq8180. doi: 10.1126/science.abq8180
S. Huang, A. Jia, W. Song, et al., Science 377 (2022) eabq3297. doi: 10.1126/science.abq3297
K. Essuman, J. Milbrandt, J.L. Dangl, et al., Science 377 (2022) eabo0001. doi: 10.1126/science.abo0001
Y. Wu, W. Xu, G. Zhao, et al., Science 386 (2024) 1405–1412. doi: 10.1126/science.adr2138
H. Yu, W. Xu, S. Chen, et al., Science 386 (2024) 1413–1420. doi: 10.1126/science.adr3150
E.V. Efimtseva, A.A. Shelkunova, S.N. Mikhailov, et al., Nucleos. Nucleot. Nucl. 22 (2003) 1109–1111. doi: 10.1081/NCN-120022748
Y. Ahmadibeni, K. Parang, Angew. Chem. Int. Ed. 56 (2007) 4739–4743. doi: 10.1002/anie.200605029
L. Liang, Y. Meng, X. Chang, et al., J. Med. Chem. 68 (2025) 68 1994–2007. doi: 10.1021/acs.jmedchem.4c02769
H.A.V. Kistemaker, L.N. Lameijer, N.J. Meeuwenoord, et al., Angew. Chem. Int. Ed. 54 (2015) 4915–4918. doi: 10.1002/anie.201412283
M.J. Lambrecht, M. Brichacek, E. Barkauskaite, et al., J. Am. Chem. Soc. 137 (2015) 3558–3564. doi: 10.1021/ja512528p
E.S. Tan, K.A. Krukenberg, T.J. Mitchison, Anal. Biochem. 428 (2012) 126–136. doi: 10.1016/j.ab.2012.06.015
G.J. van der Heden van Noort, H.S. Overkleeft, G.A. van der Marel, et al., Org. Lett. 13 (2011) 2920–2923. doi: 10.1021/ol200971z
S.N. Mikhailov, I.V. Kulikova, K. Nauwelaerts, Tetrahedron 64 (2008) 2871–2876. doi: 10.1016/j.tet.2008.01.028
J. Ludwig, Acta Biochim. Biophys. Acad. Sci. Hung. 16 (1981) 131–133.
J. Ludwig, F. Eckstein, J. Org. Chem. 54 (1989) 631–635. doi: 10.1021/jo00264a024
Y. Feng, J. Yang, C. Cai, et al., Chin. Chem. Lett. 33 (2022) 4878–4881. doi: 10.1016/j.cclet.2022.02.071
G.K. Wagner, T. Pesnot, R.A. Field, Nat. Prod. Rep. 26 (2009) 1172–1194. doi: 10.1039/b909621n
Y. Ahmadibeni, K. Parang, Org. Lett. 7 (2005) 5589–5592. doi: 10.1021/ol0521432
Q. Sun, S. Gong, J. Sun, J. Org. Chem. 79 (2013) 8417–8426. doi: 10.1021/jo4011156
G. Ofir, E. Herbst, M. Baroz, et al., Nature 600 (2021) 116–120. doi: 10.1038/s41586-021-04098-7
A. Bernheim, A. Millman, G. Ofir, et al., Nature 589 (2021) 120–124. doi: 10.1038/s41586-020-2762-2
Q. Ye, R.K. Lau, I.T. Mathews, et al., Mol. Cell 77 (2020) 709–722. doi: 10.1016/j.molcel.2019.12.009
D. Cohen, S. Melamed, A. Millman, et al., Nature 574 (2019) 691–695. doi: 10.1038/s41586-019-1605-5

Figure 1 (A) TIR-catalyzed synthesis and hydrolysis, four important immune molecules. (B) Retrosynthetic analysis of ADPr-ATP.

Scheme 1 Synthesis of compound 13. Reagents and conditions: (a) TIDPSCl2, pyridine, r.t., overnight, 75%; (b) 1-O-acetyl-2, 3, 5-tri-O-benzoyl-β-D-ribofuranose, SnCl4, DCE, r.t., 30 h, 70%; (c) NH3/MeOH, r.t., 24 h, 81%; (d) DMTrCl, pyridine, r.t., 2 h; then BzCl, DMAP, Et3N, DCM, r.t., 5 h; (e) p-TsOH·H2O, CHCl3/MeOH, −20 ℃ to 0 ℃, 5 min, two-step yield 76%; (f) dibenzyl diisopropylphosphoramidite, DCI, t-BuOOH, DCM/MeCN, r.t., 5 h, 96%.

Scheme 2 Exploration of the synthesis of compound 16. Reagents and conditions: (a) TBAF, THF, r.t., 10 min, 81%; (b) POCl3, proton sponge, PO(OMe)3, −10 ℃ to 15 ℃; and then (n-Bu3NH)2H2P2O4, PO(OMe)3, −10 ℃ to 15 ℃, 13%; (c) NH3/MeOH, r.t., 8 h.

Scheme 3 (A) Synthesis of compounds 2 and 3. (B) Exploration to synthesize compound 24. Reagents and conditions: (a) DMTrCl, pyridine, r.t., 2 h; and then BzCl, DMAP, Et3N, DCM, r.t., 1 h; (b) p-TsOH·H2O, CHCl3/MeOH, r.t., 10 min, two-step yield 68%; (c) 2-Cyanoethyl-N, N-diisopropylamidochlorophosphite, Et3N, DCM, r.t., 30 min; and then tetrazole, H2O, DCM, r.t., 10 min; and then morpholine, Et3N, CCl4, DCM, 0 ℃, 30 min, 89%; (d) Pd/C, H2, t-BuOH/H2O, (e) DMTrCl, pyridine, r.t., 2 h; and then BzCl, DMAP, Et3N, DCM, r.t., 5 h; (f) p-TsOH·H2O, CHCl3/MeOH, r.t., 10 min, two-step yield 91%; (g) 2-cyanoethyl-N, N-diisopropylamidochlorophosphite, DIPEA, THF, −78 ℃-r.t., 2 h, 87%. (h) DCI, 2, t-BuOOH, DCM/MeCN, r.t., 5 h; (i) N-chlorosuccinimide, DIPEA, CH3CN; (j) carbonyldiimidazole, Et3N, Pyrdine, 2 h; and then 2, ZnCl2, DMF.

Scheme 4 (A) Synthesis of ADPR-ATP. (B) Synthesis of pRib-AMP. (C) Synthesis of pRib-AMP prodrug. Reagents and conditions: (a) NH3/MeOH, r.t., 24 h, 60% from compound 2. (b) Tris(tetrabutylammonium)hydrogen pyrophosphate, DCI, DMSO, r.t., 16 h, 79%. (c) Dibenzyl diisopropy-lphosphoramidite, DCI, t-BuOOH, DCM/MeCN, r.t., 5 h. (d) Pd/C, H2, t-BuOH/H2O. (e) Isopropyl ((S)-(perfluorophenoxy)(phenoxy)phosphoryl)-L-alaninate, t-BuMgCl, THF, r.t., 65%. (f) TBAF, THF, −60 ℃, 73%. (g) Isopropyl ((S)-(perfluorophenoxy)(phenoxy)phosphoryl)-L-alaninate, t-BuMgCl, THF, r.t., 56%.

Figure 2 Toxicity testing of compound 30 in (A) A549 cells, (B) at different concentrations, and (C) in MDCK cells. (D) The inhibitory effect of compound 30 on the infectivity of influenza A virus (n = 3). (E) The qPCR results of viral RNA replication. (F) The effect of compound treatment on the protein content of H1N1 virus M1.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载: