
Figure 1.
SEM images of (a) Co3O4, (b) g-C3N4, and (c) Co3O4/g-C3N4. TEM images of (d) Co3O4, (e) g-C3N4, and (f) Co3O4/g-C3N4. (g) HRTEM image and (h) elemental mapping of Co3O4/g-C3N4.
Chlorinated antibiotics electrocatalytic dechlorination by construction of Co3O4/g-C3N4 heterojunctions for stable atomic hydrogen production
Ge Song , Huizhong Wu , Chaohui Zhang , Xuechun Wang , Shuaishuai Li , Jiangli Sun , Xiuwu Zhang , Minghua Zhou

A variety of chlorinated antibiotics, including thiamphenicol (TAP), chloramphenicol (CAP), and florfenicol (FLO), serve as broad-spectrum antimicrobial agents that have been overused [1]. However, the high bond energy and toxicity of C-Cl bonds pose great challenges to their removal through biodegradation, advanced oxidation processes, photocatalysis, and other methods [2]. In contrast, electrocatalytic dechlorination, which offers high efficiency, simplicity of operation, and absence of secondary pollution, has garnered widespread research interest [3]. In addition to the direct transfer of electrons, the generation of atomic hydrogen (H*) as a reducing agent to attack C-Cl bonds enables dechlorination and detoxification in electrocatalytic dechlorination, which is more efficient [4]. The ability of catalysts to produce H* is a key factor in determining the dechlorination efficiency [5]. Although palladium (Pd)-based catalysts are highly effective, their high-cost limits practical applications. Therefore, the development of non-noble metal catalysts is of paramount importance.
Studies have shown that Co3O4 can produce H* in electrochemical nitrate reduction, which has the advantages of cheap price and good catalytic performance [6, 7]. However, the stability of the Co3O4 needs to be improved due to the risk of cobalt ion leaching into the solution, leading to a secondary pollution. Strengthening the stability through metal-support interactions is an effective strategy, and the choice of support is crucial [8]. Zhang et al. demonstrated that Pd particles supported on reduced graphene oxide exhibited smaller volumes and larger active surface areas, endowing them with enhanced electrocatalytic activity [9]. Additionally, semiconductors with appropriate band structures can form heterojunctions with metals, facilitating the spontaneous transfer of electrons across the interface with a more negative Fermi level [10, 11]. Jiang et al. applied this principle in the modification of Pd-based catalysts, constructing a Pd-polymer carbon nitride (PCN) heterojunction, which led to an enhancement in performance [12]. However, the mechanism of dechlorination facilitated by heterojunctions requires further investigation. To this end, constructing a heterojunction by combining the readily available and cost-effective Co3O4 with the support not only enhances catalyst stability but also allows for the modulation of the electronic structure.
Furthermore, graphitic carbon nitride (g-C3N4) is renowned for its exceptional chemical and thermal stability, coupled with a low cost, non-toxic nature, and an abundance of reactive sites that enhance its catalytic performance. The tunability of its bandgap endows it with broad applicability across various domains, including energy conversion, environmental remediation, and catalysis. Beyond its role as a catalyst support, the distinctive electronic structure of g-C3N4 allows for its integration with semiconductors featuring larger bandgaps to fabricate heterojunction composites. Such composites offer a promising strategy for the enhancement of charge separation and the expansion of surface area, which are critical for improving the efficiency of catalytic processes. Co3O4, as a transition metal oxide semiconductor, exhibits excellent band-edge alignment with g-C3N4, making the coupling of Co3O4 and g-C3N4 an optimal choice for constructing heterojunctions to enhance catalytic activity. Moreover, both g-C3N4 and Co3O4 possess the advantages of widely available precursors and ease of preparation. In recent years, Co3O4/g-C3N4 heterojunctions have been extensively studied for applications in electrochemical sensing of environmental phenolic hormones, supercapacitors, photocatalytic treatment of wastewater, and photocatalytic water splitting for hydrogen production [13]. However, it has not yet been applied to electrocatalytic dechlorination.
Therefore, this work was aimed to investigate the impact of the Co3O4/g-C3N4 heterojunction on dechlorination performance and rate, as well as to elucidate the underlying mechanisms and assess the stability of the system for potential applications in electrocatalytic dechlorination processes. The morphology and composition of the catalysts were characterized using scanning electron microscope (SEM), transmission electron microscopy (TEM), and X-ray diffractometer (XRD), while the formation of heterojunctions was confirmed through X-ray photoelectron spectroscopy (XPS), photoluminescence spectroscopy (PL), and UV–visible diffuse reflectance spectra (UV–vis DRS). The dechlorination performance of the heterojunctions towards chlorinated organic compounds was validated, and the generation of H*, electron transfer within the heterojunctions, and adsorption of pollutants and degradation intermediates were discussed using a combination of experiments and density functional theory (DFT) calculations. Furthermore, the potential applications of Co3O4/g-C3N4 in removal of typical chloramphenicol antibiotics from aquatic environments were explored.
The morphologies of Co3O4, g-C3N4, and Co3O4/g-C3N4 were investigated to verify the formation of Co3O4/g-C3N4 heterojunctions. The SEM images revealed that Co3O4 exhibited aggregated nanoparticle structures (Fig. 1a), while g-C3N4 displayed a layered structure (Fig. 1b). The surface of Co3O4/g-C3N4 was rough, with Co3O4 nanoparticles growing on the nanosheets of g-C3N4 (Fig. 1c). Figs. 1d-f were TEM images, illustrating the aggregation of Co3O4 nanoparticles (Fig. 1d) and the porous structure of g-C3N4 nanosheets (Fig. 1e), while Fig. 1f showed the loading of Co3O4 nanoparticles on the porous sheet structure, which proved the formation of the Co3O4 and g-C3N4 composite. From the HRTEM analysis (Fig. 1g), clear heterointerface was observed. The lattice spacing of 0.244 nm and 0.286 nm was observed on Co3O4/g-C3N4, which corresponded to the (311) and (220) plane of Co3O4. The elemental mapping of Co3O4/g-C3N4 in Fig. 1h demonstrated the uniform distribution of Co, O, C, and N, confirming the successful synthesis of the composite material.
The catalysts structures were characterized by XRD, FTIR, and XPS. The XRD pattern of g-C3N4 displayed two characteristic peaks (Fig. 2a), where the weak peak at 12.9° was ascribed to the regular alignment of triazine units (100), and the intense peak at 27.3° corresponded to the assembly of conjugated aromatic structures (002) [14]. Pure Co3O4 exhibited diffraction peaks at 19.00°, 31.27°, 36.84°, 38.54°, 44.80°, 55.65°, 59.35°, and 65.22°, corresponding to the (111), (220), (311), (222), (400), (422), (511), and (440) crystal planes, respectively (JCPDS No. 78–1969). The absence of characteristic peaks related to metallic Co, CoO, or Co2O3 in the XRD pattern indicated the synthesis of high-purity Co3O4. The XRD pattern of Co3O4/g-C3N4 displayed a peak at 27.3° corresponding to the (002) plane of g-C3N4 and a peak at 36.84° corresponding to the (311) plane of Co3O4, confirming the distribution of Co3O4 throughout the g-C3N4 matrix [15].
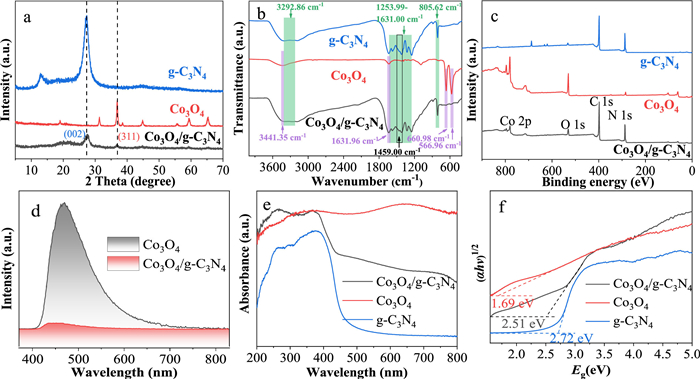
The FTIR spectra of the three materials are depicted in Fig. 2b. The peak at 566.96 cm-1 in the FTIR spectrum of Co3O4 corresponded to the Co-O stretching vibration, while the peak at 660.98 cm-1 was attributed to the bridging vibrations of O-Co-O. The peaks observed at 3441.35 cm-1 and 1631.96 cm-1 were indicative of the O-H stretching and bending vibrations associated with water molecules adsorbed on the oxide surface [16]. The FTIR spectrum of g-C3N4 indicated a sharp peak at 805.62 cm-1 that corresponded to the vibration of the tris-s-triazine structure. The characteristic stretching vibrations of aromatic carbon and C-N heterocyclic units, which were typical features, were represented by multiple peaks in the region of 1253.99 1631.00 cm-1 [17]. The broad peak at 3292.86 cm-1 was attributed to the stretching vibrations of N-H bonds. In the FTIR spectrum of Co3O4/g-C3N4, in addition to the distinct characteristic peaks of g-C3N4, the subtle peaks observed in the 500–700 cm-1 region also indicated the stretching vibrations of Co-O and O-Co-O bonds. In particular, compared to Co3O4 and g-C3N4, a new peak appeared at 1459.00 and was attributed to Co-O-C [18]. These peaks provided evidence of the successful incorporation of Co3O4 into the g-C3N4 matrix, which was crucial for the formation of the heterojunction.
Fig. 2c is the XPS survey spectra of g-C3N4, Co3O4, and Co3O4/g-C3N4. The primary constituents of g-C3N4 were C and N, while Co3O4 was composed of Co and O. The presence of Co 2p, O 1s, C 1s, and N 1s signals in the Co3O4/g-C3N4 confirmed the successful fabrication of the Co3O4/g-C3N4 heterojunction. The high-resolution XPS spectrum of Co 2p for Co3O4/g-C3N4 in Fig. S1a (Supporting information) revealed four peaks at 779.2, 780.6, 794.2, and 795.9 eV, which were attributed to Co3+ 2p3/2, Co2+ 2p3/2, Co3+ 2p1/2, and Co2+ 2p1/2, respectively, along with their corresponding satellite peaks. Specifically, the Co 2p peaks of Co3O4/g-C3N4 exhibited a negative shift relative to those of Co3O4 alone, indicating the presence of electronic interactions between Co3O4 and g-C3N4 [19]. In the high-resolution O 1s XPS spectrum of Co3O4 (Fig. S1b in Supporting information), the peaks with binding energies of 530.1 and 531.9 eV correspond to Co-O-Co and O-H caused by surface adsorption of water, respectively [20]. The emergence of a Co-O-C peak in the O 1s spectrum of Co3O4/g-C3N4, along with a shift to lower binding energies, confirmed the successful synthesis of the Co3O4/g-C3N4 heterojunction. The C 1s spectrum of Co3O4/g-C3N4 could be deconvoluted into three peaks at 285.2, 287.4, and 287.8 eV, which correspond to C-C, C-NHx, and N-C=N, respectively (Fig. S1c in Supporting information) [21]. Additionally, the C 1s spectrum of Co3O4/g-C3N4 exhibited a positive shift compared to that of g-C3N4 alone. The N 1s spectrum of Co3O4/g-C3N4 was divided into three peaks at 397.9, 399.5, and 400.2 eV, which were assigned to C-N=C, N-(C)3, and C-NHx, respectively (Fig. S1d in Supporting information) [22]. The shift of N 1s spectrum to higher binding energies relative to the g-C3N4 indicated a reduction in the electron density around nitrogen, suggesting a redistribution of electrons between Co3O4 and g-C3N4. Overall, compared to Co3O4 and g-C3N4, the Co 2p and O 1s spectra in the Co3O4/g-C3N4 heterojunction exhibited a shift towards lower binding energies, while the C 1s and N 1s spectra displayed a movement to higher binding energies. This suggested that upon the formation of the heterojunction, electrons from g-C3N4 migrated to Co3O4, indicating a transfer of electronic charge across the interface [23].
Fig. 2d presented the PL spectra of Co3O4 and the Co3O4/g-C3N4 heterojunction, with a peak in the range of 400–600 nm. The introduction of g-C3N4 into Co3O4 did not alter the peak position, but significantly reduced the peak intensity. This observation indicated the formation of a heterojunction between Co3O4 and g-C3N4 [24]. To elucidate the impact of heterojunction formation on the band structure, the UV–vis DRS spectra were investigated. As depicted in Fig. 2e, upon the introduction of Co3O4, the Co3O4/g-C3N4 composite exhibited an increased absorption intensity and a broader absorption range compared to g-C3N4 alone. This enhancement in absorption was indicative of the successful formation of a heterojunction between Co3O4 and g-C3N4, which gave rise to a pronounced interfacial effect. This interfacial interaction was proposed to bolster the catalytic stability of the material [25]. Based on the spectra, the band gap of the materials was determined using the Tauc plot method. The band gaps for Co3O4, g-C3N4, and Co3O4/g-C3N4 were determined to be 1.69, 2.72, and 2.51 eV, respectively (Fig. 2f). These values were consistent with those reported in the literature and suggested that the formation of the heterojunction had influenced the band gap [26].
To elucidate the impact of heterojunctions on the dechlorination efficacy, a series of Co3O4/g-C3N4 were synthesized with varying ratios of melamine to cobalt acetate (20:1, 10:1, 5:1, and 1:1), denoted as Co3O4/g-C3N4–20, Co3O4/g-C3N4–10, Co3O4/g-C3N4–5, Co3O4/g-C3N4–1, respectively. Text S2 and Figs. S2 and S3 (Supporting information) provided XRD, XPS, and UV–vis DRS spectra. As illustrated in Fig. 3a, Co3O4/g-C3N-x could achieve the removal of TAP due to the formation of heterojunctions, with the removal efficiencies ranging from 80.4% to 96.8%. Fig. S4a (Supporting information) demonstrated that the rate constant exhibited a volcano-shaped curve, increasing with the ratio of melamine to cobalt acetate. Co3O4/g-C3N4–10 reached the peak of the volcano with the highest rate constant (0.0584 min-1) and highest dechlorination efficiency (96.6%) (Fig. S4b in Supporting information). In particular, Co3O4/g-C3N4 represented a ratio of 10:1, at which characterization tests and performance comparisons were performed. Fig. S4c (Supporting information) indicated that although the leached cobalt increased from 2.8 µg/L to 10.1 µg/L with the increase in the ratio, it remained far below the Emission Standard of Pollutants for Copper, Nickel, Cobalt Industry of China (Co ≤ 1 mg/L).

To validate the electrocatalytic dechlorination performance of the Co3O4/g-C3N4 heterojunction, its catalytic activity was compared with that of Co3O4 and g-C3N4 catalysts. As depicted in Fig. 3b, the Co3O4/g-C3N4 heterojunction was able to remove 93.6% of TAP within 45 min, whereas the Co3O4 and g-C3N4 catalysts achieved only 67.9% and 61.8% removal after 45 min, respectively, indicating that the formation of the heterojunction significantly enhanced the dechlorination process. Fig. S5a (Supporting information) demonstrated that the process obeyed the pseudo-first-order kinetic model, with the rate constant k for Co3O4/g-C3N4 (0.0584 min-1) being 2.4 and 2.8 times that of Co3O4 (0.0246 min-1) and g-C3N4 (0.0206 min-1), respectively. Fig. S5b (Supporting information) illustrated that the dechlorination efficiency of Co3O4/g-C3N4 could reach 96.6%, surpassing that of Co3O4 (68.1%) and g-C3N4 (56.3%). The dechlorination properties of g-C3N4 might result from direct electron transfer.
To demonstrate the superior dechlorination capability of heterojunctions, the electrochemical performance of Co3O4/g-C3N4 and Co3O4 were evaluated. Electrochemical impedance (EIS) can reflect the conductivity of the electrode [27]. Rct represents the charge transfer resistance between the electrode surface and the electrolyte solution. The Nyquist plots (Fig. 3c) indicated that the Rct of Co3O4/g-C3N4 (130.5 Ω) was lower than that of Co3O4 (150.3 Ω) and g-C3N4 (193.0 Ω), suggesting that the introduction of the heterojunction facilitated electron transfer. Cyclic voltammetry (CV) was conducted near the open-circuit potential (OCP) at various scan rates to calculate electrochemical double layer capacitance (Cdl) (Fig. 3d and Fig. S6 in Supporting information) [28]. The Cdl of Co3O4/g-C3N4 was 8.76 mF/cm2, which was 2.4 times that of Co3O4 (3.68 mF/cm2). The Cdl is directly proportional to the electrochemical active surface area (ECSA) [29]. Consequently, the ECSA of Co3O4/g-C3N4 was superior to that of Co3O4, indicating a greater number of catalytic active sites. The formation of Co3O4/g-C3N4 heterojunction enhanced the electron transfer rate and ECSA compared to Co3O4 alone, which was beneficial for the dechlorination reaction. To demonstrate the formation of Co3O4/g-C3N4 heterojunction, g-C3N4 and Co3O4 were physically mixed at a ratio of 10:1 (Co3O4 + g-C3N4) and compared with Co3O4/g-C3N4. Text S3 and Figs. S7 and S8 (Supporting information) showed the synthesis of Co3O4/g-C3N4 heterojunction was essential.
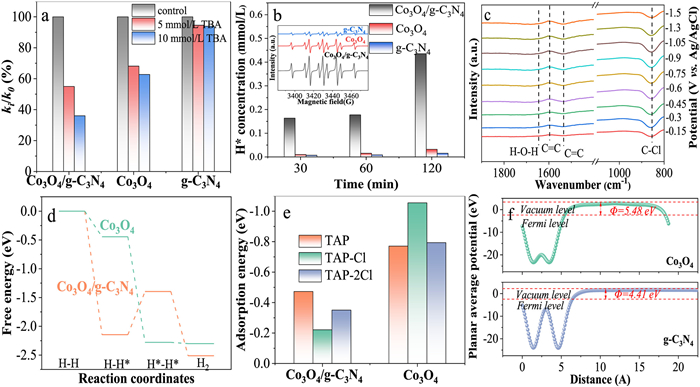
To further assess the catalyst's ability to generate H*, linear sweep voltammetry (LSV) test results indicated that the current density of Co3O4/g-C3N4 was much higher than that of Co3O4 (Fig. 3e). For instance, the current density of Co3O4/g-C3N4 (1.05 mA/cm2) was 3.4 times that of Co3O4 (0.31 mA/cm2) at −1.0 V, suggesting a higher rate of H* generation for Co3O4/g-C3N4 [30]. The generation of H* is closely associated with the current density. To elucidate the kinetics of H* formation, Fig. 3f illustrated the removal of TAP at various current densities. As the current density increased from 1 mA/cm2 to 10 mA/cm2, the TAP removal efficiency rose from 43.6% to 98.1%, and the rate constant was enhanced by 9.5 times (Fig. S9a in Supporting information). This suggested that a higher current density led to the generation of a greater amount of H*, thereby improving the dechlorination performance. However, when the current density was further increased to 12.5 mA/cm2, the rate constant declined. This was attributed to the aggravation of the hydrogen evolution side reaction at high current densities, which led to a decrease in dechlorination performance [31]. As depicted in Fig. S9b (Supporting information), the dechlorination efficiency initially increased and then decreased with the augmentation of the current density, reaching a maximum of 10 mA/cm2. H* plays an important role in the dechlorination process.
Text S4 and Fig. S10 (Supporting information) elucidated the degradation pathway of TAP and assessed the toxicity of TAP and its degradation intermediate. The toxicity of TAP was significantly reduced after the electrocatalytic dechlorination reaction on Co3O4/g-C3N4. Electrocatalytic dechlorination could be categorized into indirect dechlorination mediated by H* and direct reduction through electron transfer [32]. The mechanisms of Co3O4/g-C3N4, whether direct or indirect dechlorination, were investigated. Quenching experiments were conducted to determine the role of H*, utilizing tert‑butanol (TBA) as a scavenger for H*. As shown in Fig. S11 (Supporting information), the more TBA added, the more obvious the inhibition effect. With the addition of 10 mmol/L TBA, the TAP removal efficiency decreased by 24.8% (Fig. S11a), the rate constant dropped from 0.0584 min-1 to 0.0320 min-1 (Fig. S11b), and the dechlorination efficiency declined by 32% (Fig. S11c), thereby demonstrating the role of H* of Co3O4/g-C3N4 in the electrocatalytic dechlorination process. Since TBA also acted as the •OH scavenger, the cumulative concentration of •OH was determined to be 0 to exclude the interference from •OH (Fig. S11d). Co3O4 and g-C3N4 exhibited markedly different behaviors upon the addition of TBA. After adding 5 mmol/L TBA and 10 mmol/L TBA, TAP removal efficiencies of Co3O4 decreased from 79.1% to 66.8% and 63.3%, indicating that H* played a smaller role in Co3O4 than Co3O4/g-C3N4 (Fig. S12 in Supporting information). The formation of Co3O4/g-C3N4 heterojunction enhanced the generation of H*. The addition of TBA had little inhibitory effect on g-C3N4 (Fig. S13 in Supporting information). Upon the addition of 5 mmol/L and 10 mmol/L TBA, the removal efficiency of TAP by g-C3N4 decreased only by 0.29% and 0.31%, respectively, while the rate constant dropped from 0.0207 min-1 to 0.0195 min-1 and 0.0194 min-1. Therefore, the contribution of indirect reduction by H* of g-C3N4 was insignificant, with the primary mechanism being direct reduction. Studies have indicated the presence of electron transfer of g-C3N4 [33, 34]. Fig. S13c showed that TAP removal by g-C3N4 was largely due to the direct reduction of the cathode. To this end, the contribution of H* was assessed by evaluating the ratio of rate constants with and without the addition of TBA, denoted as ki/k0 (Fig. 4a) [35]. When 10 mmol/L TBA was added, the contribution of H* to removal efficiency was found to be in the order of Co3O4/g-C3N4 (64.0%) > Co3O4 (37.3%) > g-C3N4 (6.1%). The active species of the system were verified by electron paramagnetic resonance (EPR) with DMPO as the trapping agent (the inset of Fig. 4b). EPR spectra revealed nine characteristic peaks of the DMPO-H* adduct, with the signal intensities being: Co3O4/g-C3N4 > Co3O4 > g-C3N4, which was consistent with previous analysis. As shown in Fig. 4b, the concentration of H* increased over time, with the H* cumulative concentration of Co3O4/g-C3N4 reaching 0.44 mmol/L at 120 min, which was 13.6 and 28.2 times higher than that of Co3O4 and g-C3N4, respectively. This provided direct evidence supporting the generation of H* facilitated by the formation of Co3O4/g-C3N4 heterojunctions. By comparing EPR spectra with and without the addition of TAP, a decrease in the intensity of the DMPO-H* signal was observed upon TAP addition (Fig. S14 in Supporting information), which was attributed to the consumption of H*. However, the DMPO-H* signal remained strong even after the addition of TAP, indicating that Co3O4/g-C3N4 generated a sufficient amount of H*, and H* was present in excess. To further verify the role of H*, LSV curves of Co3O4/g-C3N4 with and without TAP was compared (Fig. S15 in Supporting information), in which they tended to flatten with TAP, indicating that part of H* was consumed during the dechlorination process, which was consistent with EPR results.
In-situ FTIR spectroscopy was employed to monitor the reaction intermediates during the dechlorination [36, 37]. The peaks observed at 1597 cm-1 and 1535 cm-1 in Fig. 4c corresponded to the C=C skeletal vibrations of the benzene ring, indicating the adsorption of TAP on the electrode surface. The presence of a peak at 1647 cm-1, corresponding to the bending mode of water molecules, could be ascribed to the electrolytic cleavage of water into H* at the electrode interface, thereby confirming the generation of H*. A distinct peak appearing at 852 cm-1 corresponded to the C-Cl stretching vibration, which demonstrated the excellent dechlorination performance of Co3O4/g-C3N4. Fig. S16 (Supporting information) displayed the in-situ FTIR spectrum for Co3O4, wherein the C-Cl bond exhibited a significant reduction in intensity compared to Co3O4/g-C3N4, thereby proving the superior dechlorination capability of Co3O4/g-C3N4.
Fig. S17 (Supporting information) investigated the relationship between the dechlorination performance of Co3O4/g-C3N4 and the initial concentration of TAP. TAP removal efficiency exceeded 94.3% for TAP concentrations between 1–50 mg/L, and even at a concentration of 100 mg/L, the removal efficiency could still reach 82.9% (Fig. S17a). The active sites on the catalyst surface were limited. At low pollutant concentrations, these sites could fully interact with pollutants. However, at high concentrations, excess pollutants saturated the active sites, preventing some pollutants from accessing them and reducing dechlorination efficiency [38]. It was noteworthy that the rate constant at a TAP concentration of 1 mg/L (0.090 min-1) was 1.5, 1.6, 1.9, and 3.1 times higher than those at concentrations of 5 mg/L (0.059 min-1), 10 mg/L (0.056 min-1), 50 mg/L (0.047 min-1), and 100 mg/L (0.029 min-1), respectively (Fig. S17b). Co3O4/g-C3N4 was applicable over a broad range of pollutant concentrations, particularly effective for low-concentration pollutants. Fig. S17c showed the dechlorination efficiencies were 97.3%, 100%, 96.6%, 92.8%, and 69.0%, respectively, at the concentration of 1, 5, 10, 50, and 100 mg/L. To investigate the origin of the heterojunction effect, the Langmuir-Hinshelwood (L-H) model [39, 40] was used to describe heterogeneous electrocatalytic dechlorination kinetics of Co3O4/g-C3N4 (Fig. S18 in Supporting information). Fig. S18b demonstrated a good linear relationship between 1/r0 and 1/C0 (r2 = 0.998), indicating that the process was consistent with the l-H model. The key step in the electrocatalytic dechlorination was the reaction between adsorbed TAP and H*.
DFT calculations were employed to further validate the reaction mechanism. The process of generating H* through hydrogen ions or water was carried out. Fig. S19a (Supporting information) demonstrated that Co3O4/g-C3N4 exhibited a lower hydrogen adsorption free energy. Furthermore, Co3O4/g-C3N4 presented a lower energy barrier for water dissociation than Co3O4 alone, which suggested a more rapid Volmer step, as depicted in Fig. S19b (Supporting information). The energy barrier for the formation of H* (ΔGH*) was utilized to assess the capability of generating H* [36]. The more negative ΔGH* value of Co3O4/g-C3N4 (−3.8 eV) compared to that of Co3O4 (−3.5 eV) indicated a lower energy barrier for Co3O4/g-C3N4, which was favorable for the generation of H* (Fig. S19c in Supporting information). Fig. 4d investigated H* transformation on Co3O4/g-C3N4 and Co3O4. Initially, an atomic hydrogen adsorbed onto the catalyst surface. Subsequently, the second atomic hydrogen generated approached the first atomic hydrogen, with the two hydrogen atoms coming infinitely close to each other, eventually transforming into hydrogen gas and desorbing from the catalyst surface. In the process of forming hydrogen gas, Co3O4/g-C3N4 experienced the highest energy barrier, at which point the adsorbed atomic hydrogen remained stable on the catalyst surface, inhibiting the generation of hydrogen gas. Fig. S19d (Supporting information) presented the projected density of states (PDOS) for the d orbitals of the Co3O4/g-C3N4 and Co3O4, reflecting the distribution of electrons within the d orbitals of the materials [41, 42]. The d-band center (εd) of Co3O4/g-C3N4 (−1.515 eV) was higher than that of Co3O4 (−1.731 eV), indicating that Co3O4/g-C3N4 has a stronger binding affinity for H* compared to Co3O4 alone [30]. Generally, the rate of electrochemical reactions is largely dependent on the adsorption activation of the substrate and the desorption of product molecules. Therefore, the adsorption energies of TAP and its degradation products on Co3O4/g-C3N4 and Co3O4 were calculated. Fig. 4e illustrated that the adsorption energy of TAP on Co3O4 (−0.77 eV) was lower than that of its degradation products TAP-Cl (−1.05 eV) and TAP-2Cl (−0.79 eV). This suggested that degradation products might compete with reactants for active sites, leading to a decrease in dechlorination performance. However, upon the formation of Co3O4/g-C3N4 heterojunctions, the adsorption energy of TAP (−0.47 eV) was enhanced relative to that of the products TAP-Cl (−0.22 eV) and TAP-2Cl (−0.35 eV), indicating that the heterojunction facilitated the adsorption of TAP and the desorption of products, thereby enhancing the performance of Co3O4/g-C3N4.
Charge density difference was employed to probe changes in the electronic structure to determine charge transfer. Fig. S20a (Supporting information) indicated a decrease in electron density on g-C3N4 and an increase on Co3O4, suggesting that upon the formation of the heterojunction, g-C3N4 lost electrons which were transferred to Co3O4. Fig. S20b (Supporting information) illustrated that the Fermi level of Co3O4/g-C3N4 undergoes a positive shift relative to the Fermi levels of Co3O4 and g-C3N4, indicating electron transfer resulting from the formation of the heterojunction, which was consistent with the findings from the charge density difference analysis. Electrons transfer from g-C3N4, which possessed a more negative Fermi level to Co3O4 [43]. The work function corresponds to the energy required to remove an electron from the Fermi level to the vacuum level, reflecting the potential energy barrier for electron egress from the solid [44]. The work function was calculated to verify the direction of electron transfer (Fig. 4f). The higher work function of Co3O4 (5.48 eV) relative to g-C3N4 (4.41 eV) indicated that upon the formation of Co3O4/g-C3N4 heterojunctions, electrons transferred from g-C3N4 to Co3O4 until the Fermi levels aligned, creating an interfacial electric field. Consequently, a possible mechanism for the electrocatalytic dechlorination by Co3O4/g-C3N4 was proposed (Text S5 and Fig. S21 in Supporting information). Co3O4/g-C3N4 was superior to Co3O4 in terms of dechlorination efficiency, rate constant and H* contribution.
The pH value exerted a significant influence on the electrocatalytic dechlorination process. As depicted in Fig. 5a, the removal efficiency of TAP within the pH range of 3–11 had little difference, with over 95% removal achieved within 60 min. The rate constants were in the range of 0.053–0.065 min-1 (Fig. S22a in Supporting information), and the dechlorination efficiencies all reached 93% (Fig. S22b in Supporting information). In particular, the TAP removal efficiencies of Co3O4/g-C3N4 increased compared with Co3O4 at different pH. The rate constants were 4.2, 4.0, 2.4, 3.5 and 3.0 times of Co3O4, respectively. Co3O4 had poor stability under acidic conditions and was prone to leach. Co3O4/g-C3N4 increased the active site, and enhanced stability due to the construction of heterojunctions, thus reducing the dependence on pH. The leached cobalt of Co3O4/g-C3N4 was between 2.3 µg/L and 4.9 µg/L (Fig. S22c in Supporting information). This indicated that the Co3O4/g-C3N4 was less affected by pH and could play a role in a wide pH range.
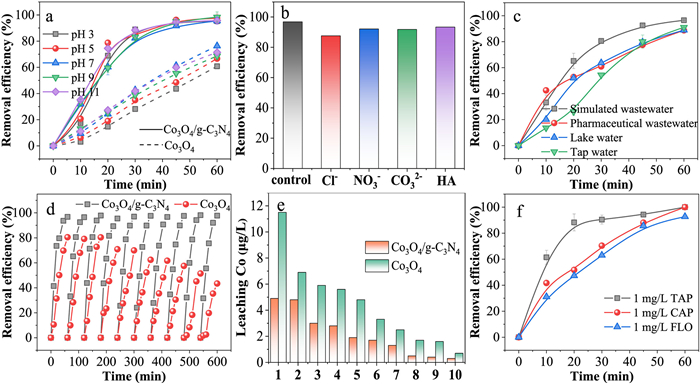
Additionally, Co3O4/g-C3N4 demonstrated effective dechlorination performance in the presence of coexisting ions and in real water bodies (Figs. 5b and c, Text S6, Fig. S23, and Table S1 in Supporting information). Reusability is an important parameter to evaluate catalyst performance. As depicted in Fig. 5d, over ten cycles, the removal efficiency of Co3O4/g-C3N4 for TAP was consistently maintained between 95.5% and 98.2%, with rate constants ranging from 0.0529 min-1 to 0.0621 min-1 (Fig. S24a in Supporting information), and dechlorination efficiencies falling within the range of 93.8%–96.6% (Fig. S24b in Supporting information). In particular, Co3O4/g-C3N4 significantly improved reusability compared to Co3O4. After ten cycles, the performance of Co3O4 declined, with the TAP removal efficiency decreasing from 80.0% to 43.4%, the rate constant diminishing from 0.0258 min-1 to 0.0087 min-1, and the dechlorination rate dropping from 68.1% to 39.6%. Besides, Fig. 5e indicated that the leaching of cobalt from the Co3O4/g-C3N4 was lower compared to the leaching of Co3O4, with a range of 0.3–4.9 µg/L. The high stability of Co3O4/g-C3N4 was attributed to the strong metal-support interactions within the heterojunction, which enhanced the structural stability and thereby reduced leaching [45]. Co3O4/g-C3N4 exhibited excellent cyclic stability.
Thiamphenicol (TAP), chloramphenicol (CAP), and florfenicol (FLO), which belong to the chloramphenicol antibiotics, are extensively utilized in veterinary medicine and aquaculture and frequently detected in aquatic environments [46]. For instance, antibiotics have been detected in surface waters of the Middle East and North Africa regions at concentrations ranging from 0.03 ng/L to 66, 400 ng/L [47]. However, low concentrations of pollutants aggravated the consumption of active species due to side reactions and were limited by mass transfer, posing significant challenges for removal [48]. Therefore, the removal of low-concentration chlorinated antibiotics (1 mg/L) was explored. Fig. 5f demonstrated that Co3O4/g-C3N4 exhibited effective removal of TAP, CAP, and FLO at low concentrations, achieving over 92.7% removal within 60 min. The rate constant k followed the order of TAP (0.0896 min-1) > CAP (0.0444 min-1) > FLO (0.0414 min-1) (Fig. S25 in Supporting information). Deng et al. utilized multi-walled carbon nanotube (MWCNTs) electrodes for the removal of low-concentration TAP (2 mg/L), achieving only 87% removal within 24 h [49]. Overall, Co3O4/g-C3N4 possessed efficient removal of low-concentration chlorinated antibiotics.
Additionally, we compared various processes for the removal of TAP, as shown in Table S2 (Supporting information). Biodegradation was time-consuming, requiring 36–120 h [50, 51]. Although 96.32% adsorption could be achieved within 60 min, this method required a large amount of catalyst (8 g/L) and subsequent treatment was necessary [52]. Notably, Co3O4/g-C3N4 exhibited significant advantages under similar experimental conditions over electro-Fenton [53], Fe(III)/PMS [54], UV/H2O2 [55], reduction [56] and electrocatalytic dechlorination [49, 57, 58]. Therefore, Co3O4/g-C3N4 was an excellent catalyst for electrocatalytic dechlorination.
This work constructed the Co3O4/g-C3N4 heterojunction, offering a novel insight into the electrocatalytic dechlorination of non-noble metals. The formation of the heterojunction facilitated the directional transfer of electrons from g-C3N4 to Co3O4, resulting in an electron-enriched state for the Co3O4, which promoted the formation of H*. The H* yield of Co₃O₄/g-C₃N₄ increased by 13.6 to 28.2 times, contributing 64.0% to pollutant removal. The energy barrier for H2 formation on Co3O4/g-C3N4 (0.75 eV) was higher than that on Co3O4 (−1.84 eV), supporting that it could stabilize H* and inhibit the formation of H2. Besides, Co3O4/g-C3N4 augmented pollutant adsorption while concurrently diminished the adsorption of degradation intermediates. This enhanced the dechlorination activity of the heterojunction. The Co3O4/g-C3N4 heterojunction was promising for practical applications in environmental remediation and wastewater treatment. The Co3O4/g-C3N4 heterojunction exhibited broad applicability and was minimally affected by pH and co-existing ions. The metal-support interaction endowed it with superior stability, sustaining a removal efficiency exceeding 90% even after 10 cycles, with cobalt leaching levels ranging from 0.3 µg/L to 4.9 µg/L. It was particularly effective for the dechlorination of chlorinated antibiotics, especially at low concentrations. The improved catalytic activity highlights the potential of Co3O4/g-C3N4 heterojunction as an efficient and sustainable catalyst for environmental remediation applications.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Ge Song: Writing – original draft, Formal analysis, Conceptualization. Huizhong Wu: Methodology. Chaohui Zhang: Methodology. Xuechun Wang: Validation. Shuaishuai Li: Investigation. Jiangli Sun: Formal analysis. Xiuwu Zhang: Validation. Minghua Zhou: Writing – review & editing, Project administration, Funding acquisition.
This work was financially supported by Natural Science Foundation of China (Nos. U23B20165 and 52170085), National Key R & D Program International Cooperation Project (No. 2023YFE0108100), Key Project of Natural Science Foundation of Tianjin (No. 21JCZDJC00320), and Fundamental Research Funds for the Central Universities, Nankai University.
Supplementary material associated with this article can be found, in the online version, at doi:
L.M. Nguyen, N.T.T. Nguyen, T.T.T. Nguyen, et al., Environ. Chem. Lett. 20 (2022) 1929–1963. doi: 10.1007/s10311-022-01416-x
J. Lina, K. Zhang, L. Jiang, et al., Sci. Total Environ. 850 (2022) 158059. doi: 10.1016/j.scitotenv.2022.158059
E.T. Martin, C.M. McGuire, M.S. Mubarak, et al., Chem. Rev. 116 (2016) 15198–15234. doi: 10.1021/acs.chemrev.6b00531
G. Song, H. Wu, J. Jing, et al., Environ. Sci. Technol. 57 (2023) 14482–14492. doi: 10.1021/acs.est.3c06021
J. Li, C. Zhang, Y. Li, et al., ACS Catal. 13 (2023) 9633–9655.
W. Fu, X. Du, P. Su, et al., ACS Appl. Mater. Interfaces 13 (2021) 28348–28358. doi: 10.1021/acsami.1c07063
L. Su, K. Li, H. Zhang, et al., Water Res. 120 (2017) 1–11.
A. Shan, X. Teng, Y. Zhang, et al., Nano Energy 94 (2022) 106913. doi: 10.1016/j.nanoen.2021.106913
Y. Liu, L. Liu, J. Shan, et al., J. Hazard. Mater. 290 (2015) 1–8.
K. Jiang, X. Shi, M. Chen, et al., J. Hazard. Mater. 411 (2021) 125119. doi: 10.1016/j.jhazmat.2021.125119
K. Wang, S. Shu, M. Chen, et al., Chem. Eng. J. 381 (2020) 122673. doi: 10.1016/j.cej.2019.122673
M. Chen, S. Shu, J. Li, et al., J. Hazard. Mater. 389 (2020) 121876. doi: 10.1016/j.jhazmat.2019.121876
M. Ismael, J. Alloy. Compd. 846 (2020) 156446. doi: 10.1016/j.jallcom.2020.156446
Q. Liang, Z. Li, X. Yu, et al., Adv. Mater. 27 (2015) 4634–4639. doi: 10.1002/adma.201502057
L. Yang, J. Liu, L. Yang, et al., Renew. Energy 145 (2020) 691–698. doi: 10.1016/j.renene.2019.06.072
D. Pradhan, L. Mohanty, R. Singhal, et al., Inorg. Chem. Commun. 156 (2023) 111259. doi: 10.1016/j.inoche.2023.111259
S. Suganthi, S. Vignesh, J. Kalyana Sundar, et al., Environ. Res. 235 (2023) 116574. doi: 10.1016/j.envres.2023.116574
Q. Zhang, S. Gao, Y. Guo, et al., Nat. Commun. 14 (2023) 1147. doi: 10.1038/s41467-023-36779-4
J. Fu, Q. Xu, J. Low, et al., Appl. Catal. B 243 (2019) 556–565. doi: 10.1016/j.apcatb.2018.11.011
M.M.S. Sanad, T.A. Taha, A. Helal, et al., Environ. Sci. Pollut. Res. 30 (2023) 60225–60239. doi: 10.1007/s11356-023-26767-y
H. Cao, Y. Yan, Y. Wang, et al., Carbon 201 (2023) 415–424. doi: 10.1016/j.carbon.2022.09.029
Z. Xu, J. Zhong, J. Chen, et al., Surf. Interfaces 38 (2023) 102838. doi: 10.1016/j.surfin.2023.102838
J. Sun, H. Wu, C. Fu, et al., Appl. Catal. B 351 (2024) 123976. doi: 10.1016/j.apcatb.2024.123976
I. Rabani, R. Zafar, K. Subalakshmi, et al., J. Hazard. Mater. 407 (2021) 124360. doi: 10.1016/j.jhazmat.2020.124360
F. Chang, J. Zheng, X. Wang, et al., Mater. Sci. Semicond. Process 75 (2018) 183–192. doi: 10.1016/j.mssp.2017.11.043
H. Wu, Z. Hu, R. Liang, et al., J. Hazard. Mater. 456 (2023) 131696. doi: 10.1016/j.jhazmat.2023.131696
M. Gaberscek, Nat. Commun. 12 (2021) 6513. doi: 10.1038/s41467-021-26894-5
E. Cossar, M.S.E. Houache, Z. Zhang, et al., J. Electroanal. Chem. 870 (2020) 114246. doi: 10.1016/j.jelechem.2020.114246
S.S. Jeon, P.W. Kang, M. Klingenhof, et al., ACS Catal. 13 (2023) 1186–1196. doi: 10.1021/acscatal.2c04452
Z. Lou, C. Yu, X. Wen, et al., Appl. Catal. B 317 (2022) 121730. doi: 10.1016/j.apcatb.2022.121730
Z. Lou, J. Zhou, M. Sun, et al., Chem. Eng. J. 352 (2018) 549–557. doi: 10.1016/j.cej.2018.07.057
S. Qin, C. Lei, X. Wang, et al., Cell Rep. Phys. Sci. 3 (2022) 100713.
B. Lin, H. An, X. Yan, et al., Appl. Catal. B 210 (2017) 173–183. doi: 10.1016/j.apcatb.2017.03.066
W. Xu, F. Yang, T. Ding, et al., Int. J. Hydrog. Energy 68 (2024) 1352–1360. doi: 10.1016/j.ijhydene.2024.04.323
R. Mao, C. Huang, X. Zhao, et al., Appl. Catal. B 241 (2019) 120–129. doi: 10.1016/j.apcatb.2018.09.013
P. Su, W. Fu, Z. Hu, et al., Appl. Catal. B 313 (2022) 121457. doi: 10.1016/j.apcatb.2022.121457
Q. Zhang, M. Peng, Z. Gao, et al., J. Am. Chem. Soc. 145 (2023) 4166–4176. doi: 10.1021/jacs.2c12586
H. Tang, Z. Bian, Y. Peng, et al., J. Hazard. Mater. 433 (2022) 128744. doi: 10.1016/j.jhazmat.2022.128744
Y. Peng, M. Cui, Z. Zhang, et al., ACS Catal. 9 (2019) 10803–10811. doi: 10.1021/acscatal.9b02282
J. Xu, X. Liu, G.V. Lowry, et al., ACS Appl. Mater. Interfaces 8 (2016) 7333–7342. doi: 10.1021/acsami.5b11859
S. Jiao, X. Fu, H. Huang, Adv. Funct. Mater. 32 (2021) 2107651.
D. Tian, S.R. Denny, K. Li, et al., Chem. Soc. Rev. 50 (2021) 12338–12376. doi: 10.1039/D1CS00590A
Y. Liu, J. Guo, E. Zhu, et al., Nature 557 (2018) 696–700. doi: 10.1038/s41586-018-0129-8
A. Kahn, Mater. Horiz. 3 (2016) 7–10. doi: 10.1039/C5MH00160A
R. Ma, G. Zhou, M. Gu, et al., Appl. Surf. Sci. 648 (2024) 158980. doi: 10.1016/j.apsusc.2023.158980
Y. Zhang, P. Guo, Y. Wu, et al., Environ. Toxicol. Chem. 38 (2019) 575–584.
N. Mheidli, A. Malli, F. Mansour, et al., Sci. Total Environ. 851 (2022) 158302. doi: 10.1016/j.scitotenv.2022.158302
Q. Zhang, X. Wang, J. Xie, et al., J. Hazard. Mater. 443 (2023) 130280. doi: 10.1016/j.jhazmat.2022.130280
D. Deng, F. Deng, B. Tang, et al., J. Hazard. Mater. 332 (2017) 168–175. doi: 10.1016/j.jhazmat.2017.03.013
K. Yang, S. Ren, M. Mei, et al., World J. Microbiol. Biotechnol. 38 (2022) 37. doi: 10.1007/s11274-021-03223-y
J. Zhang, X. Li, H. Lei, et al., J. Hazard. Mater. 426 (2022) 128101. doi: 10.1016/j.jhazmat.2021.128101
R. Li, W. Sun, L. Xia, et al., Molecules 27 (2022) 7980. doi: 10.3390/molecules27227980
D. Zhang, K. Yin, Y. Tang, et al., Chem. Eng. J. 427 (2022) 130996. doi: 10.1016/j.cej.2021.130996
W. Ma, R. Han, L. Zhu, et al., Desalination 565 (2023) 116859. doi: 10.1016/j.desal.2023.116859
N. Liu, S. Sijak, M. Zheng, et al., Chem. Eng. J. 260 (2015) 826–834. doi: 10.1016/j.cej.2014.09.055
W. Shen, X. Wang, F. Jia, et al., Appl. Catal. B 264 (2020) 118550. doi: 10.1016/j.apcatb.2019.118550
T. Liu, J. Luo, X. Meng, et al., J. Hazard. Mater. 358 (2018) 294–301. doi: 10.1016/j.jhazmat.2018.06.064
Q. Zheng, H. Xu, Y. Yao, et al., Angew. Chem. Int. Ed. 63 (2024) e202401386. doi: 10.1002/anie.202401386

Figure 1 SEM images of (a) Co3O4, (b) g-C3N4, and (c) Co3O4/g-C3N4. TEM images of (d) Co3O4, (e) g-C3N4, and (f) Co3O4/g-C3N4. (g) HRTEM image and (h) elemental mapping of Co3O4/g-C3N4.

Figure 2 (a) XRD patterns, (b) FT-IR spectra, (c) XPS survey spectra, (d) PL spectra, (e) UV–vis DRS spectra, and (f) band gaps of Co3O4/g-C3N4, Co3O4, and g-C3N4.

Figure 3 (a) Effects of Co3O4/g-C3N4-x on TAP removal, (b) TAP removal, and (c) Nyquist plots of Co3O4/g-C3N4, Co3O4, and g-C3N4, (d) plots of capacitive currents vs. scan rates, and (e) LSV curves of Co3O4/g-C3N4 and Co3O4. (f) Effects of current density on TAP removal of Co3O4/g-C3N4.

Figure 4 (a) Inhibition of the TAP removal with the addition of TBA. (b) H* concentration of different catalyst (the inset was EPR spectra). (c) In-situ FTIR spectra of Co3O4/g-C3N4 with TAP. (d) Free energy diagrams of H* transformation on Co3O4/g-C3N4 and Co3O4. (e) Adsorption energies of TAP and its degradation intermediates on Co3O4/g-C3N4 and Co3O4. (f) Planar average potential on the surfaces of Co3O4 and g-C3N4 by work function.

Figure 5 (a) Effects of initial pH on TAP removal of Co3O4/g-C3N4 and Co3O4. (b) TAP removal in different background constituents. (c) TAP removal in the simulated wastewater, pharmaceutical wastewater, lake water and tap water. (d) TAP removal, and (e) leaching Co in 10 cycles of Co3O4/g-C3N4 and Co3O4. (f) Removal of low concentration chlorinated antibiotics of Co3O4/g-C3N4.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载: