
Scheme 1.
Enhancing photocatalytic activity of molecular catalyst through host-guest interactions based on photosensitive metallacage.
Porphyrin metallacage-based host-guest complexation for highly efficient photocatalytic hydrogen production
Zeyuan Zhang , Zixuan Li , Chenjing Liu , Yali Hou , Ke Gao , Shijin Jian , Guoping Li , Gang He , Mingming Zhang

Photocatalytic hydrogen evolution has emerged as a key focus in sustainable energy research, offering a cost-effective and environmentally sustainable method for producing clean, renewable, and storable hydrogen fuel [1–4]. Over recent decades, a wide range of photocatalysts, including metal oxides [5–8], carbon-based materials [9–11], quantum dots [12,13], and noble metal nanoparticles [14–16], have been developed and systematically explored for hydrogen production. Among these, metal-organic complexes (MOCs), a class of molecular photocatalysts, have attracted significant attention due to their tunable catalytic activity through precise selection of metal centers and organic ligands [17–22]. However, the limited light absorption, propensity for aggregation, and rapid exciton recombination of MOCs significantly hinder their potential as highly efficient photocatalysts. The incorporation of MOCs and photosensitizers into crystalline materials such as metal-organic frameworks [23–25] and covalent-organic frameworks [26–28] has been proposed as an effective solution, but their limitations in mass transport and substrate diffusion often lead to reduced photocatalytic efficiency [29]. Thus, it remains both highly desirable and challenging to develop strategies that overcome these obstacles and enhance the overall photocatalytic performance.
Metallacages, as discrete molecular entities with enclosed cavities, provide a promising platform for integrating photosensitizers and MOCs to enable efficient photocatalysis, with their confined cavities facilitating substrate transport [30–45]. However, precise design and functionalization of MOCs and photosensitizers are essential to avoid mutual interference between the distinct coordination bonds in metallacages and MOCs [46–50]. Such functionalization, however, may alter the energy gaps of the MOCs, reducing their photocatalytic efficiency [51]. Additionally, the rigid aromatic scaffolds of these metallacages may promote intermolecular aggregation of the photocatalysts, diminishing their overall performance [52]. Inspired by natural enzymatic systems, where catalytic centers are confined within nanopockets, the encapsulation of MOCs within the cavities of photosensitive metallacages presents a promising strategy to address these challenges [53–57]. Nevertheless, designing and preparing suitable photosensitive metallacages that exhibit effective host–guest interactions with MOC-based photocatalysts remains difficult, and the development of metallacage-based host–guest systems for efficient photocatalysis has been limited. Such systems, by providing isolated environments for encapsulated photocatalysts, have the potential to significantly enhance photocatalytic efficiency.
Herein, we report the formation of four host–guest complexes between porphyrin-based multicomponent metallacages and Pt-based MOCs, with their structures unambiguously confirmed by single-crystal X-ray diffraction. Notably, the porphyrin-based metallacages not only serve as containers, preventing the aggregation of Pt-based catalysts, but also function as photosensitizers, facilitating efficient electron transfer from the metallacages to the Pt-based MOCs, resulting in highly efficient photocatalytic hydrogen production (Scheme 1). One of the host–guest complexes achieve a H2 generation rate of 19,786.5 µmol g−1 h−1, fivefold of the non-confined Pt-based catalyst. Femtosecond transient absorption (fs-TA) experiments and DFT calculations suggest that the directional photo-induced electron transfer (PET) from the porphyrin units to the Pt-based catalytic centers underpins the enhanced photocatalysis. This study presents a strategy for integrating photosensitive metallacages with active MOC-based photocatalysts through host–guest complexation, offering a pathway to advance metallacage-based systems for efficient photocatalytic hydrogen production.
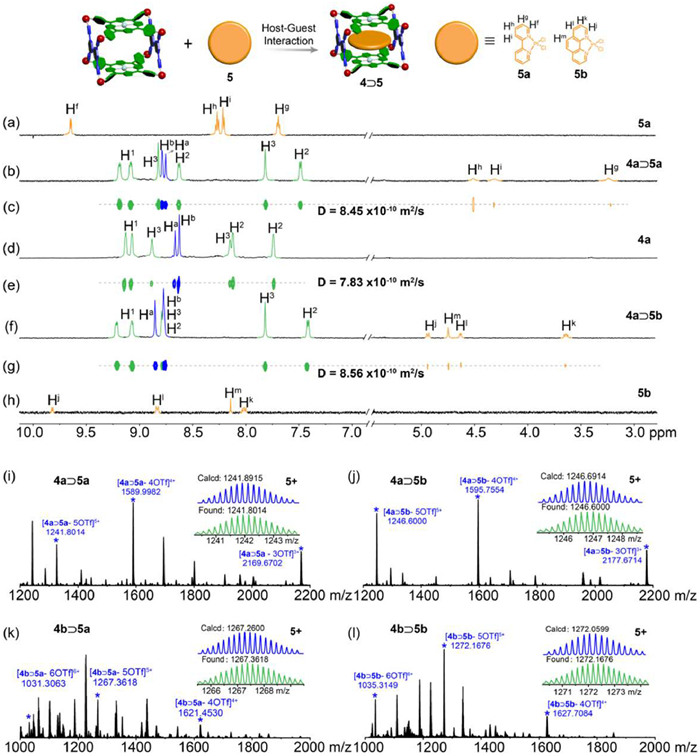
Based on the self-assembly of tetrapyridyl porphyrin (1a or 1b), cis-Pt(PEt3)2(OTf)2 (2), and a tetracarboxylic ligand (3), metallacage 4a or 4b was successfully prepared (see Supporting information for synthetic details). The two porphyrinic faces in metallacage 4a or 4b were nearly parallel, with an interplanar distance around 8.0 Å, ideal for π-π stacking with planar aromatic molecules within the cavity (Fig. S5 in Supporting information). This structural arrangement also formed two large windows, allowing guest molecules to enter and enabling the formation of stable host–guest complexes. Consequently, the complexation of metallacages 4a and 4b with two widely used Pt-based photocatalysts, PtCl2(bpy) (5a) and PtCl2(phen) (5b), was further studied. The initial assessment of this complexation was conducted using 1H NMR spectroscopy (Figs. 1a-h and Figs. S6-S9 in Supporting information), where significant upfield shifts were observed for the protons of guests, suggesting the formation of host-guest complexes. For example, in 4a⊃5b, protons Hj, Hl, Hm, and Hk of 5b shifted from 9.81, 8.83, 8.14, and 8.01 ppm to 4.94, 4.63, 4.75, and 3.64 ppm, respectively, owing to the shielding effect of the metallacage. A single diffusion coefficient (D) of 7.83 × 10–10 m2/s was observed for metallacage 4a (Fig. 1e). For host-guest complexes 4a⊃5a and 4a⊃5b, PtCl2(bpy) (5a) and PtCl2(phen) (5b) exhibited diffusion coefficients identical to that of the host, with D = 8.45 × 10–10 m2/s for 4a⊃5a, and 8.56 × 10–10 m2/s for 4a⊃5b (Figs. 1c and g), suggesting the formation of stable host-guest complexes. Based on Stokes-Einstein equation, the radii of the host-guest complexes 4a⊃5a and 4a⊃5b were calculated to be 0.75 nm and 0.74 nm, respectively. The formation of complexes 4a⊃5a, 4a⊃5b, 4b⊃5a, and 4b⊃5b was also confirmed by electrospray ionization time-of-flight mass spectrometry (ESI-TOF-MS) (Figs. 1i-l). For example, peaks at m/z 1241.8104, 1267.3618, 1246.6000 and 1272.1676 were found, corresponding to [4a⊃5a – 5OTf]5+, [4a⊃5b – 5OTf]5+, [4b⊃5a – 5OTf]5+ and [4b⊃5b – 5OTf]5+, respectively. The association constants (Ka) were further measured by UV–vis titration experiments, yielding values of (2.39 ± 0.25) × 106, (9.46 ± 0.17) × 104, (1.72 ± 0.15) × 105, and (2.25 ± 1.50) × 106 L/mol for 4a⊃5a, 4a⊃5b, 4b⊃5a, and 4b⊃5b, respectively (Figs. S14-S17 in Supporting information). These high Ka values can effectively confine the photocatalytic guests within the cavities of metallacages, preventing guest aggregation and promoting high photocatalytic activity.
Single crystals of complexes 4a⊃5a, 4a⊃5b, and 4b⊃5b, suitable for X-ray diffraction analysis, were successfully obtained through vapor diffusion of isopropyl ether into acetonitrile over two weeks. These crystals provided direct structural evidence for the formation of inclusion complexes (Fig. 2). Notably, in contrast to the empty metallacage (Fig. S5 in Supporting information), the host-guest complexes showed a marked reduction in the distance between the two porphyrin faces, decreasing from 8.1 Å to 7.5 Å (Figs. 2a-c). This contraction resulted in distances of 3.8–3.9 Å between the porphyrin faces and the guest molecules, which is conducive to π–π stacking interactions. The guest molecules within the metallacages were highly disordered, likely due to their rotational freedom within the cavities. Top views of the crystal structures revealed that the guest molecules aligned themselves to optimize interactions with the metallacages (Figs. 2d-f), either translationally or rotationally, to maximize the number of binding sites in metallacages, thereby strengthening the π–π stacking interactions. Additionally, [C–H···Cl] hydrogen bonds were observed between the β-pyridyl protons of the porphyrin faces and the chlorine atoms of the guests (Figs. 2g-i). These hydrogen bonds, in conjunction with the π-π stacking interactions, contributed to the high association constants observed. The robust host-guest complexation is expected to enhance the PET from the photosensitive metallacages to the Pt-based photocatalysts, thereby increasing the photocatalytic activity of the system.

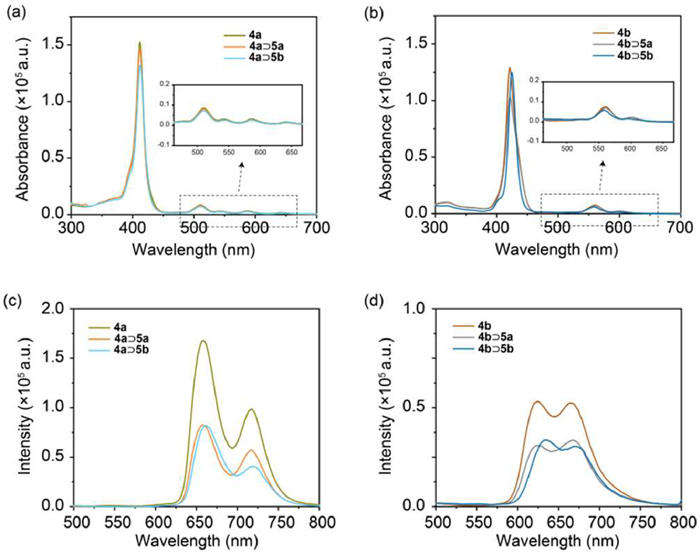
The complexation of metallacages 4a and 4b with different guest molecules, 5a and 5b, led to notable alterations in their photophysical properties. Since light absorption plays an important role of in photocatalytic reaction, we first collected the UV–vis absorption spectra of the two metallacages and their inclusion complexes (Figs. 3a and b). The UV–vis absorption spectra of 4a and its corresponding host-guest complexes, 4a⊃5a and 4a⊃5b, revealed a prominent Soret band centered at 412 nm, along with four Q bands centered at 640, 585, 543 and 510 nm (Fig. 3a), consistent with the absorption profiles of porphyrin derivatives. After the encapsulation of guest molecules, only the slight decrease in Soret band was observed. For Zn-porphyrin-based metallacage 4b, one Soret band centered at 423 nm and two Q bands centered at 560 and 600 nm were observed (Fig. 3b). Similar decrease was also found for complexes 4b⊃5a and 4b⊃5b compared with 4b Metallacage 4a exhibited two emission bands centered at 658 and 718 nm (Fig. 3c), respectively, originating from the emission of porphyrin units. The fluorescence intensity decreased by approximately 50% in complexes 4a⊃5a and 4a⊃5b A similar reduction in the two emission bands, centered at 625 and 668 nm, was observed for host-guest complexes 4b⊃5a and 4b⊃5b compared to metallacage 4b (Fig. 3d). These changes in absorption and emission indicate the potential PET process from the metallacages to the encapsulated Pt complexes.
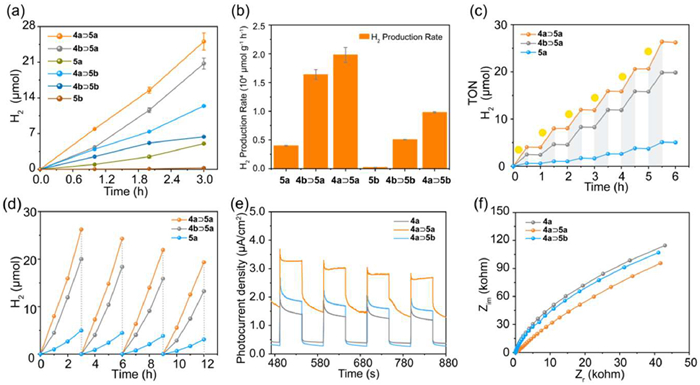
Photocatalytic hydrogen evolution experiments were carried out for complexes 4a⊃5a, 4a⊃5b, 4b⊃5a, 4b⊃5b, 4a, 4b, 5a, and 5b (0.6 µmol/L) in acetonitrile containing 1.5% acetic acid as the hydrogen source. The reactions were conducted in 10 mL crimp top vials under irradiation from a Xenon lamp at a radiation intensity of 100 mW/cm2. 1,3-Dimethyl-2-phenyl-2,3-dihydro-1H-benzimidazole (BIH) was employed as a sacrificial electron donor owing to its efficiency in photocatalytic hydrogen production (Fig. S19 in Supporting information). Metallacages 4a and 4b showed negligible hydrogen production, owing to the lack of photocatalysts (Fig. S20 in Supporting information). Complex 5a exhibited significantly higher hydrogen evolution activity than complex 5b, consistent with previous reports [58]. The mixture of porphyrin ligand 1a or 1b with 5a exhibited improved hydrogen production compared with 5a, but showed much lower hydrogen production compared with host-guest complexes 4a⊃5a and 4b⊃5a. Moreover, 4a⊃5a and 4b⊃5a demonstrated superior photocatalytic performance compared to 4a⊃5b and 4b⊃5b, respectively. Hydrogen production from complexes 5a, 4a⊃5a, and 4b⊃5a increased over time, reaching 5.0, 25.1, and 20.7 µmol, respectively, after 3 h of irradiation. In contrast, complexes 5b, 4a⊃5b, and 4b⊃5b produced only 0.3, 12.4, and 6.4 µmol of hydrogen (Fig. 4a). These findings suggest that the host-guest complexation significantly enhances the photocatalytic performance of Pt-based catalysts, with the metallacage 4a acting as a more effective photosensitizer than metallacage 4b, likely due to its superior light absorption (Figs. 3a and b). It is worth mentioning that the hydrogen generation rate for complex 4a⊃5a was calculated to be 19,786.5 µmol g−1 h−1, which was among the highest values for metallacage-based photocatalytic systems (Table S4 in Supporting information) [59–61]. The apparent quantum yield (AQY) of 4a⊃5a was and 1.3% (Fig. 4b). Light on-off experiments confirmed the photocatalytic nature of hydrogen generation, with H2 production for complexes 5a, 4a⊃5a, and 4b⊃5a increasing under light irradiation and halting in the dark (Fig. 4c). After 6 h of intermittent illumination, the total H2 production reached 26.3 µmol. Stability tests showed that after four cycles, the photocatalytic performance of 4a⊃5a and 4b⊃5a decreased slightly but still maintained at a high level (Fig. 4d). Furthermore, the photocurrent response of 4a⊃5a was significantly higher than that of 4a and 4a⊃5b, indicating enhanced charge separation efficiency (Fig. 4e). Correspondingly, the charge transfer resistance of complex 4a⊃5a was lower than that of metallacage 4a and complex 4a⊃5b (Fig. 4f), confirming improved charge transfer in 4a⊃5a compared with the other two systems. Similarly, 4b⊃5a showed better photocurrent response and lower charge transfer resistance than 5b and 4b⊃5b (Figs. S21 and S22 in Supporting information). These results demonstrate that the formation of stable host-guest complexes not only enhances the light absorption and prevents the aggregation of photocatalysts, but also facilitates directional electron transfer from the photosensitizer to the photocatalyst, leading to efficient photocatalysis.
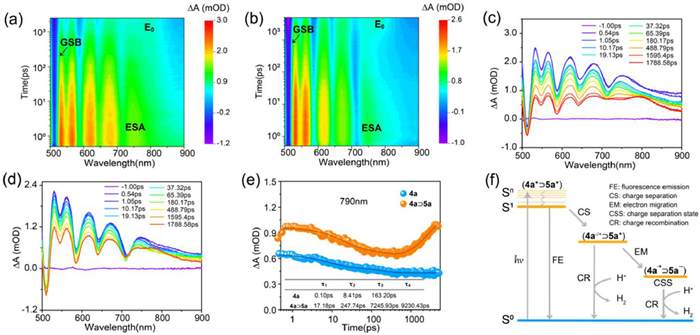
To further explore the photocatalytic process, femtosecond transient absorption spectroscopy (fs-TAS) was utilized to investigate the dynamics of excited states. The transient absorption spectra of complexes 4a and 4a⊃5a exhibited similar overall patterns, however, with subtle differences at various delay times (Figs. 5a and b). Both systems revealed a narrow negative peak at 513 nm, indicative of ground-state bleaching (GSB) of the metallacage, attributed to the Q-band absorption of porphyrin. Additionally, a broad positive peak at 750 nm was observed, corresponding to excited-state absorption (ESA) of the porphyrin. Within the initial sub-picosecond timeframe, a rapid blue shift was noted, signaling the formation of the singlet excited state 4a* [54]. Around 1000 ps, a new broad positive peak appeared at 790 nm, indicating a subsequent charge transfer process from the oxidized porphyrin state (Figs. 5c and d) [62]. Kinetic traces at 513 nm and 790 nm were plotted to elucidate the charge transfer dynamics of metallacage 4a and complex 4a⊃5a (Fig. 5e and Fig. S24 in Supporting information). Notably, at approximately 600 ps, the GSB peak of 4a⊃5a exhibited a new decay trend (Fig. S24), suggesting that more molecules transitioned from the ground state to engage in the charge separation process. Concurrently, the ESA peak at 750 nm gradually transitioned to a new E0 peak at 790 nm (Fig. 5e), demonstrating a distinct enhancement of charge separation compared to the empty metallacage 4a. Specifically, multi-exponential fitting revealed the presence of several species, with an additional exponential term in 4a⊃5a, indicating multiple distinct electron transfer events (Fig. 5f). The efficient electron transfer increased the lifetime of the charge-separated state and effectively enhanced the photocatalytic process. The host-guest complexation decreases the distance between the porphyrin-based metallacage and the Pt-based photocatalyst, establishing an optimal spatial arrangement for effective electron transfer and charge separation, which is responsible for the improved photocatalytic efficiency.
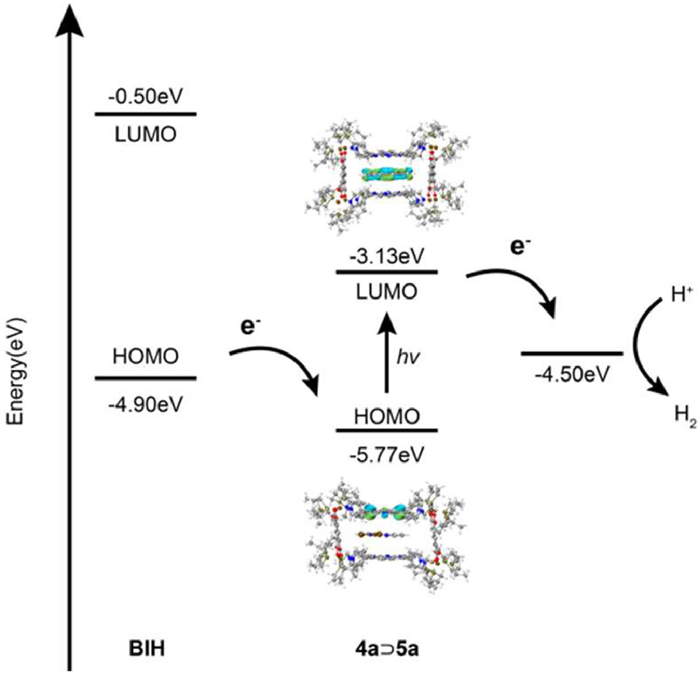
To gain further insight into the mechanism of photocatalytic hydrogen evolution, density-functional theory (DFT) calculations were performed to get the highest occupied and lowest unoccupied molecular orbital (HOMO/LUMO) energy profiles for complexes 5a, 4a⊃5a, 5b, and 4a⊃5b (Fig. 6 and Fig. S28 in Supporting information). Notably, the LUMO levels of these complexes were more negative than the redox potential of H+/H2 (−4.50 eV), whereas their HOMO levels were more positive than this redox potential (Fig. S31 in Supporting information). Therefore, these complexes have the potential to serve as photocatalysts for hydrogen evolution. Specifically, upon encapsulation within the metallacage 4a, the band gaps of the Pt-based photocatalysts, 5a and 5b, decreased from 4.01 eV to 2.64 eV, and from 4.00 eV to 2.43 eV, respectively. This reduction in band gaps suggested that the host-guest complexes were more easily to be photoexcited, potentially leading to higher photocatalytic activity. For complex 4a⊃5a, the HOMO was primarily located on the porphyrin units of the metallacage, while the LUMO was predominantly distributed at the Pt catalytic center, suggesting that PET from the metallacage to the Pt catalytic center could occur. Conversely, in the complex 4a⊃5b, both the HOMO and LUMO are distributed between the porphyrin units of the metallacage and the Pt catalytic center. This is likely attributed to the extended conjugated system in complex 5b compared to 5a, which increases the electron cloud density to levels similar to that of porphyrins. Consequently, the lack of significant PET between the metallacage and the Pt catalytic center results in a lower catalytic efficiency relative to 4a⊃5a. Moreover, the HOMO energy level of the electron donor BIH (−4.90 eV) was higher than that of 4a⊃5a (−5.77 eV), suggesting that BIH could efficiently transfer electrons to 4a⊃5a and sustain the hydrogen evolution process. Based on these calculations and the photophysical experiments, the proposed catalytic mechanism for 4a⊃5a is as follows (Fig. 6): Under simulated sunlight, the porphyrin units of the metallacage are photoexcited, subsequently generating excitons. The holes are then quenched by the sacrificial electron donor BIH, while the electrons are transferred to the Pt catalytic center, facilitated by the spatial confinement of 5a within the cavity of metallacage 4a. Finally, the electrons reduce H+ at the Pt catalytic center, producing H2.
In conclusion, we have demonstrated the successful integration of Pt-based MOCs into porphyrin-based metallacages, forming host–guest complexes that significantly enhance the performance photocatalytic hydrogen evolution. The metallacages not only prevent the aggregation of the MOCs but also act as photosensitizers, facilitating efficient electron transfer. Notably, one of the complexes achieved a hydrogen generation rate much higher than the non-confined Pt-based catalyst, driven by efficient PET from the porphyrin faces of the metallacages to the Pt catalytic centers. This work highlights the potential of using metallacage-based host-guest complexation to address key challenges in MOC-based photocatalysis, particularly in enhancing charge transfer, substrate transport, and catalytic efficiency. These findings highlight the potential of metallacage-based systems for advancing photocatalytic applications, providing a valuable pathway for the development of next-generation catalysts for sustainable hydrogen production through solar energy conversion.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Zeyuan Zhang: Data curation, Conceptualization. Zixuan Li: Writing – original draft, Data curation. Chenjing Liu: Software, Data curation. Yali Hou: Software, Data curation. Ke Gao: Data curation. Shijin Jian: Software, Data curation. Guoping Li: Data curation. Gang He: Conceptualization. Mingming Zhang: Conceptualization.
This work was supported by the National Natural Science Foundation of China (Nos. 22171219 22222112, and 22301238) and Xi'an Association for Science and Technology Youth Talent Support Program (No. 20240345). We thank Dr. Gang Chang at the Instrument Analysis Center and Dr. Aqun Zheng and Junjie Zhang at the Experimental Chemistry Center of Xi'an Jiaotong University for NMR and fluorescence measurements. The authors also acknowledge the mass spectrometry characterization by the Molecular Scale Laboratory.
Supplementary material associated with this article can be found, in the online version, at doi:
D. Han, X. Liu, S. Wu, Chem. Soc. Rev. 51 (2022) 7138–7169. doi: 10.1039/d2cs00460g
D. Xu, S.N. Zhang, J.S. Chen, X.H. Li, Chem. Rev. 123 (2023) 1–30. doi: 10.1021/acs.chemrev.2c00426
J. Lv, J. Xie, A.G.A. Mohamed, et al., Nat. Rev. Chem. 7 (2023) 91–105.
H.C. Li, M. Zhang, Q. Lv, et al., Chin. Chem. Lett. 36 (2025) 110579. doi: 10.1016/j.cclet.2024.110579
T. Wang, J. Cao, J. Li, D. Li, Z. Ao, Chin. Chem. Lett. 36 (2025) 110078. doi: 10.1016/j.cclet.2024.110078
B. He, Z. Wang, P. Xiao, et al., Adv. Mater. 34 (2022) 2203225. doi: 10.1002/adma.202203225
W.H. Lee, C.W. Lee, G.D. Cha, et al., Nat. Nanotechnol. 18 (2023) 754–762. doi: 10.1038/s41565-023-01385-4
X. Ruan, C. Huang, H. Cheng, et al., Adv. Mater. 35 (2023) 2209141. doi: 10.1002/adma.202209141
Q. Wang, T. Hisatomi, Q. Jia, et al., Nat. Mater. 15 (2016) 611–615. doi: 10.1038/nmat4589
S. Wang, Y. Gao, S. Miao, et al., J. Am. Chem. Soc. 139 (2017) 11771–11778. doi: 10.1021/jacs.7b04470
T. Takata, J. Jiang, Y. Sakata, et al., Nature 581 (2020) 411–414. doi: 10.1038/s41586-020-2278-9
X.B. Li, C.H. Tung, L.Z. Wu, Nat. Rev. Chem. 2 (2018) 160–173. doi: 10.1038/s41570-018-0024-8
J. Kosco, M. Bidwell, H. Cha, et al., Nat. Mater. 19 (2020) 559–565. doi: 10.1038/s41563-019-0591-1
X. Li, W. Bi, L. Zhang, et al., Adv. Mater. 28 (2016) 2427–2431. doi: 10.1002/adma.201505281
P. Dong, Y. Wang, A. Zhang, et al., ACS Catal. 11 (2021) 13266–13279. doi: 10.1021/acscatal.1c03441
Z. Huang, C. Guo, Q. Zheng, et al., Chin. Chem. Lett. 35 (2024) 109580. doi: 10.1016/j.cclet.2024.109580
D. Shi, R. Zheng, M.J. Sun, et al., Angew. Chem. Int. Ed. 56 (2017) 14637–14641. doi: 10.1002/anie.201709869
S. Guo, L.H. Kong, P. Wang, et al., Angew. Chem. Int. Ed. 61 (2022) e202206193. doi: 10.1002/anie.202206193
Y. Wang, R. Tang, D. Wang, et al., Inorg. Chem. 62 (2023) 1786–1790. doi: 10.1021/acs.inorgchem.2c01206
Y. Zhang, X. Yan, L. Shi, et al., Inorg. Chem. 60 (2021) 7627–7631. doi: 10.1021/acs.inorgchem.1c00962
Y. Wang, C. Wang, R. Long, et al., Chem. Commun. 55 (2019) 5167–5170. doi: 10.1039/c9cc02173f
Y. Hou, R. Shi, H. Yuan, et al., Chin. Chem. Lett. 34 (2023) 107688. doi: 10.1016/j.cclet.2022.07.031
A. Dhakshinamoorthy, Z. Li, H. Garcia, Chem. Soc. Rev. 47 (2018) 8134–8172. doi: 10.1039/c8cs00256h
A. Bavykina, N. Kolobov, I.S. Khan, et al., Chem. Rev. 120 (2020) 8468–8535. doi: 10.1021/acs.chemrev.9b00685
X. Zhang, S. Tong, D. Huang, et al., Coord. Chem. Rev. 448 (2021) 214177. doi: 10.1016/j.ccr.2021.214177
H. Wang, H. Wang, Z. Wang, et al., Chem. Soc. Rev. 49 (2020) 4135–4165. doi: 10.1039/d0cs00278j
Y.N. Gong, X. Guan, H.L. Jiang, Coord. Chem. Rev. 475 (2023) 214889. doi: 10.1016/j.ccr.2022.214889
D.V. Wagle, H. Zhao, G.A. Baker, Acc. Chem. Res. 47 (2014) 2299–2308. doi: 10.1021/ar5000488
Q. Zhu, Y. Zheng, Z. Zhang, Y. Chen, Nat. Protoc. 18 (2023) 3080–3125. doi: 10.1038/s41596-023-00868-x
A.K. Bar, R. Chakrabarty, G. Mostafa, P.S. Mukherjee, Angew. Chem. Int. Ed. 47 (2008) 8455–8459. doi: 10.1002/anie.200803543
Y. Shi, I. Sánchez-Molina, C. Cao, T.R. Cook, P.J. Stang, Proc. Natl. Acad. Sci. U. S. A. 111 (2014) 9390–9395. doi: 10.1073/pnas.1408905111
S. Chen, K. Li, F. Zhao, et al., Nat. Commun. 7 (2016) 13169. doi: 10.1038/ncomms13169
D. Preston, J.E.M. Lewis, J.D. Crowley, J. Am. Chem. Soc. 139 (2017) 2379–2386. doi: 10.1021/jacs.6b11982
G. Yu, S. Yu, M.L. Saha, et al., Nat. Commun. 9 (2018) 4335. doi: 10.1038/s41467-018-06574-7
C.M. Hong, M. Morimoto, E.A. Kapustin, et al., J. Am. Chem. Soc. 140 (2018) 6591–6595. doi: 10.1021/jacs.8b01701
J. Wei, L. Zhao, C. He, et al., J. Am. Chem. Soc. 141 (2019) 12707–12716. doi: 10.1021/jacs.9b05351
L.J. Jongkind, J.A.A.W. Elemans, J.N.H. Reek, Angew. Chem. Int. Ed. 58 (2019) 2696–2699. doi: 10.1002/anie.201812610
L. Zhao, J. Cai, Y. Li, J. Wei, C. Duan, Nat. Commun. 11 (2020) 2903. doi: 10.1038/s41467-020-16714-7
Y. Sun, C. Chen, J. Liu, et al., J. Am. Chem. Soc. 142 (2020) 17903–17907. doi: 10.1021/jacs.0c08058
N. Kishida, K. Matsumoto, Y. Tanaka, et al., J. Am. Chem. Soc. 142 (2020) 9599–9603. doi: 10.1021/jacs.0c02932
L.S. Lisboa, D. Preston, C.J. McAdam, et al., Angew. Chem. Int. Ed. 61 (2022) e202201700. doi: 10.1002/anie.202201700
Y. Yang, X. Jing, Y. Shi, Y. Wu, C. Duan, J. Am. Chem. Soc. 145 (2023) 10136–10148. doi: 10.1021/jacs.3c00626
D. Chakraborty, N. Kaur, J. Sahoo, et al., J. Am. Chem. Soc. 146 (2024) 24901–24910. doi: 10.1021/jacs.4c05899
Q. Feng, N. Li, Z. Zhang, et al., Chin. Chem. Lett. 34 (2023) 108439. doi: 10.1016/j.cclet.2023.108439
R. Li, H. Zhang, Y. Hou, et al., Nat. Commun. 16 (2025) 2733. doi: 10.1038/s41467-025-57822-6
C. Mu, Z. Zhang, Y. Hou, et al., Angew. Chem. Int. Ed. 60 (2021) 12293–12297. doi: 10.1002/anie.202100463
Y. Hou, Z. Zhang, L. Ma, et al., CCS Chem. 4 (2022) 2604–2611. doi: 10.31635/ccschem.021.202101382
Z. Zhang, L. Ma, F. Fang, et al., JACS Au 2 (2022) 1479–1487. doi: 10.1021/jacsau.2c00245
Y. Hou, Z. Zhang, L. Ma, et al., CCS Chem. 4 (2022) 2604–2611. doi: 10.31635/ccschem.021.202101382
K. Gao, Y. Cheng, Z. Zhang, et al., Angew. Chem. Int. Ed. 63 (2024) e202319488. doi: 10.1002/anie.202319488
Y.H. Luo, L.Z. Dong, J. Liu, S.L. Li, Y.Q. Lan, Coord. Chem. Rev. 390 (2019) 86–126. doi: 10.1016/j.ccr.2019.03.019
H.S. Lee, S. Jee, R. Kim, et al., Energy Environ. Sci. 13 (2020) 519–526. doi: 10.1039/c9ee02619c
Y. Yang, F.H. Arnold, Acc. Chem. Res. 54 (2021) 1209–1225. doi: 10.1021/acs.accounts.0c00591
R. Zhu, S. Yu, X. Yang, et al., Chin. Chem. Lett. 35 (2024) 109539. doi: 10.1016/j.cclet.2024.109539
Z. Chen, S. Li, Q. Mo, L. Zhang, C.Y. Su, Chin. Chem. Lett. 34 (2023) 108196. doi: 10.1016/j.cclet.2023.108196
Y. Hou, C. Mu, Y. Shi, et al., Aggregate 5 (2024) e628. doi: 10.1002/agt2.628
R. Zhang, D. Hu, Y. Fu, et al., Aggregate 5 (2024) e408. doi: 10.1002/agt2.408
K. Sakai, H. Ozawa, Coord. Chem. Rev. 251 (2007) 2753–2766. doi: 10.1016/j.ccr.2007.08.014
X. Jing, C. He, Y. Yang, C. Duan, J. Am. Chem. Soc. 137 (2015) 3967–3974. doi: 10.1021/jacs.5b00832
C. Ji, W. Wang, E.S.M. El-Sayed, et al., Appl. Catal. B: Environ. 285 (2021) 119782. doi: 10.1016/j.apcatb.2020.119782
C. Mu, L. Zhang, G. Li, et al., Angew. Chem. Int. Ed. 62 (2023) e202311137. doi: 10.1002/anie.202311137
M. Ortiz, S. Cho, J. Niklas, et al., J. Am. Chem. Soc. 139 (2017) 4286–4289. doi: 10.1021/jacs.7b00220

Scheme 1 Enhancing photocatalytic activity of molecular catalyst through host-guest interactions based on photosensitive metallacage.

Figure 1 1H NMR spectra (600 MHz, CD3CN, 295 K) of 5a (a), 4a⊃5a (b), 4a (d), 4a⊃5b (f), and 5b (h). 1H DOSY spectrum (600 MHz, CD3CN, 295 K) recorded for 4a⊃5a (c), 4a (e), 4a⊃5b (g). ESI-TOF-MS spectra of 4a⊃5a (i), 4a⊃5b (j), 4b⊃5a (k) and 4b⊃5b (l).

Figure 2 Crystal structures of (a, d, g) 4a⊃5a (CCDC 2388923), (b, e, h) 4a⊃5b (CCDC 2388922) and (c, f, i) 4b⊃5b (CCDC 2388892). Hydrogen atoms, triethylphosphine units, counterions, and solvent molecules were omitted for clarity.

Figure 3 (a, b) UV–vis absorption (c = 2.50 µmol/L) and (c, d) fluorescence spectra (c = 10.00 µmol/L, λex = 405 nm) of complexes 4a, 4a⊃5a, 4a⊃5b, 4b, 4b⊃5a, and 4b⊃5b in CH3CN.

Figure 4 (a) Time-dependent H2 evolution of different complexes under a xenon lamp. (b) H2 evolution rate of different complexes. (c) Light on-off experiments for H2 reduction by 4a⊃5a, 4b⊃5a and 5a. (d) Recycle tests of 4a⊃5a, 4b⊃5a and 5a in four consecutive runs. (e) Representative photocurrent responses of 4a, 4a⊃5a and 4a⊃5b on a carbon paper electrode with the interval of 50 s, which were recorded in sodium sulfate solution (0.1 mol/L), with Ag/AgCl as a reference electrode, and platinum as a counter electrode, white light, 400 mW/cm2. (f) Charge transfer resistance of 4a, 4a⊃5a and 4a⊃5b, white light, 400 mW/cm2.

Figure 5 Femtosecond transient absorption spectra (fs-TA) excited at 405 nm of (a) 4a and (b) 4a⊃5a in acetonitrile. fs-TA at the indicated delay times (λex = 405 nm) of (c) 4a and (d) 4a⊃5a in acetonitrile. (e) Experimental decay curves of 4a and 4a⊃5a in transient absorption probed with fitting kinetic traces at 790 nm. (f) Schematic of charge transfer processes in 4a⊃5a after irradiation.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
