School of Environmental Science and Technology, Key Laboratory of Industrial Ecology and Environmental Engineering (Ministry of Education), Dalian University of Technology, Dalian 116024, China
b.
Department of Civil Engineering, The University of Hong Kong, Hong Kong SAR 999077, China
c.
Department of Civil Engineering, Faculty of Engineering, University of Peradeniya, Peradeniya 20400, Sri Lanka
Received Date:
08 January 2025 Accepted Date:
20 May 2025 Revised Date:
20 March 2025 Available Online:
15 February 2026
Abstract:
Peroxymonosulfate (PMS)-based advanced oxidation technology has been proven to be a viable option for the decontamination of organic pollutants from water bodies. Advanced catalyst design is essential to this technology. Herein, a vanadium-doped LaFeO3 perovskite (LFO-V) featuring asymmetric Fe-O-V sites was rationally designed. Thanks to orbital electron interaction between Fe and V atoms, the modified electronic structure elevated electron density near the Fermi energy level while reducing the energy barrier toward effective PMS activation. This facilitated concurrent PMS reduction at the Fe sites to generate SO4•- and •OH (57.7%), and PMS oxidation at V sites to produce 1O2 (42.3%). The LFO-V/PMS system demonstrated excellent tetracycline (TC) degradation performance with a 2-fold enhancement in rate constant compared to that of pristine LFO. Further, the LFO-V maintained long-term stability, and the toxicity of degradation intermediates was evaluated through microbial metabolomics. This work establishes an effective route to regulate the PMS activation pathways through precise electronic structure modulation, advancing the rational design of advanced Fenton-like catalysts.
Developing efficient and affordable water purification technologies remain a critical challenge for sustainable societies [1]. Peroxymonosulfate (PMS)-based advanced oxidation processes (AOPs) have been gained significant attention for their ability to degrade pollutants through the generation of highly reactive oxygen species (ROS) [2], including radical species (SO4•- and •OH) and non-radical species (1O2) [3]. The high redox potentials of SO4•- (E0 = 2.5 to 3.1 VNHE) and •OH (E0 = 1.9 to 2.7 VNHE) achieve superior mineralization efficiency but suffer from poor selectivity and rapid quenching by ubiquitous matrix components, (e.g., dissolved organic matter, chloride, and bicarbonate) [4-6]. In contrast, 1O2 exhibits enhanced selectivity and environmental persistence yet demonstrates compromised mineralization capacity [7, 8]. Thus, combining radical and non-radical pathways may integrate improved selectivity and mineralization capability.
Asymmetric triple-atomic sites have been proposed to endow dual reactive sites with oxygen-bridged to simultaneous generate both radical and non-radical species [9]. Initially, PMS initially undergoes chemisorption on low-valent metal sites, followed by a reduction reaction that leads to the generation of SO4•- and •OH. Subsequently, PMS re-adsorbs onto the high-valence metal sites, leading to the production of 1O2 through an oxidative reaction [10, 11]. Crucially, the bridging oxygen atom serves as an ultrafast electron highway to promote rapid electron transfer. Zhou et al. [12] engineered asymmetric Co-O-Bi sites to achieve efficient water purification via radical and non-radical species cooperativity. Zhan et al. [13] developed Fe-O-Cu sites to achieve pollutants degradation rate up to 7.7 times higher than that of conventional Fe-O sites, thanked to the coupling effect of radical and non-radical species. Thus, it is crucial to construct asymmetric triple-atomic sites for enabling effective pollutants removal via PMS activation.
Electronic structure modulation has been proposed recently, rearranging the electron density of active sites near the Fermi energy level to tune the adsorption strength of reaction intermediates, thus improving the overall activity [14, 15]. Li et al. [16] synthesized vanadium (V)-doped Co2P4O12 to adjust the electron density around the Fermi energy level. The interaction of orbital electrons led to an upward shift of the Co d-band center, which achieved superior electrocatalytic activity for water and urea splitting at low overpotentials. Meanwhile, the high valence V(V) also displayed an oxidative capacity for partial catalytic reactions [17]. Hence, constructing triple-atomic sites with V may further optimize the electronic density near the Fermi energy level, providing additional sites for PMS oxidation. As the most important parent perovskite oxide, LaFeO3 exhibits outstanding structural tunability and chemical stability, making it widely utilized in AOPs for wastewater treatment [18-20]. The design of asymmetric Fe-O-V triple-atomic sites by V-doped LaFeO3 may be an ideal platform to fine-tune the electron density of dual metal sites near the Fermi energy level to upgrade the yield of radical and non-radical species.
Inspired by the above discussion, we first constructed asymmetric Fe-O-V sites on V-doped LaFeO3 (LFO-V) (Fig. S1 in Supporting information). The orbital electron interaction between V and Fe shifted the d-band center of LFO-V upward, reducing the energy barrier for PMS activation. This enabled efficient PMS oxidation at the V sites and PMS reduction at the Fe sites. Then, tetracycline (TC) was selected as the model pollutant to evaluate the catalytic performance of catalysts. This work not only provides rational design principles for heterogeneous catalysts, but also offers valuable insights into the mechanism of PMS activation.
As illustrated in Fig. S2 (Supporting information), new peaks appeared at 2θ values of 27.7° and 30.1° for the LFO-V0.2 catalysts with V doping, indicating the formation of LaVO4 [21]. Moreover, LFO-V0.2 exhibited a higher specific surface area (29.31 m2/g) and larger pore size (17.41 nm) compared to LFO (13.68 m2/g and 3.05 nm). High resolution transmission electron microscope (HRTEM) images of LFO-V0.2 displayed lattice fringe of 0.27 and 0.32 nm, corresponding to the (121) crystallographic plane of LaFeO3 and the (120) crystallographic plane of LaVO4 (Fig. S3 in Supporting information) [21]. As shown in Fig. S4 (Supporting information), the Fe 2p3/2 spectra exhibited two peaks at 710.5 and 709.2 eV, which were attributed to Fe(II) and Fe(III) species, respectively [22]. After V doping, the Fe(II) content in LFO-V0.2 (57.2%) was higher than that in LFO (46.5%), suggesting that V doping facilitated electron transfer from V to Fe. Moreover, the V 2p spectra showed two peaks at 516.2 and 517.5 eV, corresponding to V(IV) (41.6%) and V(V) (58.4%) [23]. Previous studies have indicated that the high-valence V enhances PMS oxidation, while low-valence V promotes PMS and Fe(III) reduction [24]. Then, the O 1s spectrum was deconvoluted into four peaks at 528.5, 529.6, 530.4 and 532.1 eV, which were assigned to metal-oxygen bonds (OL), deficient oxygen (OV), adsorbed hydroxy (OA), and water (OH), respectively [25]. Notably, the higher intensity of OV in LFO-V0.2 indicated the formation of abundant oxygen vacancies by V doping.
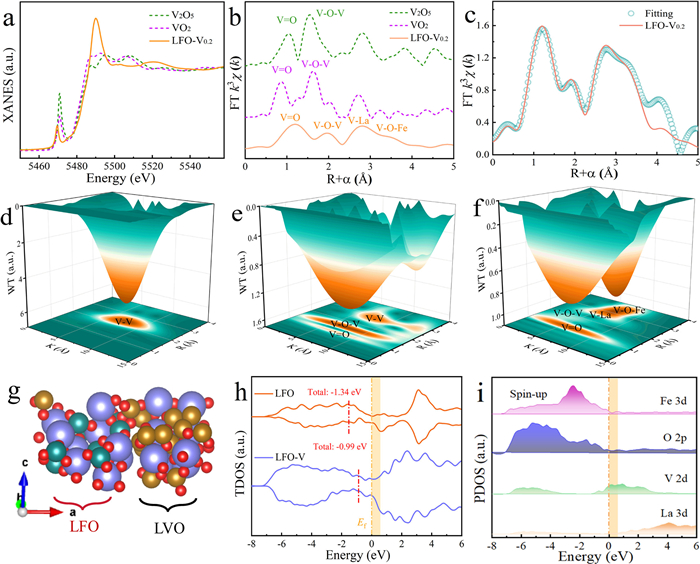
X-ray absorption fine structure (XAFS) analysis was conducted to examine the electronic structure and local coordination environment of the LFO-V0.2 catalyst. Fig. 1a showed the normalized X-ray absorption near edge structure (XANES) of the LFO-V0.2 with VO2 and V2O5 as references. The V K-edge intensity for LFO-V0.2 closely matched that of V2O5, suggesting an average oxidation state of +5 for V in LFO-V0.2. Next, the atomic configuration of V species in LFO-V0.2 was determined using extended X-ray absorption fine structure (EXAFS, Fig. 1b). For V2O5, peaks at approximately 1.0 Å and 1.5 Å were assigned to V=O and V-O-V bonds, while for VO2, the peaks at around 0.9 Å and 1.6 Å were similarly assigned to V=O and V-O-V bonds [26]. In case of LFO-V0.2, the first shell of V=O bond at ~1.6 Å, the second shell of V-O-V bond at ~1.9 Å, the third shell of V-La bond at ~3.1 Å, and the fourth shell of V-O-Fe bond at ~3.4 Å were clearly observed. Moreover, the fitted R-space EXAFS spectrum for LFO-V0.2 revealed that each V atom was coordinated with eight oxygen atoms, indicating a coordination number of 8 for the V-O-Fe bond (Fig. 1c and Table S1 in Supporting information). Compared to V foil and V2O5 (Fig. 1d and e), the wavelet transform (WT) contour plots further supported the co-existence of V=O, V-O-V, V-La and V-O-Fe coordination bonds in LFO-V0.2 (Fig. 1f) [27]. Thus, the incorporation of V into LFO was expected to induce localized structural perturbations, which would enhance the catalytic activity of LFO-V0.2 for effective PMS activation.
Figure 1
Figure 1.
(a) Normalized V K-edge XANES spectra and (b) EXAFS spectra of V2O5, VO2 and LFO-V0.2. (c) The EXAFS fitting curves of LFO-V0.2 at R space, and (d) the WT contour plots of (d) V foil, (e) V2O5 and (f) LFO-V0.2. (g) The atomic structure model, (h) the total density of state of LFO and LFO-V, and (i) the project density of state of Fe 3d, O 2p, V 2p and La 3d orbitals in the LFO-V.
Density functional theory (DFT) calculations were performed to further investigate the electric structure of LFO and LFO-V catalysts. The molecular geometry structure of LFO-V were illustrated in Fig. 1g with LaFeO3 in the left panel and LaVO4 in the right panel. The total density of states (TDOS) of LFO and LFO-V was presented in Fig. 1h. Compared to LFO (with a d-band center of −1.34 eV), LFO-V exhibited a d-band center at −0.99 eV, which was closer to the Fermi energy level. This shift was due to the V-doping, which contributed empty orbitals that enhanced the intensity of electronic states near the Fermi energy level [28]. The partial density of states (PDOS) suggested that the p and d orbitals primarily contributed to the charge density of O, Fe, and V atoms (Fig. 1i and Fig. S5 in Supporting information). Notably, the La atom exhibited negligible charge density near the Fermi energy level, implying minimal involvement in interfacial reactions. A decrease in the intensity of the O electronic state was observed in LFO-V compared to LFO, which could be attributed to the formation of oxygen vacancies [29]. In contrast, the V atom exhibited empty orbitals near the Fermi energy level, indicating its ability to accept electrons and participate in PMS oxidation. Additionally, the spin-up electrons of Fe increased around the Fermi energy level, providing a higher density of electron sites for PMS reduction [30].
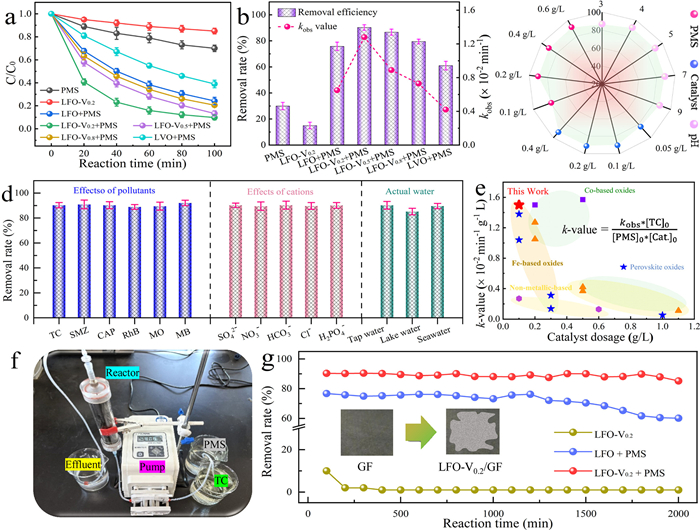
TC was selected as the target contaminant to evaluate the catalytic performance. As depicted in Fig. 2a, PMS activation was investigated in different systems without adjusting pH (pH 5.8). Notably, TC adsorption on LFO-V0.2 was negligible, and PMS exhibited weak oxidation capacity in the absence of catalysts. In contrast, 90.3% of TC was removed within 100 min in the LFO-V0.2/PMS system, indicating the generation of a significant amount of ROS. The TC degradation efficiency for LFO, LFO-V0.5, LFO-V0.8 and LaVO4 was 75.8%, 86.7%, 79.5% and 61.0%, respectively. Clearly, the LFO-V0.2 showed the best performance for TC degradation compared to the other catalysts, primarily due to the formation of asymmetric Fe-O-V sites and favorable interactions between V and Fe. The pseudo-first-order kinetic constant (kobs) for TC degradation was subsequently calculated (Fig. 2b). LFO-V0.2 exhibited the highest value of 1.28 × 10–2 min-1, nearly 2-fold higher than that of LFO (0.65 × 10–2 min-1). A 28.2% total organic carbon (TOC) removal was achieved within 100 min for LFO-V0.2 (Fig. S6a in Supporting information), demonstrating a 3-fold higher mineralization compared to LFO (with 9.5% TOC removal). The optimal parameters (pH value, catalyst and PMS dosage) were also discussed in Fig. 2c. Increasing the solution pH from 3.0 to 9.0 had a minor effect on TC degradation. In contrast, TC degradation was significantly enhanced with increasing catalyst or PMS dosage. A lower dosage resulted in insufficient active sites or oxidants, thus hindering TC removal [31]. Moreover, the LFO-V0.2 catalyst demonstrated excellent reusability with minimal ion leaching of Fe (0.21 mg/L) and V (0.52 mg/L). A slight decline in catalytic activity was observed after five successive runs (Fig. S6b in Supporting information). X-ray diffraction (XRD) patterns (Fig. S6c in Supporting information) and FTIR spectra (Fig. S6d in Supporting information) confirmed the stability of the LFO-V0.2 catalyst. Additionally, the degradation intermediates of TC in the LFO-V0.2/PMS system were explored (Fig. S7 in Supporting information), and the toxicity of TC was significantly reduced by detected the Escherichia coli metabolism (Figs. S8 and S9 in Supporting information).
Figure 2
Figure 2.
(a) TC degradation efficiency under different conditions and (b) the corresponding pseudo-first-order kinetic constant, (c) comparison of catalyst dosage, PMS dosage and pH value, and (d) degradation efficiency for different pollutants, anions and actual water solutions in the LFO-V0.2/PMS system, (e) comparison of the k-value of various catalytic materials in TC removal, (f) photograph of the wastewater treatment experimental equipment, and (g) TC removal efficiency using continuous reactor (Insert: the catalyst-loaded graphite felt filter).
Apart from TC, the LFO-V0.2/PMS system also effectively removed other refractory organic pollutants with 100 min reaction, e.g., sulfamethoxazole (SMZ), chloramphenicol (CAP), rhodamine B (RhB), methyl orange (MO), and methylene blue (MB) (Fig. 2d). Moreover, the LFO-V0.2/PMS system exhibited excellent resistance to interference from background ions (Cl−, H2PO4−, SO42−, NO3− and HCO3−), and successfully degraded TC in real water sources (tap, lake and seawater). From Fig. 2e and Table S2 (Supporting information), the k-value (× 10–2 min−1 g−1 L) of LFO-V0.2 demonstrated superior catalytic performance compared to other PMS systems. Fig. 2f showed the flow diagrams for wastewater treatment utilizing the LFO-V0.2 catalyst. To further test the robustness of the catalyst, 500 mg of LFO-V0.2 powder was immobilized onto a graphite felt carrier for long-term treatment of TC-laden wastewater. The TC removal efficiency remained above 90% for up to 2000 min in the LFO-V0.2/PMS system at a flow rate of 1 mL/min (Fig. 2g), while the LFO/PMS system achieved only 70% removal, indicating the LFO-V0.2/graphite felt unit could consistently and efficiently activate PMS.
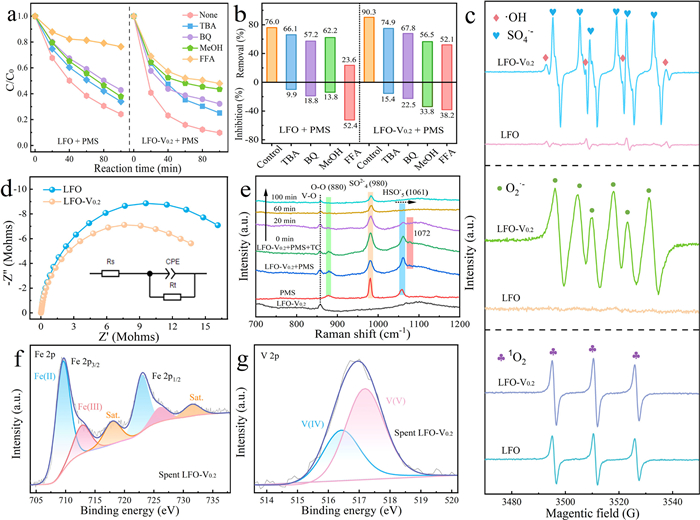
Subsequently, methanol (MeOH), tert‑butanol (TBA), benzoquinone (BQ) and furfuryl alcohol (FFA) were selected as scavengers for SO4•- and •OH, O2•- and 1O2, respectively (Figs. 3a and b). The addition of FFA caused in a significant reduction in TC degradation, with inhibition rates of 52.4% and 38.2% for the LFO/PMS and the LFO-V0.2/PMS systems. In contrast, MeOH, TBA and BQ resulted in only slight inhibition (13.8%, 9.9% and 18.8%), implying that radicals played a minimal role in TC removal rarely in the LFO/PMS. However, in the LFO-V0.2/PMS system, the inhibition rates of MeOH (33.8%), BQ (22.5%) and TBA (15.4%) significantly increased, indicating that non-radicals (1O2) were the primary contributors in the LFO/PMS system, while both radicals (SO4•-, •OH and O2•-) and non-radicals (1O2) played a dominant role in the LFO-V0.2/PMS system. As shown in Fig. 3c, strong electron spin resonance (ESR) signals corresponding to DMPO-SO4•- and DMPO-•OH adducts confirmed the formation of SO4•- and •OH. More pronounced signals in the LFO-V0.2/PMS system indicated a higher production of SO4•-, emphasizing that asymmetric Fe-O-V sites facilitated rapid electron transfer, enhancing PMS activation to generate a higher concentration of radicals. However, when DMPO was used to trap O2•- in MeOH, the DMPO-O2•- adduct was barely detected in the LFO-V0.2/PMS system, implying that O2•- was primarily produced by PMS oxidation at Fe-O-V sites, which promoted the formation of 1O2. Afterwards, the stronger intensity of the TEMP-1O2 adduct in the LFO-V0.2/PMS system further supported this conclusion. These findings suggested that both reduction and oxidation of PMS occurred simultaneously at asymmetric Fe-O-V sites, promoting the generation of radical and non-radical.
Figure 3
Figure 3.
(a, b) Comparison of TC removal efficiency under various quencher conditions. (c) ESR spectra of SO4•-, •OH or 1O2 using DMPO or TEMP as a trapping agent. (d) EIS spectra of various samples. (e) In-situ Raman spectra of various oxidation systems. (f, g) High-resolution Fe 2p and V 2p spectra of spent catalysts.
The electrochemical impedance spectroscopy (EIS) spectra suggested that LFO-V0.2 exhibited the smallest semicircular radius, inferring higher electrical conductivity and better charge transfer capacity (Fig. 3d) [32]. As shown in Fig. 3e, the characteristic peaks at 1060, 980 and 880 cm-1 were assigned to HSO5-, SO42- and O-O bond of PMS, respectively. The peak of HSO5- in the LFO-V0.2/PMS/TC system shifted slightly towards higher wavenumbers (from 1060 cm-1 to 1072 cm-1), indicating an increased electron density due to electron transfer [33]. Subsequently, a faster decline on the peaks of HSO5- and O-O bond, indicating the efficient activation of PMS to generate ROS with the time increased (from 0 to 100 min) in the LFO-V0.2/PMS/TC system. Following PMS activation, the Fe(II) and V(V) content increased notably in the spent LFO-V0.2 (Figs. 3f and g), suggesting that Fe may acquire electrons from V via an O-bridge. This behavior helped maintain Fe in a low valence for PMS reduction and V in a high valence for PMS oxidation. Besides, the O 1s results displayed that OL participated in electron transfer as a bridge during PMS activation (Fig. S10a in Supporting information). No significant changes were identified for the La species during the catalytic reaction (Fig. S10b in Supporting information). Then, a possible ROS generation pathway for the LFO-V0.2/PMS/TC system was proposed. Initially, ≡Fe(II) and ≡V(V) served as electron donors to activate PMS to generate SO4•− and SO5•-, while ≡Fe(III) and ≡V(IV) were produced via electron loss (Eqs. S3 and S4 in Supporting information). Subsequently, ≡V(IV) activated PMS to derive additional SO4•−, and the redox interaction between ≡V(IV) and ≡Fe(III) further facilitated ROS generation (Eqs. S5 and S6 in Supporting information). Since SO4•− and SO5•- were highly reactive species, they could derive series ROS. For example, part of the SO4•− and SO5•- could react with H2O to generate •OH and O2•- (Eqs. S7 and S8 in Supporting information) [24]. Meanwhile, OV also reacted with PMS to produce O2•- (Eq. S9 in Supporting information), which could further convert to 1O2 through subsequent reactions (Eqs. S10 and S11 in Supporting information). Ultimately, the ROS could attack TC and mineralize intermediates to CO2, H2O and inorganics (Eq. S12 in Supporting information).
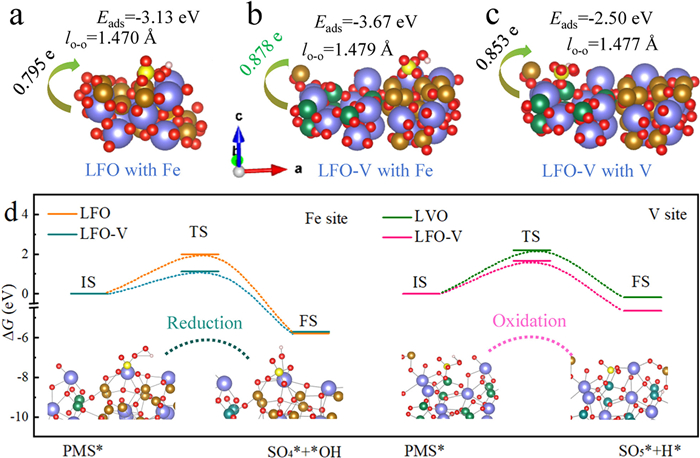
The adsorption behavior of PMS on the Fe sites of LFO and LFO-V, and the V site of LFO-V was discussed in Figs. 4a and b, the adsorption energy (ΔEads) of O-O bond on the Fe sites of LFO-V (−3.67 eV) was stronger than that on LFO (−3.13 eV), suggesting that the asymmetric Fe-O-V sites facilitated PMS activation. Moreover, the ΔEads of the O-O bond at the V site of LFO-V was also investigated (Fig. 4c), and the relatively weak ΔEads (−2.50 eV) also proved its catalytic activity on the V site. However, the lengths of O-O bonds at different sites were 1.470 Å (Fe site of LFO), 1.479 Å (Fe site of LFO-V), and 1.477 Å (V site of LFO-V), indicating that PMS activation was more likely to occur at the asymmetric Fe-O-V sites. Charge density difference analysis confirmed that more electrons transferred through the asymmetric Fe-O-V sites (Fig. S11 in Supporting information). Meanwhile, Bader charge calculations also demonstrated that PMS obtained more electrons from Fe (0.878 e) and V (0.853 e) sites of LFO-V compared to LFO (0.795 e), highlighting the higher electron donation capacity of the asymmetric Fe-O-V sites. The calculated transition state energy for different sites was discussed in Fig. 4d, PMS adsorbed on the Fe sites of LFO and LFO-V to form SO4•- and •OH, while the V site of LFO-V transformed PMS into SO5•- and H+. The energy barrier for LFO-V (1.98 eV) was lower than that for LFO (1.12 eV), indicating that the O-O bond dissociated more easily at the asymmetric Fe-O-V sites. The energy barrier at the V site of LFO-V (1.66 eV) was lower than that at the V site of LaVO4 (2.19 eV), further supporting enhanced activation at the asymmetric Fe-O-V sites. These results demonstrated that the higher adsorption energy, O-O stretching, interfacial charge transfer, and lower energy barrier at the asymmetric Fe-O-V sites significantly facilitated ROS generation via PMS activation.
Figure 4
Figure 4.
The optimized adsorption configurations, O—O bond stretching and Bader charge of PMS molecules on the surface of (a) LFO with Fe site, (b) LFO-V with Fe site and (c) LFO-V with V site. (d) The calculated energy for the transition state based on PMS transformation to •OH/SO4•− on Fe site of LFO and LFO-V, and H+/SO5•− on V site of LFO-V (inset corresponding intermediate structures).
In summary, the LFO-V0.2 catalyst demonstrated excellent PMS activation capability, enabling the coupling of both radical and non-radical species for efficient pollutants elimination. DFT calculations revealed that the creation of asymmetric Fe-O-V sites increased the electron density near the Fermi energy level, which facilitated interfacial electron transfer and lowered the reaction energy barrier. These results contributed to efficient oxidation and reduction of PMS. The generation of SO4•-, •OH and 1O2 played a crucial role in pollutants degradation. Consequently, the mineralization efficiency of TC in the LFO-V0.2/PMS system was 3-fold higher than that in LFO/PMS, and then the system showed promising potential for continuous operation over 2000 min. This work provides the foundation for the rational design of environmental catalysts for sustainable water purification.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was financially supported by the National Natural Science Foundation of China (Nos. W2412093 and 52170068) and the Fundamental Research Funds for the Central Universities (No. DUT24RC(3)079).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111355.
[1]
Y. Liu, P. Wang, L. Xie, et al., Angew. Chem. Int. Ed. 63 (2024) e202319470. doi: 10.1002/anie.202319470
[2]
P. Zhang, Y. Yang, X. Duan, Y. Liu, S. Wang, ACS Catal. 11 (2021) 11129–11159. doi: 10.1021/acscatal.1c03099
Z. Zhao, P. Wang, C. Song, et al., Angew. Chem. Int. Ed. 62 (2023) e202216403. doi: 10.1002/anie.202216403
[33]
Y. Jiang, X. Weng, Y. Hu, et al., J. Environ. Chem. Eng. 12 (2024) 111941. doi: 10.1016/j.jece.2024.111941
Figure 1
(a) Normalized V K-edge XANES spectra and (b) EXAFS spectra of V2O5, VO2 and LFO-V0.2. (c) The EXAFS fitting curves of LFO-V0.2 at R space, and (d) the WT contour plots of (d) V foil, (e) V2O5 and (f) LFO-V0.2. (g) The atomic structure model, (h) the total density of state of LFO and LFO-V, and (i) the project density of state of Fe 3d, O 2p, V 2p and La 3d orbitals in the LFO-V.
Figure 2
(a) TC degradation efficiency under different conditions and (b) the corresponding pseudo-first-order kinetic constant, (c) comparison of catalyst dosage, PMS dosage and pH value, and (d) degradation efficiency for different pollutants, anions and actual water solutions in the LFO-V0.2/PMS system, (e) comparison of the k-value of various catalytic materials in TC removal, (f) photograph of the wastewater treatment experimental equipment, and (g) TC removal efficiency using continuous reactor (Insert: the catalyst-loaded graphite felt filter).
Figure 3
(a, b) Comparison of TC removal efficiency under various quencher conditions. (c) ESR spectra of SO4•-, •OH or 1O2 using DMPO or TEMP as a trapping agent. (d) EIS spectra of various samples. (e) In-situ Raman spectra of various oxidation systems. (f, g) High-resolution Fe 2p and V 2p spectra of spent catalysts.
Figure 4
The optimized adsorption configurations, O—O bond stretching and Bader charge of PMS molecules on the surface of (a) LFO with Fe site, (b) LFO-V with Fe site and (c) LFO-V with V site. (d) The calculated energy for the transition state based on PMS transformation to •OH/SO4•− on Fe site of LFO and LFO-V, and H+/SO5•− on V site of LFO-V (inset corresponding intermediate structures).

 DownLoad:
DownLoad:

 下载:
下载:





