Received Date:
23 September 2024 Accepted Date:
26 May 2025 Revised Date:
21 May 2025 Available Online:
15 February 2026
Abstract:
Vacuum-ultraviolet (VUV) radiation is high-energy UV radiation with a wavelength of 100–200 nm capable of decomposing/mineralizing hazardous emerging organic pollutants (EPs) in water through direct photolysis and/or by generating reactive free radicals (RFRs) during photolysis. However, due to the unsatisfactory photoelectric conversion rate, strong absorption by oxygen and water molecules, and other characteristics of VUV radiation, its application and development are hindered, leading to misconceptions regarding high energy consumption and insufficient free radical yield. The objectives of our assessment in this review are as follows: The illumination of the photochemical characteristics of VUV and the reactivity of aqueous solutions. Summarization of accurate UV dose and energy evaluation criteria. Comparison and analysis of the photochemical mechanisms and reaction kinetics of different types of EPs via VUV direct photolysis, as well as the interference origins of typical substrates in water for VUV decontamination. We found that quantities typically reported in VUV photochemical reactions of engineered systems are underreported in low-pressure mercury lamp (LPUV) photochemical reactions, especially a quantitative indicator of the species or energy that induces a chemical reaction. The absence of these quantities has made it difficult to assess the fundamental performance of VUV photolysis fully compared with that of UV-C. Some studies have sought to optimize VUV-advanced reduction processes (VUV-ARP) or VUV reactor treatment of these contaminants; however, an abundant evaluation of the reaction origins and processes between VUV-derived main RFRs and reactants (H2O, O2, organic matter, inorganic ions, etc.) is essential, cause these scientific elements will provide the possibility to break the application gap for VUV in the field of EPs treating. Overall, the data compilation, analysis, and research recommendations provided in this review will form the basis for all photochemical reactions initiated by VUV radiation with water as the backing agent.
Emerging organic pollutants (EPs) are known for their ability to accumulate in biological organisms, travel long distances in the environment, and pose challenges in detection. These substances can disrupt human endocrine, reproductive, and immune systems, and are linked to various diseases, including cancer and neurological disorders. EPs, which include persistent organic pollutants (POPs), endocrine-disrupting chemicals (EDPs), pharmaceutical [1], and personal care products (PPCPs), have drawn significant global attention, leading to efforts in research and technology aimed at mitigating their environmental impact. To address the risks associated with persistent organic pollutants and safeguard both the environment and human health, the international community established the “Stockholm Convention on Persistent Organic Pollutants” (referred to as the “Convention”) in 2001. The Convention entered into force on 17 May 2004 and, as of July 2023, has 186 contracting parties. The 12 types of POPs that are most harmful to human health and the natural environment and were initially eliminated under the Convention include aldrin, dieldrin, endrin, dichloro-diphenyl trichloroethane (DDT), heptachlor, chlordane, dechlorane, toxaphene, hexachlorobenzene, polychlorinated biphenyls, and dioxins. Through systematic screening and expansion, the list of substances controlled by the Convention has been incrementally expanded to include chlordecone, hexabromobiphenyl, hexabromodiphenyl ether, heptabromodiphenyl ether, lindane, pentachlorobenzene, tetrabromodiphenyl ether, pentabromodiphenyl ether, sulfan, hexabromocyclododecane, perfluorooctane sulfonic acid and its salts, and perfluorooctane sulfonyl fluoride. The Convention underscores the importance of implementing measures to mitigate the adverse effects of POPs across their life cycle. Governance of EPs plays a crucial role in fostering a global community with a shared vision for the future of humanity.
EPs enter the human body through aerosols in domestic and drinking water. Current mainstream treatment methods, such as coagulation-precipitation-filtration in sewage and water supply plants, are ineffective against EPs [1–3]. EPs can only be eliminated by advanced treatment technologies like ozone-activated carbon, membrane filtration, chlorine oxidation, and ultraviolet radiation. Chemical reagent-based oxidation technologies (chlorine, H2O2, permanganate, persulfate, etc.) can cause structural damage and complete mineralization of EPs [4–6], and are currently the most promising for EPs treatment. However, the fourth generation of water treatment technology aims to produce green, high-quality water while preserving its original properties without adding additional soluble chemical reagents. Chemical oxidation technologies, in contrast, contradict this concept. Despite the increasing focus on water treatment oxidants in recent years, articles have not adequately addressed the secondary risks or potential issues in advanced treatment processes. In light of this, ultraviolet (UV) technologies, which do not require auxiliary reagents, offer significant advantages. They can decompose targets through direct photolysis of EPs or by stimulating oxygen or water molecules to produce oxidizing or reducing active substances.
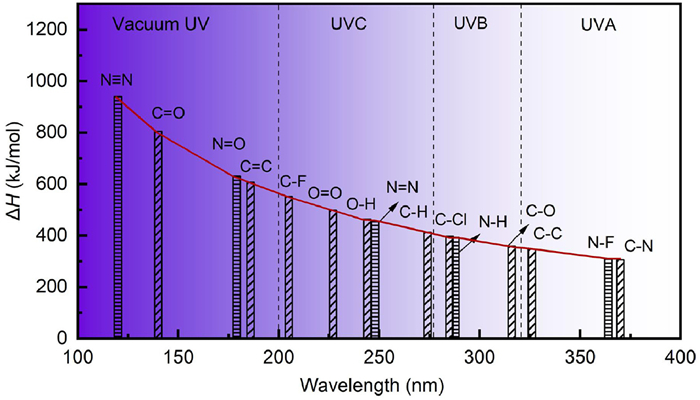
The Commission Internationale de l’Eclairage (CIE) classifies UV radiation into UV-A (315–400 nm), UV-B (280–315 nm), and UV-C (100–280 nm). UV-C comprises vacuum ultraviolet (VUV, 100–200 nm), Far-UV-C (200–230 nm), and inactivated-UV-C (250–280 nm). Photochemical reactions primarily commence through photon absorption from the UV electromagnetic spectrum. While bond energies within molecules vary due to factors like participating atoms, oxidation states, and neighboring functional groups, typical bond energy values can be summarized [7]. Fig. 1 illustrates the bond energy of a typical chemical bond in relation to photon radiant energy and the corresponding wavelength range. Reactions occur when photon energy surpasses the bond energy (ΔH) of a compound. Photon energy decreases with increasing wavelength, indicating higher ultraviolet light energy with shorter wavelengths. Consequently, the high energy efficiency of VUV enables the photodissociation or indirect decomposition of stable EPs, including “forever chemicals” such as perfluorinated compounds, dodecyl dimethyl benzyl ammonium chloride, fluoroquinolone antibiotics, clotrimazole, dimethoate, imipramine [8–11].
Figure 1
Figure 1.
The bond energy values of a typical chemical bond corresponding to the photon radiant energy value, indicated at the bottom of the graph, are wavelength ranges corresponding to definitions of radiation types. Source: Based on Photolysis and the Hydroxyl Radical. http://butane.chem.uiuc.edu/pshapley/environmental/l14/1.html.
In light-driven VUV’s water purification technology, the effectiveness relies on the type of light source utilized. Excimer lamps and low-pressure mercury vapor lamps (LPUV) are the two common light sources in VUV photolysis. Excimer and excimer complex light sources, powered by inert gas/halogen or noble gas/halogen exciters, represent an innovative generation of VUV light sources. In contrast to the development of noncoherent excimer radiation sources, LPUV lamps encased in synthetic quartz envelopes, emitting at 185 nm, have been extensively employed to explore oxidative/reduction reactions of organic pollutants in aqueous systems and the dissolution of total organic carbon (TOC) [12]. However, research on the mechanistic aspects of VUV-photochemically induced reactions is significantly restricted due to the non-monochromatic nature of LPUV radiation sources, predominantly emitting at 254 nm while 185 nm accounts for only 10% of the total output power. Meanwhile, the high requirement for the purity of the lamp casing material to ensure VUV penetration makes the application of LPUV in laboratory research and water plants mostly limited to UVC. Nevertheless, the potential of VUV’s high ionization energy (6–12 eV) for the thorough, efficient, green removal of ultra-stable EPs in water cannot be ignored.
Currently, most water treatment studies utilizing LPUV as a light source have encountered difficulties in reaching the water interface or accurately discerning the performances and principles of VUV and UVC wavelengths. While some studies have attempted to enhance VUV-advanced reduction processes (VUV-ARP) or treatment of contaminants using VUV reactors, a comprehensive and precise assessment of the origins and processes of reactions between VUV-derived main RFRs and reactants (e.g., H2O, O2, organic matter, inorganic ions) is essential. This paper summarizes the advanced treatment process of EPs in VUV-irradiated water, primarily covering the following aspects:
(1) The fundamental optical properties and source of VUV radiation, as well as its behavior within water, including energy transmission in the remote water layer (>5 mm) triggered by secondary radicals.
(2) Precise assessment methods for UV dose and criteria for selecting different VUV systems.
(3) Comparing and analyzing the photochemical mechanisms and reaction kinetics of diverse EPs via VUV direct photolysis, as well as establishing reliable methods for identifying critical active species.
(4) Interference phenomena and factors contributing to the presence of common background substrates in water during VUV decontamination.
This review presents an insightful and comprehensive analysis of the mechanisms underlying EPS reduction through VUV technology, offering guidance for the strategic design of VUV systems in advanced water purification setups, ensuring increased efficiency and selectivity.
2.
Background of VUV
2.1
Optical characteristics
The exchange of energy between VUV-emitted photon energy and substances is strictly governed by environmental conditions. VUV irradiation can trigger homolysis and photochemical ionization of water molecules, swiftly generating •OH oxidizers, hydrated electrons (eaq–), and •H reductants (Eqs. 1 and 2) [13]. The absorption coefficient of water is α = 1.8 cm–1 at 185 nm [14] with 90% of the 185 nm irradiation being absorbed by H2O within the initial 0.55 cm of the water layer [15]. Moreover, VUV radiation absorption by oxygen induces the photolysis of oxygen molecules. This process leads to the formation of ozone through the reaction of oxygen atoms with other oxygen molecules (Eqs. 3 and 4). Such reactions typically entail the transfer of kinetic energy, absorbed by the intermediate X medium without direct involvement in the reaction [16]. Consequently, VUV light exhibits extremely limited penetration ability in air and necessitates usage within vacuum environments. The introduction of N2 into VUV processes is common practice to maintain normal efficiency, air pressure, and system temperature.
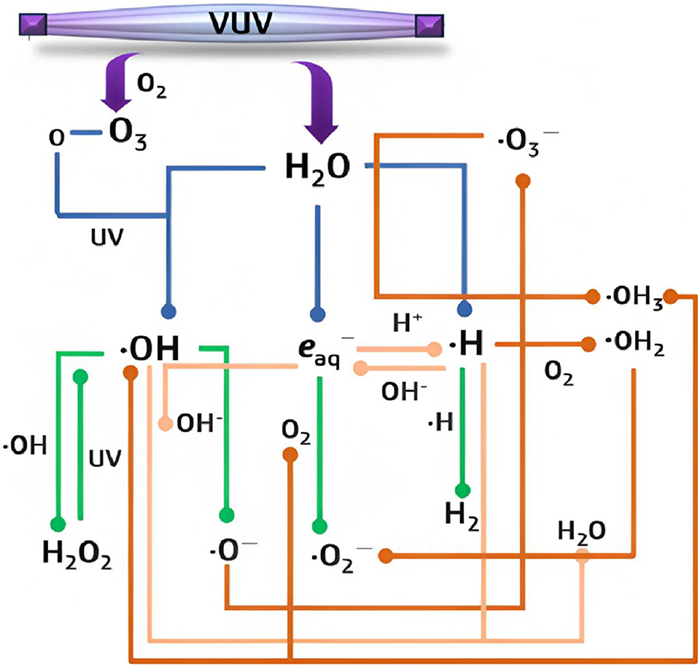
•OH, eaq– and •H are the primary active agents produced during the VUV photolysis of water. The quantum yield (Φ) of eaq– is significantly lower than that of •OH; therefore, •OH is often considered the dominant species in terms of its contribution to the mechanism [13]. The presence of eaq– in aqueous solutions is mildly influenced by pH levels. Under acidic conditions (pH < 4), eaq– is almost completely neutralized by H+ at a high reaction rate of 2.3 × 1010 L mol-1 s-1. On the other hand, under alkaline conditions (pH > 9.0), the primary reactions of generated •H and OH– help to replenish the eaq– that has been consumed or degraded (Eqs. 5 and 6) [13,17,18]. The breakdown of •OH, •H, and eaq– is further facilitated by a series of reactions with oxide radical anions (•O–), superoxide radicals (•O2–), H2O2, H2, and H2O, depending on the actual pH of the reaction system (Eqs. 7–10). Furthermore, •H and •OH can trigger a series of diffusion-controlled secondary reactions, leading to the formation of more stable ions and molecules (Eqs. 11–13 and Fig. 2). The subsequent reactions of these primary agents result in the formation of ozonide radicals (•HO3), hydroperoxyl radicals (•HO2), hydroxide (HO–), hydroperoxide anions (HO2–), and protons (H+) [13]. Protonated •HO3 can quickly decompose into O2 and •OH; similarly, •OH2 more readily breaks down to •O2–. These extensive reactions are halted by the introduction of substances that react more rapidly with free radicals.
Figure 2.
The formation and interconversion of key and secondary RFRs in aqueous solution and O2 photolysis under VUV irradiation. The heterochromatic lines represent different paths: The blue lines represent the formation of primary free radicals, the green lines represent the decay of key RFRs, the light brown lines represent the key RFR interactions, and the dark brown line represents the formation of secondary RFRs.
2.2
Types and characteristics of VUV light sources
In recent years, considerable research attention has been directed towards the advancement of long-lasting, effective, and highly stable VUV light sources [19–21]. These studies primarily fall into two main categories. The first category includes the widely used LPUV, characterized by low-pressure gas discharge, which emits an atomic line spectrum featuring principal wavelengths of 254 nm and 185 nm. LPUV radiation can be divided into two types: O3-production, and non-O3-production, depending on the quartz lamp used. As mentioned before, O2 reacts with photons emitted by 185 nm light to generate O3, and the adverse effect on quartz lamps due to ozone production results in a 50% reduction in the penetration rate of 185 nm, while high-purity synthetic quartz glass is used to cover the lamp, which can penetrate the light tube by 185 nm [22]. Additionally, the LPUV optimum working temperature is 40 ℃, and a lower temperature may cause part of the mercury vapor to coagulate on the lamp tube. Conversely, a higher temperature (>40 ℃) enhances the self-absorption capacity of mercury vapor [22]. Therefore, the output efficiency of a VUV lamp depends on the temperature inside the tube.
The other type is excimer lamps, the radiation band of which is 100–200 nm and is based on rare gases/halogen activators; these lamps represent a new generation of light sources and have developed rapidly in recent decades. The functional gases and corresponding optical properties suitable for producing VUV radiation are listed in Table S1 (Supporting information). Currently, excimer lamps are generally considered to contain excited dimers (identical atoms or molecules) and excited complexes (heterodimers). The irradiation spectra of rare gas/halogen excimer lamps have extremely narrow emission bands, which are often referred to as “quasi-monochromatic light sources” [23]. The most common excimer lamps used are xenon (Xe2*) [24], KrCl [19], XeCl [21], and XeBr [25], whose emission wavelengths are 172, 222, 308, and 282 nm, respectively. Xe2* lamps are the most popular VUV light source and have been widely used in the study of water purification. Generally, the radiation efficiency of these excimer lamps can reach 5% to 18% [23,26,27]. The performance advantages of excimer lamps include a high average power coefficient, high photon emission energy, quasi-monochromatic radiation, high spectral power, low heating requirements for radiation surfaces, no fixed geometry, and no preheating time under VUV and UV irradiation. Multiple wavelengths of UV radiation can be obtained by exciting different types of interacting excimers simultaneously [28].
2.3
VUV reactor/reaction equipment
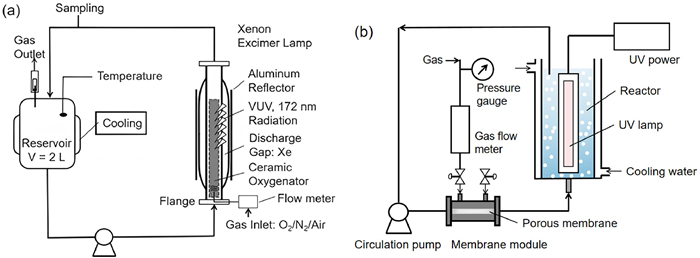
Depending on the target of processing and the application context, VUV reactors or reaction systems typically come in two types: collimated parallel beam instruments (CPIs) and circulation systems (CSs). The UV dose, which is crucial for assessing UV reactor performance, results from multiplying irradiation time by irradiation intensity (explained below). The collimator in CPIs converts scattered light into parallel light, serving as an effective tool (or platform) for accurately determining or confirming the UV dose. CPIs are rarely used in the laboratory; although CPIs have been used in a few investigations, time is still used as a standard for evaluating reaction properties, which is a misunderstanding because of the lack of calculations of UV doses. CPIs are not as widely used as CSs, but there is little difference in the design of CPIs according to diverse reports, except for the power of the configured lamps. Lian et al. [29] explored the UV photolysis kinetics of eight sulfonamide antibiotics (SA) in CPI and fine-tuned the fluence using real-time monitoring with a micro-fluorescence silica detector. However, other studies have placed more emphasis on the performance of VUV parallel optical systems driven by optics. Xiao et al. [30] investigated the contribution of primary active species under parallel light irradiation by a VUV/UV/Cl system to EP degradation. Chintalapati [31] highlighted the vital role of VUV-induced deep oxidation in MC-LR degradation in a CPI system equipped with a VUV/UV light source. Nong et al. [15] documented variations in active species at different water depths under parallel VUV irradiation, observing an 80% decrease in VUV oxidation as the water depth increased to 4 mm.
CSs are configured into sequencing batch and batch types based on the volume of water requiring treatment. Light sources are positioned within the center of the tubular UV photoreactor, where VUV/UV light interacts with substances in the solution through contact between the sleeve and the circulating water interface, the structural design of the applied semi-batch VUV reactors is shown in Fig. 3 [28]. Simultaneously, the flowing solution removes the heat generated during the irradiation process. Alekseev et al. [32] established a continuous flow reactor utilizing a single Xe2* lamp to irradiate aqueous solution and gas under a pressure of 40 atm (1 atm = 101.325 kPa), with a radiated surface area of approximately 700 cm2. Their investigation revealed that the average VUV radiated power decreased with increasing lamp temperature. Notably, in the VUV/UV CS photoreactor, the degradation rate of EPs is limited by mass transfer in the UV-irradiated reaction zone. The size of the mixing and circulation zones is a crucial factor in controlling the efficiency and energy consumption of the treatment process. Bagheri and Moheseni [33] underscored the impact of flow rate on VUV reactions’ performance, focusing on the distribution of •OH in VUV/UV sequencing batch CS reactors. They found that the volume-weighted average •OH concentration at 0.5 L/min was double that at 6.7 L/min.
Theoretically, the cumulative UV flux rate in the time range is the UV fluence (UV dose), which is the standard for evaluating UV efficiency. Chemical photometers are materials (or systems) utilized to quantify the UV flux rate via quantitative photochemical reactions. Chemical photometers, capable of detecting photons absorbed by the solution, come into play when only the product and photometers exit the target solution after photon absorption [34]. Actinometric procedures represent one of the standard methods for measuring the UV flux rate, as the photochemical behavior of a chemical remains constant and can replicate the reaction [34]. To date, the UV flux rates of LPUV and 172 nm have been determined precisely via chemical photometers by selecting specific absorption solutions for the two types of light [35,36].
Among them, potassium iodide (SSKI oral solution) is the most common absorption solution, for which the incident fluence can be measured from an LPUV that provides >85% of its energy at 254 nm [37]. Uridine-based chemical actinometers were developed more than a decade ago because of their stability, availability, and reproducibility [36]. Long et al. [38] effectively determined the luminous irradiation flux rate of 185 nm VUV by using the absorption properties of methanol, particularly in the presence of two bands at 185 nm and 254 nm. A DMAB (4,4′-dimethylazobenzene)-based actinometric procedure was also used because of its stability, convenient availability, and reusability [39]. The cis-trans photoisomerization of cis-cyclooctene and the photolysis of ethanol (Farkas actinometer) are viable options as actinometers. Imoberdorf et al. [40] employed the cis-trans photoisomerization of cis-cyclooctene to determine the radiation flux at 185 nm, resulting in an irradiation duration of 25 s, corresponding to a VUV dose of 44.8 J/m2.
3.2
UV dose calculation table
The International Ultraviolet Association (IUVA), committed to advancing ultraviolet technologies, published a guidance document titled “Protocol for the Determination of Fluence (UV Dose) Using A Low-Pressure or Low-Pressure High-Output UV Lamp in Bench-Scale Collimated Beam Ultraviolet Experiments”. This document provides a worksheet for calculating the UV dose for LPUV. By inputting the target dose and setting the system structure, operational parameters, and influencing factors in the table, researchers can determine the corresponding irradiation time before each experiment. The theoretical foundation of this calculation method, drawn from the work by Bolton and Linden [41], is explained step-by-step to aid experimental measurement. These calculation tables are accessible either from the authors directly or within the member zone of the IUVA website.
In the calculation table, Bolton [41] introduced several correction factors to adjust deviation values (Eq. 14). These factors, including the PF (petri factor), WF (water factor), DF (divergence factor), and RF (reflection factor), play crucial roles. The PF corrects irradiation at the center of the dish to ensure an average fluence across the surface area. WF accounts for absorption and the path length of water in the dish, impacting irradiation through the water, which can be absorbed, consequently reducing irradiation [41]. The DF is related to the distance between the lamp and solution surface and the path length in water. Finite distances may lead to incomplete collimation and divergence of the beam. Finally, RF means that light passes through one medium to another, and the refractive index will change [41]. In this method, these four factors and the readings of the center irradiation intensity are multiplied to obtain the average irradiation intensity.
where E0 is radiometer meter reading at the center of the dish, PF is the ratio of the average incident irradiance over the area of the Petri dish to the irradiance at the center of the dish, RF is the fraction of the incident beam that enters the water; WF is crucial to consider the decrease in irradiance caused by absorption as the beam traverses through the water when water absorbs UV radiation at a certain wavelength, DF needs to be considered because the beam is not perfectly collimated and diverges significantly for finite distances of the dish from the UV lamp. The average germicidal fluence (UV dose) (J/m2 or mJ/cm2) is then given by the product of E′avg and the exposure time t (s) [41].
3.3
Computational fluid dynamics (CFD)
The photochemical reactor system operation is affected by the water solution concentration and water matrix, the rate constants of photons absorbed by target contaminants, and the dose distribution, which are generally determined by the fluence rate distribution and fluid mechanical behavior within the reactor [42]. To successfully simulate the properties of photochemical reactor systems, overcoming the abovementioned problems is necessary. Utilizing high-performance computing tools for numerical simulation offers an efficient method to calculate the UV dose at dynamic reaction sites. Among manufacturers, the CFD model is widely utilized for optimizing the disinfection efficiency of UV systems.
The basic process of the CFD simulation is shown in Fig. S2 (Supporting information). (1) Establishing the calculation domain and grid. According to the structure and size of the VUV/UV reactor, a three-dimensional model was established to subdivide the reactor into numerous small control bodies. (2) The ideal fluid is selected, and the governing equation is established. According to the properties of the medium, the appropriate equation of state was selected, and the Navier–Stokes equation and the mass and heat transfer equation were established. (3) Confirm the boundary conditions. The inflow and outflow boundary conditions and interface conditions at the boundary of the computational domain are specified. (4) A numerical method is selected to solve the governing equations. The finite volume method or finite element method is used for discretization and solution. (5) Lumiere program inspection and verification.
Wright et al. [43] outlined several crucial aspects of CFD modeling, focusing on the transient dose, flow patterns, and particle trajectories. Their findings emphasized the significance of the Reynolds stress model in accurately predicting velocity and dose, highlighting the necessity for higher-order momentum differences. Bagheri and Mohseni [44] developed a comprehensive CFD model for simulating VUV/UV photoreactors used in water treatment. This model considered over 40 parameters, including fluid dynamics, material mass transfer, and irradiance distribution at wavelengths of 185 nm and 254 nm. Their study underscored the critical role of mass transfer near the quartz sleeve in limiting the degradation rate of micropollutants within the VUV/UV photoreactor. Peralta et al. [45] investigated the mechanism behind enhancing customized secondary flows in spiral microcapillary membrane (MCF) VUV photoreactors through CFD analysis, particularly in removing EPs (e.g., antiviral agents: acyclovir, stavudine and zidovudine, and biocidal antifungal agents: methylisothiazolinone, benzothiazolinone and isoxazole). Their findings revealed that the second-order reaction rate constant of the six EPs with •OH increased (ranging from 4.4% to 37.9%) in helical MCF photoreactors compared to assumptions made for MCF photoreactors with plug flow.
4.
EPs abatement by VUV technology
VUV technology is currently predominantly applied in water treatment for two main purposes: Breaking down organic pollutants and sterilization. It offers remarkable advantages in completely decomposing EPs in water, especially those composed of highly stable organic compounds. These persistent EPs cover a range of substances, including antibiotics (e.g., norfloxacin [46], cloxacillin [47], tetracycline [48], sulfamethoxazole), persistent organic pollutants (e.g., perfluoro caprylic acid [49], perfluorooctane sulfonate [17], carbamazepine, methyl tert-butyl [50], GenX ether, atrazine [51], p-chlorobenzoic acid [52], and endocrine disruptors (e.g., acetaminophen [53], bisphenol A [54], metformin (Glucophage) [55], trimethoprim). Key research areas include studying the kinetics and reactivity of active substances, understanding the interference characteristics of inorganic ions, and elucidating the degradation pathways and intermediates involved in EPs decomposition.
Szabó et al. [56] discovered that 254 nm UV radiation has a significant effect on ketoprofen degradation, while 185 nm VUV radiation accelerated the degradation rate of ibuprofen. The predominant role in the degradation of phenols is played by the •OH produced by the VUV system [57,58]. It was proposed by Wu et al. [59] that SO42– was first released due to •OH attack on the S=P bond of dimethoate in a VUV/UV system, which governed the dimethoate degradation efficiency. The effect of neglected excitation on the degradation of organic phosphorus pesticides was studied by Liu et al. [60], who reported that organic phosphorus pesticides were transformed into an excited state after VUV irradiation and that the number of selective free radical attack sites changed. Yan et al. [61] utilized the VUV/UV process to treat fluorine-containing pharmaceutical wastewater, resulting in an increase in the biodegradability index of 4-fluorophenol from 0.24 to 0.47 after VUV/UV irradiation. Gonçalves et al. [62] revealed that the toxicity of a ketoconazole solution was significantly reduced via VUV/UV treatment.
4.1
Direct photolysis of VUV
Some EPs have the capacity for direct photolysis through UV photon absorption or by reacting when other compounds in an excited state transfer energy (sensitized photolysis). For direct photolysis to occur, there must be an overlap or partial overlap between the absorption spectrum of organic matter and the UV spectrum reaching the water layer [63]. Hence, the feasibility of direct photolysis is closely linked to the molecular structure of organic matter, including light-absorbing functional groups and intramolecular hydrogen bonds, as well as factors like pH and metal ions that can alter molecular structures. PFCs, such as PFOS, are exemplary refractory EPs susceptible to direct photolysis by VUV [49]. Gu et al. [17] highlighted the significant role of direct VUV photolysis in PFOS degradation under slightly acidic conditions. After absorbing 185 nm photons, PFOA and other PFCs transition to an unstable excited state, facilitating reactions like fragmentation and gradual degradation (Eqs. 15–18) [49,64]. Xie et al. [8] suggested that VUV’s destructive effect on the toxic imipramine originates from the compound’s own excited state post-light absorption.
The molar absorption coefficient (ε) and quantum yield (Φ) are essential parameters for evaluating how organics utilize photon energy in VUV/UV systems [65]. ε indicates how well a molecular structure absorbs specific UV radiation wavelengths, while Φ represents the ratio of converted product moles to absorbed photons. Currently, data on UV absorption (usually within 200–420 nm) and the use of various chemicals in water, including different EPs [11], oxidants [66], and oxidizing free radicals [67], have been collected. However, due to the absence of a key absorbance index (А) of organics in the VUV range, there is often a lack of ε indicators in academic reports on VUV treatment of EPs. Only a few reports include ε values for organic matter, typically using them as indicators instead of processing targets (Table 1). The calculation formula for Φ is provided in Supporting information. The Φ of organic matter does not seem to correlate with the degradation rate (k) and is likely influenced by the А of the substance in aqueous solution. However, sparse data make it challenging to assess these parameters. Notably, photolysis rates vary significantly among chemicals with the same functional group, such as CIP, atrazine, and cefalexin with amine groups (–NH–). Furthermore, extensive research has explored the direct photolysis behavior of other EPs under VUV. The development of a microfluidic photocatalytic reaction system can enhance the efficiency of VUV utilization in water molecules to some extent and provide insights into the impact of VUV direct photolysis on removing iodinated EPs. Pourakbar et al. [68] treated the antibiotic amoxicillin (AMX) with a VUV/UV system; 254 nm UV radiation required about 50 min to degrade the AMX by approximately 90%, whereas 185 nm VUV irradiation took only 3 min to completely degrade the AMX. Wen et al. [69] explored the decomposition of sulfamethazine (SMN) by VUV/UV photo-Fenton (VPF) and reported that the absorption energy of VUV photons from SMN reached 50.9% at a high concentration (90 µmol/L). Li et al. [6] compared VUV/UV, UV photolysis, and adding reagent systems for the treatment of chloramphenicol (CA) and reported that direct VUV photolysis is still the main degradation pathway for CA.
$
\mathrm{PFOA}+h v \rightarrow \mathrm{C}_7 \mathrm{~F}_{15}^{\cdot}+{}^{\cdot} \mathrm{COOH}
$
Annotation: * present the data given by the original texts, others are calculated by the data in the original texts.
4.2
Oxidation substances
In addition to the direct photolysis of VUV [70–79], •OH produced by water decomposition induced by VUV can react nonselectively with EPs of large or small molecules, which is another essential tool for attacking organic compounds that cannot be preferentially photolyzed. The main reaction pathways of •OH include hydrogen substitution, addition reactions and radical ring-opening reactions. The rate constant of the reaction with organic matter is 106 to 1010 L mol-1 s–1 [68], and the lifetime of the reaction in natural water generally does not exceed 10–4 s. The steady-state surface concentration of •OH in natural water was (0.15–50) × 10‒17 mol/L under typical sunlight radiation levels [80]. However, there is a lack of reference data for the steady-state concentration under VUV irradiation. Huang et al. [81] demonstrated that •OH’s oxidation contribution was exceptionally high, reaching 92.7%, in the degradation process of methyl isothiazolinone (MIT) using a VUV/UV system. The degradation of sulfadimidine (SMN) and amoxicillin (AMX) at low concentrations (0.02 µmol/L) relies entirely on •OH’s role in the VUV/UV system [68].
Due to •OH’s high reactivity with various organic compounds, different probe molecules, such as terephthalate (TPA) [82], benzoic acid (BA) [66], and coumarin (COU) [83], are utilized for its detection. Among these probe molecules, COU is more suitable for the quantitative detection of •OH in complex aqueous solutions due to its sensitivity, linearity, and response range.
4.3
Reduction substances
4.3.1
Primary reductive products
The primary reducing free radicals produced by VUV photolysis of water are the hydrogen atom (•H) and hydrated electrons (eaq–). •H is a strongly reducing species (E0 = −2.3 V in acid solution) and is formally the conjugate acid of eaq– (E0 = −2.9 V) [84]. •H abstracts hydrogen from saturated organic molecules and adds to structures (Eqs. 19 and 20). Its chemical reactivity resembles that of •OH radicals in its reactions with multiple organic substrates, although it is less reactive and more selective [13]. Although •H has a higher yield than eaq–, it can easily recombine into H2 (Eq. 13), and its solubility in water is low. Neglecting the reaction process of •H with oxygen-containing intermediates, the reactivity of eaq– with organic matter deserves attention.
eaq– is a potent reducing agent with a reduction potential of E0 = −2.9 V. The increase in eaq– yield in alkaline solutions indicates that its formation originates from the photolysis of OH– [85] and the contribution of •H (Eq. 6). However, some researchers have observed a gradual rise in eaq– with a quantum yield of 0.05 ± 0.01 for excitation wavelengths ranging from 175 nm to 200 nm. Moreover, for pH values between 4 and 9, the eaq– production remains consistent, irrespective of pH [86,87]. The low quantum yields of eaq– (Φeaq– < 0.04) in acidic environments may be attributed to rapid recombination with H3O+ in the absence of scavengers (Eq. 5). However, the mechanism of eaq– production is a matter of debate. One viewpoint suggests that photonically excited H2O may transfer an electron to the bulk, facilitated by the energy generated from the hydration of both H2O+ and electrons [85]. Consequently, alkaline solutions saturated with H2 are considered nearly ideal for the photonic production of eaq– due to the increased yield resulting from the reactivity of •OH and H2 (Eq. 21). Nevertheless, it is undeniable that HO– excitation electron transfer below 200 nm [88] is significant. Therefore, charge transfer from HO– to bulk H2O may represent another essential photochemical pathway for eaq– formation, according to these research findings.
As a nucleophile, eaq– demonstrates significant reactivity with organic compounds containing electron-absorbing groups (M) (Eq. 22). Halogenated hydrocarbons’ M– (RX–, X = F, Cl, Br, or I) swiftly eliminate halides (X–) (Eq. 23), confirming their active reactivity with eaq– [89]. Within a homologous series of halogenated aliphatic compounds, reactivity with eaq– typically follows the sequence of $ \mathrm{F} \ll \mathrm{Cl}<\mathrm{Br}<\mathrm{I} $. For instance, the relative reactivity of fluoroacetate, chloroacetate, bromoacetate, and iodoacetate is 0.002 < 1.0:6.2:12.0 [90]. Li et al. [91] reported that the degradation of monochloroacetic acid (MCAA) proceeded via reductive dechlorination induced by eaq– (Φeaq– = 0.116 ± 0.002 mol/Einstein), and all the chlorine atoms in MCAA were released as chloride ions in the pH range of 6.0–8.7. The dual roles of pH were to regulate the S(Ⅳ) species distribution and to control the interconversion between eaq– and •H.
Another typical reaction characteristic is that eaq– is not very reactive toward alkanes, while the reactivity of eaq– strongly depends on the substituents attached to the hydrocarbon chain, and its high reactivity should be noted. For example, for alkylchlorides, CCl4 reacts very slowly with •OH radicals for decomposition into different intermediate products (TPs). Conversely, dechlorination by eaq– is almost diffusion controlled (Eqs. 24 and 25). The depletion of CCl4 observed during VUV photolysis is mainly attributed to the direct reaction with eaq– in the absence of other organic substrates [92].
In a reductive reaction medium, pathways leading to the efficient mineralization of atrazine are favored, where reactions with eaq– compete with those induced by •OH [85]. Effective reactions of eaq– with similar compounds containing amino groups, such as uracil and cytosine derivatives, have been reported, with rate constants of 3 × 108 and 1 × 1010 L mol–1 s–1, respectively [13]. These reactions result in the formation of hydrogen adducts.
4.3.3
Identification of eaq–
The foundation of eaq– reaction kinetics relies on the necessity for empty orbitals to accommodate incoming electrons [93]. This distinguishes it from other highly reactive free radicals (•OH, •H, etc.) due to differences in redox potential. Challenges in identifying eaq– include the dense arrangement of water molecules, capturing metastable complexes, and tracking electron numbers. Consequently, precise detection methods for eaq– are still confined to absorption spectroscopy and chemical reagent product detection.
4.3.3.1
Absorption spectroscopy
Due to its strong absorption in the near-infrared band (700 nm), transient absorption spectroscopy (TA) is the most direct detection method. Huang et al. [94] demonstrated a prominent absorption peak at 690 nm using a 4.0 × 10–5 mol/L K4Fe(CN)6 solution exposed to flash light. Electron spin resonance (ESR) or electron paramagnetic resonance (EPR) techniques allow visualization of free radical signals by extending the adduct’s lifespan with a trapping agent. DMPO (5,5-dimethyl-1-pyrroline N-oxide) and TEMP (2,2,6,6-tetramethylpiperidine) are popular spin traps for free radicals [9,95,96]. The pseudo-first-order termination reaction of the protonated DMPO-hydrated electron adduct (DMPO–H) with DMPO–OH was observed for the first time by Madden [97]. The rate constant for the reaction of DMPO–H with DMPO–OH is (2.44 ± 0.22) × 102 L mol–1 s–1. However, signals from DMPO–H and TEMPX can sometimes be misleading in situ production tests of eaq– as DMPO–H/TEMPX can also be obtained through single-/multistep one-electron oxidation of DMPO without the participation of eaq–. Accurate detection of eaq– typically involves multiple spectral synergies and is verified through chemical reagent methods. Liu et al. [9] explored eaq– formation via ESR and TA measurements, then utilized a typical eaq– probe, CdSO4, as a specific scavenger for eaq– quenching experiments.
Moreover, the absorption characteristics of eaq– can be analyzed using diverse models and methodologies. Julien et al. [98] conducted a study investigating the influence of nonreactive metal cations on the manifestation of eaq–. They measured visible absorption spectra using concentrated aqueous solutions containing eight chlorides and five perchlorates of varying valences. They observed that the increased mobility of the absorption bands was not solely correlated with salt concentration but also with the cationic charge. Their findings indicate that under high pH conditions (corresponding to a high concentration of current mainstream treatment methods or K+), eaq– is not present in a free state but forms an ion pair in solution.
4.3.3.2
Chemical reagent detection
Similar to the process of contaminant removal by eaq– from water, eaq– can also interact with certain probe compounds containing specific groups, like formic acid groups. Detection of eaq– can be achieved by observing intermediate products or changes in spectral absorption rates at 700 nm. Ransom et al. [99] determined the rate constant (k) for the reaction between tert-butyl formate (TBF) and eaq– (k = (5.48 ± 0.09) × 108 L mol–1 s–1), which closely matches the rate constant of •OH (k = (5.37 ± 0.07) × 108 L mol–1 s–1). Stephen et al. [100] studied the reaction of eaq– with halogenated nitromethane in water, determining the reaction rate constant by fitting a first-order kinetic decay parameter of the substance and monitoring absorbance at 700 nm.
Moreover, CuSO4 and N2O are identified as quenchers of eaq–, demonstrating high second-order reaction kinetics of 6.4 × 1010 and 9 × 1019 L mol–1 s–1, respectively [101]. Detecting the reaction effect post-quenching allows for either the exclusion or confirmation of the presence of eaq–. Unlike probe compound indicators, quenching methods lack precise control over the dosage of chemical reagents, leading to frequent questioning of their accuracy due to experimental observations suggesting that the oxidizer bulk is quenched before free radicals are extinguished during quenching [102].
5.
Factors affecting EPs destruction in the VUV
5.1
Temperature
The optimal operating temperature for LPUV is 40 ℃, with lower temperatures potentially causing some mercury vapor to condense on the lampshade. Conversely, temperatures exceeding 40 ℃ enhance the self-absorption ability of mercury vapor, thereby affecting the VUV/UV output of LPUV. It is important to highlight that the absorbance coefficient of water at 184.9 nm ranges from 1.46 cm–1 to 1.80 cm–1 at 25 ℃, while research by Kröckel and Schmidt [103] indicates a specific absorbance coefficient of 1.62 cm–1 at 184.9 nm. However, the absorbance coefficient at 187 nm exhibits a linear relationship with temperature. For instance, as the temperature rises from 10 ℃ to 30 ℃, the absorbance coefficient increases from 0.45 cm–1 to 0.67 cm–1 [104]. Consequently, it is essential to consider the impact of temperature on the absorption of VUV by purified water, which affects the homolytic cleavage and photochemical ionization of water molecules.
5.2
pH and dissolved oxygen (DO)
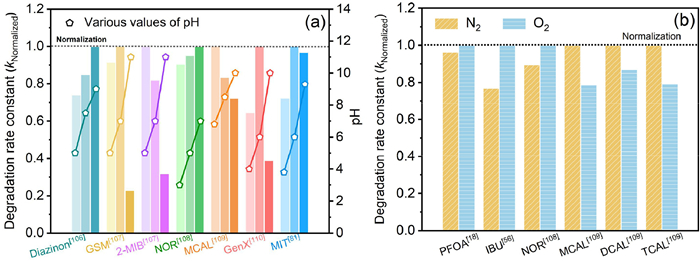
In photoinduced chemical reactions, the rate constants for protonation and deprotonation of target compounds vary [105], as do the pH-dependent structures of the active components containing •OH, •H, and eaq– (Fig. 4a) [106–110]. The concentration of •OH at pH 11 is lower compared to pH 3.4, resulting in a faster removal rate of many EPs under acidic conditions than alkalis [13]. This phenomenon is observed in citric acid under 172 nm VUV irradiation [111] and cloxacillin under 185 nm [112]. The •OH reaction rates for complete deprotonation (6.4 × 109 L mol–1 s–1) at pH 0) and single protonation (1.1 × 1010 L mol–1 s–1 at pH 6.8) of gallic acid differ, but the pH in the range of 2.5–7.5 shows no significant effect under 172 nm VUV irradiation [13]. Another aspect affected by pH in EPs VUV removal is the reduction potential of •OH, with the oxidation potential being 2.57 V at pH 0 and 2.18 V at pH 7. Consequently, this leads to a decrease in the decomposition rates of 1,4-dioxins, bromine, and 2-MIB as pH increases within the range of 5.5–8.0 [113].
Figure 4
Figure 4.
The pH (a) and DO (b) dependence of EPs degradation rate in the process of VUV185 treatment.
Both •H and eaq– are easily captured by DO, and the transformation of organic matter in DO-containing solutions primarily occurs through reactions with •OH (Eqs. 7 and 8). In highly basic solutions, DO can also capture •OH, generating a substantial number of ozone anion radicals, thus the presence of DO will affect the degradation rate of EPs (Fig. 4b) [18,56,108,109]. Laszlo and Dombi [114] employed [Fe(CN)6]4– and [Fe(CN)6]3– to evaluate the oxidation and reduction properties of purified water under 172 nm irradiation. While there was no significant difference between the oxidation and reduction rates in an anaerobic solution, the presence of DO notably increased the oxidation rate of [Fe(CN)6]4– while diminishing its reduction properties.
The photolysis of purified water without DO under VUV generates negligible H2O2. In the presence of DO, H2O2 can also be created via Eq. 26, whereas at higher pH, H2O2 generation reaches a maximum at pH ≈ pKa [115]. The increased yield of H2O2 induced by DO strongly interferes with the VUV photolysis of many EPs. Azrague et al. [116] discovered that the presence of H2O2 boosted the degradation rate of oxalic acid in a VUV system by 25%. Interestingly, after the reaction ceased, the quantity of H2O2 returned to its initial level in the original aqueous sample. Robl et al. [112] supported this finding, indicating that •HO2, generated through the accumulation of DO and organic peroxygen radicals, played a pivotal role in removing methanol from aqueous solutions via VUV treatment. Furthermore, multiple studies have indicated that the increase in H2O2 levels is a result of the increased concentration of •HO2/•O2 formed during the oxidative decomposition of organic compounds [117].
The reaction rate of NO3– with •OH is almost negligible, but it is highly sensitive to reactions with reducing components. NO3– reacts with eaq– at a diffusion-controlled rate, and the rate constant of the reaction with •H is approximately three orders of magnitude lower than that of the reaction with the product of •(NO3H)– (Eqs. 27 and 28). NO3– competes effectively with DO for eaq– with the products of •(NO3)2– [90], which is a critical factor that leads to an evident decrease of an order of magnitude in PFOS in the VUV treatment with the addition of NO3– [17]. •(NO3H)– and •(NO3)2– can reach an acid‒base equilibrium at pKa ≈ 7.5, and •(NO3H)– may further resolve to •NO2 in the absence of other substrates, dimerize to N2O4 and/or dismutate to NO3– and NO2– (Eqs. 29–32), which is exactly the photolysis source of the uncommon NO2– in water, especially in situ under LPUV light. NO2– reacts with almost all the free radicals of photolysis water under VUV at diffusion-controlled rates (Eqs. 33–35). In addition, NO2– can be rapidly eliminated by ozone produced at 172 nm and/or by LPUV in combination with ozone [118].
Cl- is also a kind of representative photons and RS competing species, and the chlorine free radical (Cl•) generated by its reaction with •OH has high reactivity with the majority of organic compounds. For instance, the presence of Cl- significantly increases the level of decomposition of organohalogens. Gu et al. observed that Cl- inhibited the conversion of 1,1,1-trichloroethane in VUV/UV-radiated solutions [119].
Natural organic matters (NOM), which includes aromatic and oxygen-containing functional groups (such as humic acid and fulvic acid), is a typical •OH scavenger and UV absorber [5,120]. The high-molecular-weight conjugated compounds within hydrophobic NOM components undergo continuous decomposition, competing for photon energy or free radicals (i.e., •OH, •H, eaq–, •O2–) with EPs or other target substances, resulting in the formation of degradable charged (hydrophilic) and neutral (hydrophilic) structures. Conversely, low-molecular-weight NOM, due to its non-photodecomposition, can only undergo mineralization through reaction with the •OH generated from the decomposition of water molecules under VUV radiation. In the degradation of tris(2-chloroethyl)phosphate (TCPL), the addition of humic acid (HA) to the aqueous solution led to a continual decrease in the reaction effect until the added amount of HA reached 46% of the TCPL concentration [76]. Dong et al. [121] observed significant inhibition of the direct photolysis of florfenicol (FLO) once the concentration of HA exceeded 5 mg/L as an interference additive. This inhibition could negatively impact the yields and reactivity of eaq– and ·H with FCL. However, for NOM, humic acid has been demonstrated to aid in the photolysis of PFOS at concentrations ≤ 10 mg/L, possibly due to the electron donors of phenolic compounds enhancing the bimolecular reaction rate between eaq– and PFOS [122]. Further research is essential to unravel the pivotal role of NOM in the VUV process, particularly concerning eaq– scavenging and potential NOM-EP interactions.
6.
Challenge and prospects
Scholars have been exploring various methods to produce high-energy VUV radiation, including excimer light sources, solid-state luminescent light sources, synthetic quartz-coated low-pressure mercury lamps, pulsed radiolysis, and laser lightning photolysis. They are also investigating the mechanisms of free radicals and pathways generated by different VUV radiation sources during water photolysis. VUV radiation has shown promise in efficiently and thoroughly mineralizing recalcitrant EPs and systematically decomposing macromolecule NOM within a short time frame. Because water serves as the primary absorbent, the total yield of •OH, •H, eaq–, and other free radicals derived from water photolysis by VUV exceeds that of any other advanced oxidation technology (AOPs) [123]. Additionally, when O2 in the gas phase produces O3 through VUV irradiation, there is potential for synergistic effects between VUV photolysis and O3. This could eliminate the need for an additional O3 generator and enhance the overall decontamination and disinfection efficacy of the system. The combined VUV/O3 technology serves as a safeguard against waterborne NO2– and aids in disinfecting secondary NO2–.
Various researchers have addressed misconceptions about the high energy consumption of VUV technology at the application level, emphasizing the advantages of VUV or UV technology alone compared to UV/persulfate combinations [70]. However, the widespread adoption of VUV technology is often hindered by limitations in output power and efficiency. Depending on the brand and type of LPUV, the relative radiant exitance of the line at 185 nm can vary between 5% and 10% of that at 254 nm. Excimer lamps, unlike mercury vapor lamps, offer several benefits, such as mercury-free operation, high photon energy, clear emission lines, instant switching, and no UV power loss. Moreover, their design can be tailored to suit various reactor configurations. The rapid advancement of excimer lamps in recent years has expanded the potential applications of VUV water decontamination and multifunctional reactor development. Nonetheless, the application of VUV technology in water treatment still faces challenges due to the limited penetration depth of VUV radiation and the heterogeneous nature of VUV water-based properties [124].
When considering the potential of VUV technology for EPs abatement, we suggest focusing on the following areas:
(1) Recognizing the significance of the irradiated volume is crucial. Within this volume, the homolysis of water takes place, typically confined to a thin liquid sheet covering the surface of the tube housing the VUV radiation source. The considerable absorption cross-section of H2O at VUV wavelengths creates a distinct boundary between the irradiated and non-irradiated reaction volumes, resulting in spatially varying concentration profiles of all species participating in a simultaneous array of reactions. While reaction rates are influenced by substance concentrations, they also rely on the lifetimes and diffusion at the flow interface, especially in scenarios with multiple coexisting radicals.
(2) Establishing standardized methods for measuring UV dose during EP abatement is essential. While UV disinfection standard dose measurements are already well-developed, future efforts in VUV radiation should concentrate on devising methods capable of quantifying the degradation of organic pollutants while considering potential interference from background substrates on the effective dose. By verifying model compounds through irradiation, treatment efficiencies among studies can be compared. These measurements will also aid in simulating pollutant conversion efficiencies in various backgrounds, determining whether VUV can be implemented on the same scale as UV-AOP.
(3) The role of secondary reactive species in VUV photolysis of water is significant. There exists a plethora of bimolecular rate constants for oxidizing/reducing free radicals (such as •OH, •H, eaq–) and various organic and inorganic contaminants. This abundance leads to the formation of energy transfer and diffusion of secondary radicals in the distant water layer, typically beyond 5 mm. These secondary radicals interact with sensitive reaction rates and intermediates involving DO, NOM, nitrate, carbonate, and other inorganic ions, in conjunction with primary radicals. It is advisable for researchers to intensify their exploration of more sensitive and specific probe compounds and/or photoreceptors for secondary radicals. This approach will aid in identifying the contribution and reactivity of free radicals directly produced by VUV photolysis and their subsequent diffusion.
Despite numerous limitations, advanced water treatment using VUV technology holds promise for niche applications. One such example is the integration of activated carbon filtration technology following VUV treatment, serving as a purification mechanism for hazardous synthetic organic compounds in secondary water supply processes. To explore these VUV-AOP/ARP applications further, ongoing bench-level experiments and increased focus on pilot research are necessary. Future studies should aim to assess the placement of VUV and VUV-based treatment strategies within the broader context of contaminant management in water, particularly concerning EPs.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
E.M. Payne, B. Liu, L. Mullen, K.G. Linden, Environ. Sci. Technol. Lett. 9 (2022) 779–785. doi: 10.1021/acs.estlett.2c00472
[21]
S. Baadj, Z. Harrache, A. Belasri, Plasma. Phys. Rep. 39 (2013) 1043–1054. doi: 10.1134/S1063780X13120015
[22]
W.J. Masschelein, Ultraviolet light in water and wastewater sanitation, in: R.G. Rice (Ed.), Available Lamp (Or Burner) Technologies, LEWIS Publishers, 2013, pp. 9–54.
M. Gu, L. Liu, G. Yu, et al., Environ. Sci. Technol. 57 (2023) 15288–15297. doi: 10.1021/acs.est.3c03308
[71]
E.B. Esfahani, M. Mohseni, et al., Environ. Chem. Eng. 10 (2022) 107050. doi: 10.1016/j.jece.2021.107050
[72]
C. Duca, G. Imoberdorf, M. Mohseni, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng. 52 (2017) 524–532. doi: 10.1080/10934529.2017.1282770
D. Wang, A.L. Junker, M. Sillanpää, et al., Engineering 23 (2023) 19–23. doi: 10.1016/j.eng.2022.08.005
Figure 1
The bond energy values of a typical chemical bond corresponding to the photon radiant energy value, indicated at the bottom of the graph, are wavelength ranges corresponding to definitions of radiation types. Source: Based on Photolysis and the Hydroxyl Radical. http://butane.chem.uiuc.edu/pshapley/environmental/l14/1.html.
Figure 2
The formation and interconversion of key and secondary RFRs in aqueous solution and O2 photolysis under VUV irradiation. The heterochromatic lines represent different paths: The blue lines represent the formation of primary free radicals, the green lines represent the decay of key RFRs, the light brown lines represent the key RFR interactions, and the dark brown line represents the formation of secondary RFRs.

 DownLoad:
DownLoad:

 下载:
下载:





