
Figure 1.
Various methods of synthesizing oxygen vacancies.
A comprehensive review on oxygen vacancies modified catalysts: Synthesis, characterization, and crucial role in catalytic ozonation
Fengchen Wang , Yujia Xiang , Yuqi Zhang , Xin Zhou , Jing Zhang , Chuanshu He , Heng Zhang , Zhaokun Xiong , Peng Zhou , Hongyu Zhou , Yang Liu , Bo Lai

In recent decades, the pollution of water bodies by anthropogenic discharges of heavy metals [1,2], organic pollutants [3–6] and other pollutants [7–10] has been a cause of widespread concern worldwide. In particular, pharmaceuticals [11–13], organic dyes [14–16], and personal care products [17–19] in wastewater are extremely harmful to the environment and human health. Traditional water treatment methodologies encounter numerous constraints [20,21], thereby necessitating an expedient development of an environmentally benign water purification technology capable of eliminating contaminants. Advanced oxidation processes (AOPs) have been extensively studied in the context of wastewater treatment due to their efficacy in the removal of recalcitrant organic pollutants and their environmentally friendly treatment methodologies [22–29]. Among AOPs, heterogeneous catalytic ozonation has emerged attention as a promising approach in wastewater treatment due to its inherent advantages, such as the elimination of the requirement for additional reagents, and its adaptability across a wide pH range [30,31]. An ideal ozone catalyst should have high catalytic activity [32]. To enhance catalyst performance, several strategies are employed, including metal and non-metal doping [33–37], metal loading [38–41], the construction of homojunctions and heterojunctions [42–46], the development of specific crystal structures [47–51].
Oxygen vacancies were first proposed by Tompkins in 1960 [52], which become one of the most common anionic defects in lattice oxygen detached metal compounds [53]. In recent years, research on the catalytic ozone degradation of pollutants utilizing oxygen vacancy materials has seen a surge [54]. Introducing oxygen vacancies into the catalyst facilitates the modulation of its electronic structure, thereby augmenting electron transfer capabilities [55–57]. Furthermore, these vacancies serve as active sites during the reaction process, interacting with ozone to generate radicals and non-radicals, ultimately enhancing the system’s degradation performance for pollutants [58]. However, upon reviewing the existing literature, it is evident that while numerous studies have investigated the role of oxygen vacancies in catalytic ozone systems, a comprehensive review of the underlying mechanism of oxygen vacancies remains elusive.
In view of this, this paper reviews recent advances in the study of the role of oxygen vacancies in catalyzing ozone. It details diverse strategies employed to introduce and adjust oxygen vacancies within catalysts, such as doping, liquid-phase reduction, quenching, and mechanochemistry, and evaluates their respective strengths and weaknesses. This contributes to the understanding of the principles of oxygen vacancy generation and the currently common means of preparing oxygen vacancies. Subsequently, this section discusses the prevalent techniques employed to characterize oxygen vacancies within catalysts, such as X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), electron paramagnetic resonance (EPR), high-resolution transmission electron microscopy (HRTEM), and Raman spectroscopy, all of which are instrumental in elucidating the concentration levels of oxygen vacancies as well as their function in the catalytic system. In conclusion, we propose several primary mechanisms through which oxygen vacancies play a pivotal role in catalyzing ozone, while also highlighting the limitations of existing research on oxygen vacancies. The review in this paper provides a theoretical basis and valuable reference for designing suitable ozone catalysts for efficient degradation and studying the role of oxygen vacancies in ozone.
Metal or non-metal doping is an effective strategy to generate oxygen vacancies. For example, the doping of transition metals (Mn, Fe, Co, Ni) weakened the strength of the Pr-O bond, generated more oxygen vacancies, and effectively regulated the local electronic structure. This manipulation of the local electronic environment not only optimizes the redox potential but also enhances the catalyst’s ability to activate oxygen [59]. Oxygen vacancies at the interface with varied coordination environments were created by incorporating low-valence dopant ions with distinct d-band centers into CeO2 utilizing a hydrothermal synthesis approach. The catalyst possessing the highest concentration of interfacial oxygen vacancies demonstrated superior catalytic performance [60]. Oxygen vacancies are induced by doping Cu2+ ions in place of Zn2+ ions within the primary lattice, and the presence of these vacancies elevates the Fermi energy levels of the oxide, resulting in the formation of defect energy levels within the band gap, thereby narrowing the energy band widths and enhancing the electrochemical performance. Furthermore, theoretical calculations suggest that materials containing oxygen vacancies may exhibit enhanced lithophilic [61].
In addition to the introduction of metal ions, the introduction of non-metal ions can also increase the oxygen vacancies in the material. Wang et al. [62] introduced sodium (Na+) ions into the pore framework of an octahedral molecular sieve (OMS-2) using a straightforward solid-phase reaction technique. Studies using O2-TPD (programmed temperature-raising desorption of oxygen) and EPR, along with theoretical calculations, demonstrated that modulating the concentration of Na+ within the tunnel structure selectively enhances the formation of oxygen vacancies. Additionally, Na+ doping facilitates the regeneration of these oxygen vacancies, which is attributed to the destabilization of manganese-oxygen bonds (Mn-O) within the octahedral structure.
The oxygen-containing compound is immersed in a specific liquid-phase reducing agent, which is capable of extracting oxygen from the lattice of the oxygen-containing compound at room or elevated temperatures to produce oxygen vacancies (Fig. 1). Selecting NaH as a reducing agent facilitates the creation of numerous oxygen vacancies within TiO2 under mild conditions. This strategy not only reduces the working temperature of the material, but also greatly improves the Mg/MgH2 electron mobility through the generation and elimination of oxygen vacancies throughout the reaction process [63]. Using various alcohols as reducing agents, the reduction of these alcohols causes the cleavage of Bi-O bonds, which in turn generates oxygen vacancies. Theoretical calculations and experimental results demonstrate that these vacancies can act as trapping sites, enhancing the efficiency of photogenerated carrier separation, lowering the band gap energy, and boosting the absorption of visible light. The CO yield from the catalyst synthesized with ethylene glycol is approximately 7.2 times higher than that from a catalyst prepared using pure water [64].
It is both feasible and significant to modulate oxygen vacancies during the process of introducing oxygen vacancies through liquid-phase reduction. The materials were synthesized with controlled oxygen vacancies using a straightforward and economically viable liquid-phase reduction method at temperatures ranging from 30 ℃ to 90 ℃, with NaBH4 serving as the reducing agent. The introduction of oxygen vacancies enhances the Fermi energy level and electrical conductivity of the carbon nanotubes, surpassing that of pristine ones, and leads to a reduced effective work function [65]. Liquid-phase treatment of Co3O4 porous nanosheets with hydrazine hydrate successfully induced oxygen vacancies. Furthermore, the oxygen vacancy concentration in Co3O4 porous nanosheets was effectively controlled through adjusting the volume of hydrazine hydrate. The introduction of oxygen vacancies enhances the electrical conductivity and facilitates charge transfer and lithium-ion diffusion, significantly enhancing the electrochemical lithium storage performance regarding reversible capacity, rate performance, and cycling stability [66].
Due to the low partial pressure of oxygen in the atmosphere, metal oxides can release lattice oxygen at elevated temperatures without undergoing phase transitions. Introducing oxygen vacancies into the material can be achieved through high-temperature treatment in a controlled atmosphere, such as vacuum, CO, H2, Ar, N2, and others (Fig. 1) [67].
Three-dimensional CaTiO3 was combined with a fixed quantity of NaBH3 and subjected to annealing in an argon atmosphere at varying temperatures to introduce oxygen vacancies in a controlled fashion. The resulting oxygen vacancies are mainly present on the catalyst surface, and the oxygen vacancies enhance the catalytic activity and optimize the charge separation that occurs during the photocatalytic process, thus significantly improving the photocatalytic performance of the material [68]. Two distinct catalysts, featuring oxygen vacancies at varying positions, were synthesized through the calcination of CaTiO3 in a H2/N2 atmosphere. Both experimental and theoretical findings indicate that subsurface oxygen vacancies can change the energy band structure of CaTiO3, thereby enhancing charge separation. Additionally, surface oxygen vacancies serve as active sites that facilitate the generation of hydrogen. Comparatively, the hydrogen precipitation rates of these catalysts have been elevated by approximately 49 times when compared to the pristine CaTiO3 [69]. The material was quenched in liquid nitrogen at temperatures below −196 ℃. Oxygen vacancy defects are retained by transient cooling in an anoxic environment and become electrocatalytically active sites for oxygen evolution reactions [70].
Graphite powder serves as an economical alternative to costly reducing gases, which are primarily oxidized to carbon monoxide when graphite is exposed to temperatures in excess of 700 ℃. The fabrication of oxygen vacancies within the electrode plate film was achieved through calcination within a graphite layer. These vacancies bolster the carrier concentration and markedly enhance the charge separation between electrons and holes [71].
Mechanochemistry serves as a prevalent technique for the synthesis of materials with oxygen vacancies. The mechanical ball milling process involves the treatment of diverse materials within sealed containers, offering benefits such as ease of operation, straightforward post-treatment, cost-effectiveness, and environmental sustainability [72]. The application of shear stress and compression through ball milling induces lattice distortion within the material. This mechanical strain displaces oxygen atoms from their original lattice sites, thereby generating oxygen vacancies (Fig. 1).
Bi12SiO20 and powder (S) can be combined to form n-p heterojunction structures through a straightforward ball milling technique, which introduces surface oxygen vacancies. These vacancies can be used to greatly improve the catalytic performance by taking advantage of their strong affinity for NO, O2 and H2O as well as the favorable energy conversion during the oxidation process [73]. Cu-Ce catalysts were synthesized using the ball milling method. The catalytic activity of these catalysts towards toluene was significantly enhanced due to the accelerated backfilling and activation of gaseous oxygen facilitated by the asymmetric oxygen vacancies generated at the Cu-Ce interface [74].
Furthermore, it has been demonstrated that the oxygen vacancy concentration on the surface of the material can be effectively manipulated by altering the parameters of the ball milling process. A series of BiOCI/biochar nanocomposites enriched with oxygen vacancies were synthesized by mixing BiOCI particles with biochar through the ball milling method, while varying parameters such as milling time, milling speed, and milling ratio. The presence of oxygen vacancies alters the bond lengths, coordination modes, and other characteristics of the adsorbed molecules, thereby enhancing their affinity for these molecules. The adsorption performance of the modified materials for dyes (60%) was significantly superior to that of the original BM-BC (20%) [75].
In addition to adjusting the parameters of ball milling, it is also feasible to produce materials enriched in oxygen vacancies through the incorporation of chemical additives. The concentration of oxygen vacancies can be systematically controlled by mixing TiO2 and urea at specific molar ratios. The presence of oxygen vacancies in the materials facilitates the chemisorption of dioxygen and enhances electron transfer between defect sites and oxygen, resulting in the generation of reactive oxygen species. This mechanism contributes to improved catalytic performance in the oxidation of aromatic sulfides [76].
The introduction of a significant number of oxygen vacancies into layered double hydroxides via argon plasma etching has been shown to enhance the number of active sites, thereby improving the capacitive performance of capacitors [77]. Plasma treatment was employed to meticulously regulate the concentration and types of oxygen defects present in the nickel oxide substrate. The incorporation of oxygen vacancies has been demonstrated to facilitate charge separation and enhance electron transfer capabilities within battery systems [78]. Additionally, oxygen vacancies were successfully introduced onto bismuth oxychloride (BiOCl) nanoflakes through photodeposition techniques. The resultant abundance of oxygen vacancies on the surface effectively adsorbed reactant molecules, improved light absorption, and increased the rate of electron-hole separation, thus enhancing photocatalytic activity. Experimental results indicated that the photocatalytic reduction rate of CO2 using the modified material was 17.6 times greater than that of the unmodified BiOCl nanoflake [79]. During the synthesis process, the application of pressure generates lattice stresses that promote the release of oxygen atoms, leading to the formation of oxygen vacancies. In the case of lithium titanate (Li4Ti5O12), the concentration of oxygen vacancies was precisely controlled through a high-pressure treatment method that integrates elevated temperature and pressure. The presence of these vacancies not only improves the intrinsic conductivity of the material but also enhances its ionic diffusion capacity [80].
XRD serves as a fundamental analytical method for the examination and identification of crystal structures, offering critical insights into the atomic arrangement and symmetry within crystalline materials. As illustrated in Fig. 2a, the crystal structures of the synthesized catalysts were analyzed using XRD. At lower doping levels of TiO2, no peaks indicative of additional crystalline phases were detected, suggesting that the samples preserved their fundamental crystalline structure following the reduction treatment. Conversely, the introduction of polymetallic elements resulted in an enhanced formation of oxygen vacancies and lattice distortion. This phenomenon led to a gradual decrease in the intensity and an increase in the half-peak width of the predominant anatase (101) peak, which is situated at 25.28°, as the variety of elemental types increased [81]. The distinctive peak observed at 28.24° exhibited a slight shift towards a higher angle following the incorporation of transition metals (Fig. 2b). This alteration can be ascribed to the smaller ionic radii of the transition metal ions (Co: 0.075 nm, Mn: 0.053 nm, Fe: 0.065 nm, Ni: 0.069 nm) in comparison to those of the Pr ions (Pr3+: 0.113 nm, Pr4+: 0.096 nm). Such a lattice contraction may result in an increase in the number of oxygen vacancies [59]. When the molar ratio of Na2CO3 to SIn2O3 was maintained at 2:1, the sample S2 demonstrated enhanced purity, with no detectable impurity phases present. Conversely, at molar ratios of 1:1 and 1.5:1, the emergence of impurity phases related to In2O3 was noted. Furthermore, the peak positions (104) in the samples S1.5 and S1 exhibited a shift towards a lower angle (Fig. 2c), which is likely indicative of oxygen vacancy defects within the structural framework of the samples [82].
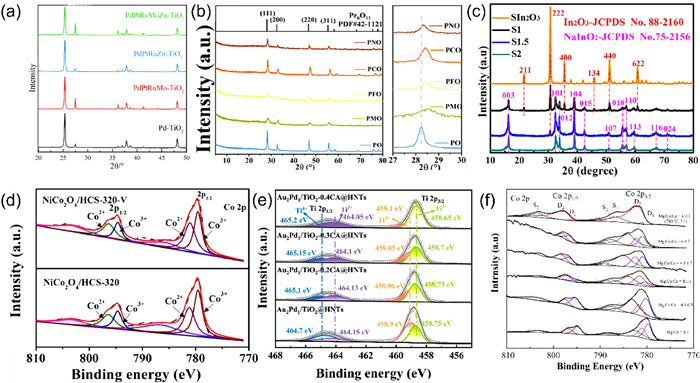
XPS is a widely utilized method for surface analysis, as it can reveal how defects within a material influence bonding energy, leading to the displacement of existing peaks or the emergence of new ones. As such, XPS is an effective technique for identifying oxygen vacancies in various materials. In this study, mesoporous NiCo2O4 nanosheets were successfully synthesized on conducting hollow carbon spheres via a straightforward hydrothermal treatment followed by a quenching process. The XPS analysis of the Co 2p orbitals, illustrated in Fig. 2d, reveals that the Co2+/Co3+ atomic ratio for the NiCo2O4/HCS-320-V sample (0.96) exceeds that of the NiCo2O4/HCS-320 sample (0.79), indicating a higher concentration of oxygen vacancies in the former. The O2 peak observed in the O 1s orbitals at 531.5 eV is associated with defect sites characterized by low oxygen coordination. Furthermore, the area percentage of the O2 peaks in the NiCo2O4/HCS-320-V sample is significantly greater than that in the NiCo2O4/HCS-320 sample, suggesting that samples subjected to quenching in an oxygen-deficient environment exhibit an increased number of oxygen vacancies. The NiCo2O4/HCS-320-V sample, with its abundant oxygen vacancies, demonstrated enhanced redox activity [83].
Novel ammonia-nitrogen ozonation catalysts were synthesized using γ-Al2O3 as the support material, with Fe and Mg as dopants, through an impregnation-calcination process. XPS analysis of the O 1s region revealed three distinct peaks at 531.02, 532.02, and 533.09 eV, which correspond to lattice oxygen (Olat), chemisorbed oxygen (Oads), and physically adsorbed oxygen, respectively. The Olat was predominantly derived from metal-oxygen bonds involving the elements O, Al, Fe, and Mg, which primarily serve as active sites for the ozonation process. The Oads is primarily associated with metal hydroxide bonding (MOH). The catalysts γ-Al2O3@Mg, γ-Al2O3@Fe, and γ-Al2O3@Fe/Mg5 exhibited Oads/(Olat + Oads) ratios of 46.52%, 46.94%, and 61.69%, respectively, indicating that γ-Al2O3@Fe/Mg5 possesses the highest oxygen vacancies content. Under optimized conditions of initial pH 7, a catalyst dosage of 112.88 g/L, and an ozone dosage of 2.4 mg/min, γ-Al2O3@Fe/Mg5 efficiently catalyzed the ozonation of ammonia and nitrogen, achieving gas-phase product selectivity ranging from 67.82% to 98.73%. Moreover, γ-Al2O3@Fe/Mg5 demonstrated stability with low ion leachability, facilitating easy precipitation and recovery from water [84].
Researchers crafted a variety of noble metal-based catalysts, identified as AumPdn/TiO2-xCA@HNTs. These were synthesized through an environmentally friendly approach, utilizing a naturally occurring and cost-effective tubular aluminosilicate clay mineral of HNTs as template by depositing a layer of TiO2 and adding citric acid (CA) to adjust oxygen vacancies prior to calcination. The chemical composition and surface properties of the Au2Pd1/TiO2-xCA@HNTs catalysts were characterized using XPS (Fig. 2e). The lower binding energy peaks in the Ti 2p spectra, observed at 464.05–464.15 eV (2p1/2) and 458.65–458.75 eV (2p3/2), indicated the presence of partially reduced Ti3+ ions. The formation of Ti3+ suggests the creation of oxygen vacancies. The ratio of Ti3+/(Ti3++ Ti4+) calculated from the peak area, serves as an indicator of the oxygen vacancies concentration within the TiO2 layer. This ratio was found to increase with the CA concentration, reaching its maximum in the Au2Pd1/TiO2–0.4CA@HNTs catalyst, which contained the highest Ti3+ concentration (64.78%). The ratio of Oads/(Oads + Olat) also reflects the concentration of oxygen vacancies, with the Au2Pd1/TiO2–0.4CA@HNTs catalyst demonstrating the highest relative concentration of oxygen vacancies at 60.48% in the O 1s XPS spectrum. These results are consistent with the findings from the Ti 2p spectral analysis. Notably, the Au2Pd1/TiO2–0.4CA@HNTs catalyst demonstrated the most efficient conversion of 5-hydroxymethylfurfural (HMF) to 2,5-furandicarboxylic acid (FDCA) in aqueous solution using air as the oxidizing agent, achieving an FDCA yield of 98.4% [85].
Ternary composite oxides of Mg-Co-Ce were synthesized utilizing a co-precipitation method followed by calcination. XPS analysis of the Co 2p spectra indicated that the ratio of Co2+/Co initially increased and subsequently decreased with an increase in Ce content within the composites, peaking at a composition of M (Mg:Co:Ce) = 4:1:1. This observation implies that Ce is integrated into the spinel structure, resulting in distortion of the crystal lattice and the formation of additional oxygen vacancies (Fig. 2f). The Mg-Co-Ce catalysts, characterized by a high density of surface oxygen vacancies, demonstrated remarkable ozone-selective catalytic activity for the oxidation of ammonia to nitrogen gas. Under optimal conditions, the ammonia removal rate for the MgO/Co3O4/CeO2 catalytic system was measured at 0.03328 min-1 (R2 = 0.99942), which is approximately 2.1 times greater than that of the MgO/Co3O4 catalytic system, which exhibited a rate of 0.01597 min-1 (R2 = 0.99813) [86].
EPR is a magnetic resonance technique that facilitates the direct detection and analysis of paramagnetic substances characterized by unpaired electrons. The introduction of unpaired electrons into a material through oxygen vacancies renders the EPR technique particularly effective for identifying these vacancies.
EPR was employed to further characterize the formation of oxygen vacancies in Fe-Co layered double hydroxide (LDH). As depicted in Fig. 3a, an asymmetric signal is distinctly observable in Fe-Co LDH at g = 2.003, attributed to the capture of unpaired electrons in oxygen vacancies. The signal intensity increases from approximately 6.98 × 103 to 1.66 × 104 as the calcination temperature rises from 0 ℃ to 300 ℃, with a notable increase observed at 200 ℃. However, when the temperature exceeds 200 ℃, the presence of oxygen vacancies may compromise the crystal structure of Fe-Co LDH, resulting in a deceleration of the increase in oxygen vacancy intensity [48]. CoxMn1-xFe2O4 was synthesized using microwave-assisted hydrothermal synthesis. To confirm the presence and quantity of oxygen vacancies, EPR spectroscopy was conducted on MFO and CMFO-0.5 samples. The analysis revealed a characteristic peak at g ≈ 2.003 in both MFO and CMFO-0.5, indicating the capture of unpaired electrons resulting from the formation of oxygen vacancies (Fig. 3b). The intensity of this peak provides evidence that CMFO-0.5 contains a substantial concentration of oxygen vacancies [49].
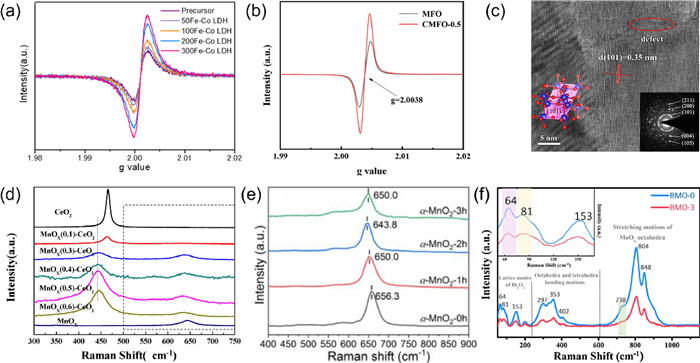
Oxygen vacancy-enriched VO-MoxCo3-xO4 catalysts were synthesized by doping a minimal amount of Mo (~0.5wt%) into the Co3O4 matrix. EPR spectroscopy was utilized to characterize the structural defects in VO-MoxCo3-xO4. The EPR spectrum of pure Co3O4 exhibited no discernible signal, indicating that it is a defect-free, pristine crystal. In contrast, the EPR spectrum of VO-MoxCo3-xO4 demonstrated a significant increase in the g-factor, measured at 2.003, which signifies that the doping process effectively introduced oxygen vacancies within the Co3O4 lattice [87]. Additionally, novel cobalt molybdate nanosheets (CoMoO4–Ov-n@GF) with tunable oxygen vacancy content were synthesized through the in-situ formation of CoMoO4 nanosheets on graphite felts (GFs), followed by annealing under a reducing atmosphere. The modulation of oxygen vacancies was investigated using EPR spectroscopy. Notably, the intensity of the characteristic peaks for CoMoO4@GF significantly increased following quenching treatment, indicating that a greater number of oxygen vacancies were generated extended treatment duration. The CoMoO4–Ov-2@GF, which exhibited optimal oxygen vacancy content, demonstrated exceptional electrocatalytic performance, as evidenced by its low overpotential (296 mV at 10 mA/cm2) and a small Tafel slope (62.4 mV/dec) in alkaline solution, comparable to that of RuO2@GF [88].
Conventional transmission electron microscopy (TEM) is limited to imaging at the surface level, which poses challenges in identifying internal structural features, such as grain spacing and atomic arrangement. In contrast, HRTEM offers a substantially enhanced resolution, facilitating the detailed analysis of intricate features in conjunction with accurate crystallographic information. As a result, HRTEM serves as a robust technique for characterizing the presence and distribution of oxygen vacancies within materials.
Oxygen vacancies were successfully induced on the surface of Ti3O5 through the incorporation of iron ions via ball milling. HRTEM analysis revealed significant lattice disorder. The interplanar distances in the Fe-Ti3O5 (110) and (020) planes were measured at 0.37 nm and 0.21 nm, respectively, which are greater than those observed in undoped Ti3O5, where the interplanar distances were 0.36 nm and 0.20 nm for the same orientations. In Fe-Ti3O5, the formation of Fe-O bonds, as opposed to Ti-O bonds, facilitates the penetration of iron and the development of surface defects, which in turn increases the lattice spacing and contributes to a low crystallinity of 84.8%. The introduction of oxygen vacancies enhances electron concentration and electronic conductivity (2.49 × 10–4 S/cm) at low temperatures. This increased conductivity, combined with the reduced crystallinity, promotes ultra-sensitive sensing across a wide temperature range prior to the metal-insulator transition [89]. MnO2 catalysts with varying concentrations of oxygen vacancies were synthesized through the doping of Cu2+. HRETEM image of the MnO2–OV3 sample exhibits a lattice stripe spacing of 0.23 nm, which is attributed to the (100) plane of the birnessite-type MnO2 structure. No copper oxide particles were identified within the sample. Notably, the lattice stripe in certain regions of the MnO2–OV3 sample appeared blurred, which may be ascribed to the presence of oxygen vacancies. The introduction of an optimal number of oxygen vacancies in MnO2 significantly enhances its low-temperature reducibility and oxygen activity, thereby improving catalytic efficiency for toluene oxidation reactions [90].
Oxygen vacancies were deliberately introduced into defect-rich Fe-doped TiO2-x nanosheets through a specific fabrication process. Citric acid served as a chelating agent to improve the dispersion of iron within the material. HRTEM image (Fig. 3c) displays a lattice spacing of 0.35 nm, which corresponds to the (101) crystal plane of anatase TiO2. The absence of observable lattice streaks associated with iron and its oxides indicates that the iron has been effectively incorporated into the TiO2 lattice without the formation of distinct phases. Furthermore, the HRTEM image reveals the presence of crystal defects (highlighted by red circles) in the Fe/TiO2-x-S composite, suggesting an increased concentration of oxygen vacancies as a result of iron doping. These Fe-doped TiO2-x nanosheets, characterized by a high density of oxygen vacancies, achieve nearly 100% conversion and exhibit significant sulfur selectivity for H2S at 210 ℃, surpassing the performance of most other Ti-based materials documented in the literature [91].
Raman spectroscopy serves as a robust analytical method for the examination of molecular structures, offering valuable insights into their vibrational and rotational properties. The vibrational modes are influenced by various chemical bonds, and the presence of material defects can alter these bonds, leading to shifts in peak positions or the appearance of new peaks. Consequently, Raman spectroscopy is an effective characterization tool for the assessment of oxygen vacancies. The controlled creation of oxygen vacancies in WO3 nanoflake photoanodes was accomplished through the application of an Ar-plasma engraving technique. The physical phase characteristics of the WO3-Vo samples were evaluated using Raman spectroscopy. In comparison to pure WO3, the peaks associated with O-W-O bending modes (266 and 320 cm-1) and stretching modes (701 and 801 cm-1) in WO3-Vo exhibit a marked reduction, suggesting a significant presence of oxygen vacancies within the WO3-Vo photoanode. These oxygen vacancies contribute to an increase in carrier density and provide additional driving forces that facilitate interfacial charge transport, thereby improving photoelectrochemical performance. Under AM 1.5 G solar irradiation, the photocurrent density of WO3-Vo attains 2.76 mA/cm2 at 1.23 V vs. RHE, which is threefold greater than that of the unmodified WO3 photoanode [92].
MnOx-CeO2 catalysts were synthesized by the citric acid complex method. Fig. 3d presents the visible Raman spectroscopic data for CeO2, the MnOx(z)-CeO2, and the MnOx catalysts. The Raman band at 465 cm-1 in MnOx(z)-CeO2 exhibits an increase in bandwidth accompanied by a decrease in peak intensity. The observed broadening of these energy bands is a size-dependent effect, attributed to non-uniform strain broadening resulting from grain size dispersion and phonon confinement. Given that Raman spectroscopy is a surface-sensitive characterization technique, the down-shifts of these bands is associated with surface oxygen vacancies [93].
The α-MnO2 samples, which were synthesized under different hydrogen reduction durations, were subjected to analysis via Raman spectroscopy (Fig. 3e). The Raman bands detected in the vicinity of 650 cm-1 are indicative of the symmetric stretching vibrations of Mn-O bonds within the MnO6 octahedral structure. The spectral data reveal that, as a result of varying hydrogen reduction times, the band at 656.3 cm-1 for α-MnO2–0 h shifts to 650 cm-1 for α-MnO2–1 h, then to 643.8 cm-1 for α-MnO2–2 h, and subsequently returns to 650 cm-1 for α-MnO2–3 h. This observed peak shift phenomenon signifies alterations in the bonding forces of Mn-O within the catalysts. Notably, α-MnO2–2 h exhibits the weakest Mn-O bonding force among the tested catalysts, which is likely to facilitate the formation of oxygen vacancies [94]. A series of Bi2MoO6 compounds were synthesized using a simple solvothermal method. Significant changes in the relative intensities of Raman modes were detected at approximately 64, 81, and 153 cm-1, which are associated with the lattice vibrations of Bi2O22+ (Fig. 3f). These alterations are likely due to the partial depletion of oxygen from the Bi-O bonds, a phenomenon induced by the high concentration of oxygen vacancies facilitated by TMEDA. The optimized Bi2MoO6 material, enhanced by the presence of oxygen vacancies, achieved a maximum ciprofloxacin degradation rate of 1.799 mg min-1 m-1, which is nearly eight and a half times greater than that of the unmodified Bi2MoO6. Following a 6-h photocatalytic treatment, the biotoxicity of ciprofloxacin and its metabolites against Escherichia coli K-12 and Saccharomyces cerevisiae was completely eliminated [95].
Previous research has indicated that the metal atoms surrounding oxygen vacancies possess enhanced electron density, thereby facilitating the redox cycling within metal oxide systems [96]. Lu et al. [58] synthesized a series of α-MnO2 catalysts through a straightforward quenching process. Characterization results, including XPS, EPR and positron annihilation lifetime spectroscopy (PALS), indicated a higher concentration of oxygen vacancies under high-temperature calcination conditions at 600 ℃. Cyclic voltammetry (CV) and hydrogen temperature-programmed reduction (H2-TPR) analyses revealed that MnO2–600, with the highest oxygen vacancy content, exhibited superior redox properties and enhanced interfacial electron transfer capacity. Additionally, MnO2–600 achieved a toluene conversion of 95% and a mineralization rate of 89.5% at 20 ℃. Zhang et al. [55] prepared Fe-LDO-BC layered double oxides by combining hydrotalcite-like materials with biochar, and used them in catalytic ozonation to remove methylene blue (MB). They conducted electrochemical measurements, including CV and AC impedance (EIS), on biochar (BC), CoFe layered double oxide (CoFe-LDO), and the composite CoFe-LDO-BC using a three-electrode setup. The results indicated that the CoFe-LDO-BC catalyst, which had a high concentration of oxygen vacancies, exhibited the highest current density and the smallest arc radius on the Nyquist plot. These oxygen vacancies facilitated electron transfer between Co(Ⅲ) and Co(Ⅱ) (Fig. 4a). Additionally, TOC analysis showed that the mineralization of methylene blue increased from 31.6% to 68.25% comparing with ozone alone in the catalytic ozonation system.
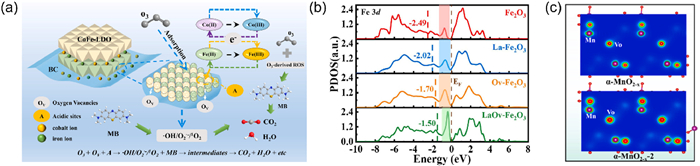
In addition to employing characterization techniques to investigate the impact of oxygen vacancies, density functional theory (DFT) calculations are frequently utilized to delve deeper into the role that oxygen vacancies play. Pan et al. [97] successfully synthesized a Fe0.8La/MCM-48 catalyst through a method that combined ball milling and calcination. XPS and EPR analyses confirmed the presence of a substantial number of oxygen vacancies within the catalyst structure. To delve into the functional significance of these oxygen vacancies, DFT calculations were employed. These computations revealed that the oxygen vacancies contribute to an optimized bonding orbital distribution around the Fermi energy level. The d-band center of Fe in the catalyst moves up to the Fermi energy level, showing a positive shift (Fig. 4b). This upward shift of the d-band center facilitates enhanced adsorption of O3. Consequently, the presence of oxygen vacancies modulates the catalyst’s electronic structure, thereby promoting the overall reaction process
The study of oxygen vacancies in catalytic ozonation usually focuses on the effect of surface oxygen vacancies and often neglects to explore the different roles that oxygen vacancies may play at different spatial locations within the material. Oxygen vacancies in the bulk of material, being situated far from the interface and unable to interact directly with the substrate, display distinct characteristics compared to those at the surface during catalytic processes. DFT calculations reveal that in α-MnO2-x-2, electrons are released from the Mn 2p orbitals into the domain, accumulating around the oxygen vacancies to create a sustained electron-rich environment (Fig. 4c). This leads to the formation of a continuous electron energy level, which facilitates the outward migration of electrons from the interior to the surface. The EIS results also support the conclusion that bulk oxygen vacancies stimulate the flow of electrons from the interior of the catalyst to the reaction interface. And the bulk oxygen vacancies that discharge electrons to the surface lack the compensation process that surface oxygen vacancies have. Therefore, these bulk oxygen vacancies are in an electron-deficient state, creating an electronic imbalance that triggers the phenomenon of electronic relaxation. This effect attracts external electrons and forms an electron transfer pathway for degrading organic pollutants [98].
Many studies have demonstrated that oxygen vacancies are active sites in catalytic materials that interact directly with ozone. This interaction facilitates the generation of radical and non-radical species, thus contributing to the overall efficiency and effectiveness of pollutant degradation.
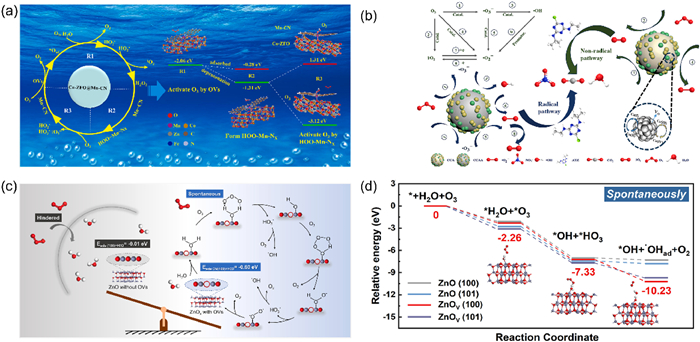
Xu et al. [99] successfully prepared Co-ZFO@Mn-CN catalysts through cobalt doping. Their EPR and XPS analyses indicated that the lattice doping of cobalt induced the formation of a significant number of oxygen vacancies. Furthermore, DFT calculations revealed that ozone adsorbs onto these oxygen vacancies. Due to the strong Lewis acidic nature of these vacancies, the O–O bond length in ozone undergoes stretching, extending from 1.284 Å to 2.212 Å. This stretching of the bond length results in the decomposition of ozone, yielding oxygen and adsorbed oxygen (*O). Subsequently, these products engage in a series of chain reactions with dissolved ozone, leading to the production of 1O2 and O2- (Fig. 5a). Ultimately, these reactions promote the degradation process. Wu et al. [100] synthesized CuO–CeO2-activated carbon loaded attapulgite (CCAA) through a straightforward hybrid calcination approach. The incorporation of CuO–CeO2 and activated carbon elevated the concentration of oxygen vacancies. EPR experiments, quenching tests, and probe experiments revealed that the primary reactive species in this system is 1O2. DFT calculations further demonstrated that the oxygen vacancies on the catalyst surface exhibit a high affinity for ozone molecules. Upon adsorption of ozone onto these vacancies, the O–O bonds in the ozone molecule spontaneously cleave, initiating the formation of surface-bound atomic oxygen and 1O2 (Fig. 5b).
In addition to traditional reactive species, Liang et al. [101] discovered that the removal of pollutants was primarily attributed to non-radicals *O, rather than the conventional •OH. Both experimental evidence and DFT calculations revealed that oxygen vacancies serve as the primary active sites for ozone adsorption and the generation of *O. The O–O bond in ozone undergoes dissociation at these vacancies, resulting in the production of *O. Shao et al. [102] similarly observed the presence of *O in the system. Through comprehensive characterizations and DFT calculations, they determined that the introduction of pyrophosphate resulted in increased charge aggregation on the adjacent metal oxides, thereby enhancing the activity of the oxygen vacancies. This alteration in the nature of the oxygen vacancies facilitated the accumulation of *O on the catalyst surface and improved the efficiency of ozone utilization in the catalytic reaction.
To examine the impact of oxygen vacancies at various locations, Shi et al. [56] prepared two Ag/CeO2 catalysts using impregnation and reduction methods. The results from EPR and Raman spectroscopy revealed that the in-situ reduction method yielded higher concentrations of surface oxygen vacancies, whereas the impregnation method generated more vacancies within the bulk phase. The alteration in peak shape observed in the in-situ Raman spectra suggested that the surface oxygen vacancies on the catalyst facilitated the adsorption of ozone and enhanced its decomposition into reactive oxygen species, including O- and O2-, during the catalytic process. Furthermore, it was noted that the bulk oxygen vacancies migrated to the surface, where they underwent reformation and participated in subsequent reactions as new surface oxygen vacancies.
It has been recognized that the hydroxyl groups (-OH) generated by the dissociation of adsorbed water on the surface of metal oxides are the key active sites for the reaction. These hydroxyl groups exhibit dual reactivity; the hydrogen atom is strongly electrophilic, while the oxygen atom is nucleophilic. Such distinct reactivity allows them to engage in reactions with ozone, facilitating the catalytic process [103]. The presence of oxygen vacancies exposes more metal sites and favors the generation of hydrated hydroxyl groups.
The catalyst α-Fe0.9Mn0.1OOH was synthesized by in situ substitution of Fe with Mn in the FeOOH structure. This substitution resulted in an enhanced presence of oxygen vacancies on the catalyst’s surface. Utilizing ATR-FTIR spectroscopy for analysis, it was observed that these oxygen vacancy-enriched catalysts exhibit a heightened affinity for water molecule adsorption. The adsorbed water molecules on the surface are more prone to undergo a reaction with ozone, leading to the generation of hydroxyl radicals [104]. To evaluate the role of oxygen vacancies within the system, a comparative analysis was conducted on the ozone concentration changes between the O3/ZnOx-500 system and alternative systems. The ozone concentration on the surface of ZnOx-500 decreased significantly, suggesting that the oxygen-enriched vacancy material can effectively promote ozone decomposition. ATR-FTIR analysis revealed a notable attenuation of the telescopic vibrational signals associated with the metal-bound hydrated-OD species during catalytic ozonation, along with an inhibitory influence of EDTA and phosphate on ZnOx-500. Consequently, these results affirm the pivotal role of oxygen vacancies in augmenting the exposure of additional metal ions. DFT calculations indicate that oxygen vacancy pairs can facilitate the formation of surface hydrated hydroxyl groups (Fig. 5c) [105]. The in-situ Raman spectra of O3/NC and O3/ZNC400 were analyzed, revealing characteristic peaks associated with *O and *O2 in both samples. However, no distinct ozone adsorption peaks were detected in the O3/ZNC400 sample. This absence may be attributed to the presence of oxygen vacancies, which facilitate the rapid decomposition of adsorbed ozone into •OH. ATR-FTIR spectroscopy demonstrated that H2O dissociates at the exposed zinc sites, leading to the formation of hydrated hydroxyl groups. The decrease in vibrational frequency during the reaction confirmed the involvement of these hydrated hydroxyl groups in the chemical process. DFT calculations suggest that oxygen vacancies are crucial for exposing zinc sites, promoting the formation of hydrated hydroxyl groups (Fig. 5d). These groups then react with ozone to produce reactive oxygen species [106].
In conclusion, oxygen vacancies are pivotal in facilitating ozone reactions. The present review initiates by outlining a variety of tactics employed to introduce and regulate oxygen vacancies within catalysts, such as doping, liquid-phase reduction, quenching, and mechanochemical techniques. The comparative benefits and drawbacks of these synthesis methodologies are meticulously examined. For instance, chemical reduction methods commonly necessitate the employment of toxic reductants. Quenching processes are characterized by their high energy demand and susceptibility to generating unwanted by-products. Ball-milling approaches, despite their ease of operation for large-scale production, suffer from protracted processing durations and the propensity to create dust particles. Although advanced techniques, including plasma-induced defect formation, exhibit considerable potential, their advancement is constrained by the dependency on sophisticated and costly apparatus. Secondarily, a range of widely employed detection techniques for identifying oxygen vacancies, such as XRD, XPS, EPR, HRTEM and Raman spectroscopy, are introduced. These diverse characterization tools can be utilized in a synergistic manner, leveraging their distinct detection principles. This collaborative approach allows for a more precise and in-depth exploration of the role of oxygen vacancies in the system. Finally, the mechanisms of the role of oxygen vacancies in catalytic ozonation are summarized (Fig. 6). These key roles include Accelerating the reaction rate by modifying the electronic structure of the catalyst and facilitating electron transfer. Increasing the active sites of the catalyst so that the active sites interact with ozone to produce more radicals or non-radicals, thus speeding up the reaction. Exposing more metal sites, which in turn promotes the generation of hydrated hydroxyl groups. These hydrated hydroxyl groups then react with ozone to produce reactive oxygen species.

Based on the current research, several suggestions for future work are put forward from the following aspects:
(1) The ideal oxygen vacancy concentration is not simply higher; an excessive number of vacancies can compromise the crystal structure’s stability. Therefore, opting for a preparation technique that is environmentally sound, cost-effective, and capable of precisely controlling the oxygen vacancy concentration is of great significance.
(2) Present characterization methods are limited to verifying the existence of oxygen vacancies, falling short in providing both quantitative and qualitative insights into their concentrations and surrounding microenvironments. There is a pressing need to refine in situ characterization technologies to achieve a deeper understanding.
(3) A significant portion of research into the function of oxygen vacancies has concentrated on assessing their impact on the overall system, yet often overlooks the nuanced mechanisms by which these vacancies operate in varying positions and within distinct microenvironments.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Fengchen Wang: Writing – original draft, Methodology, Investigation, Data curation. Yujia Xiang: Supervision, Formal analysis. Yuqi Zhang: Supervision, Methodology, Investigation. Xin Zhou: Methodology, Investigation. Jing Zhang: Writing – review & editing, Supervision, Resources. Chuanshu He: Writing – review & editing, Supervision, Resources. Heng Zhang: Writing – review & editing, Supervision, Resources. Zhaokun Xiong: Writing – review & editing, Resources. Peng Zhou: Writing – review & editing, Supervision, Resources. Hongyu Zhou: Writing – review & editing, Investigation. Yang Liu: Writing – review & editing, Supervision, Software, Resources. Bo Lai: Writing – review & editing, Supervision, Software, Resources.
The authors acknowledge financial support from the Key R&D Program of Zhejiang province (No. 2024C03136).
W. Chen, X. Li, L. Ma, Chin. J. Environ. Eng. 12 (2018) 86–92.
T. Dippong, M.A. Resz, C. Tănăselia, O. Cadar, J. Hazard. Mater. 476 (2024) 135187. doi: 10.1016/j.jhazmat.2024.135187
Y. Xiang, Y. Liu, B. Cong, et al., Water. Res. 267 (2024) 122494. doi: 10.1016/j.watres.2024.122494
B. Al-Ghafri, T. Bora, P. Sathe, S. Dobrestov, M. Al-Abri, Appl. Catal. B 233 (2018) 136–142. doi: 10.1016/j.apcatb.2018.03.095
H. Liu, C. Wang, X. Ai, et al., J. Environ. Eng. 11 (2023) 110573.
S. Silvestri, A.R. Fajardo, B.A. Iglesias, Environ. Chem. Lett. 20 (2021) 731–771.
A. Shirmardi, M.A.M. Teridi, H.R. Azimi, et al., Appl. Surf. Sci. 462 (2018) 730–738. doi: 10.1016/j.apsusc.2018.06.252
M. Bashir, M. Batool, N. Arif, et al., Coord. Chem. Rev. 492 (2023) 215286. doi: 10.1016/j.ccr.2023.215286
N. Madima, S.B. Mishra, I. Inamuddin, A.K. Mishra, Environ. Chem. Lett. 18 (2020) 1169–1191. doi: 10.1007/s10311-020-01001-0
C. Onorato, L.J. Banasiak, A.I. Schäfer, Sep. Purif. Technol. 187 (2017) 426–435. doi: 10.1016/j.seppur.2017.06.016
Z. Xiong, B. Lai, Y. Yuan, et al., Chem. Eng. J. 302 (2016) 137–145. doi: 10.1016/j.cej.2016.05.052
S. Zhu, B. Dong, Y. Yu, et al., Chem. Eng. J. 328 (2017) 527–535. doi: 10.1016/j.cej.2017.07.083
Y. Huang, M. Liang, L. Ma, et al., Environ. Pollut. 268 (2021) 115722. doi: 10.1016/j.envpol.2020.115722
H. Xue, S. Xiong, K. Mi, Y. Wang, Green Energy Environ. 8 (2023) 194–199. doi: 10.1016/j.gee.2020.09.010
L.Y. Wang, J.H. Liu, M.N. Liu, et al., Nano Energy 128 (2024) 109910. doi: 10.1016/j.nanoen.2024.109910
M.J. Shanker, Vidhishas, Environ. Chem. Lett. 15 (2017) 623–642. doi: 10.1007/s10311-017-0650-2
K.J. Harris, G. Munoz, V. Woo, S. Sauvé, A.A.J.E.s. Rand, Environ. Sci. Technol. 56 (2022) 14594–14604. doi: 10.1021/acs.est.2c02660
Jeff Lyon, JAMa 316 (2016) 1859.
Y.T. Yang, H.G. Ni, Water. Res. 236 (2023) 119981. doi: 10.1016/j.watres.2023.119981
P.V.A. Padmanabhan, K.P. Sreekumar, T.K. Thiyagarajan, R.U. Satpute, K.G.K. Warrier, Vacuum. 80 (2006) 1252–1255. doi: 10.1016/j.vacuum.2006.01.054
U.I. Gaya, A.H. Abdullah, J. Photoch. Photobio. C 9 (2008) 1–12.
S. Wacawek, H.V. Lutze, K. Grübel, V.V.T. Padil, D.D. Dionysiou, Chem. Eng. J. 330 (2017) 44–62. doi: 10.1016/j.cej.2017.07.132
Q. Yang, Y. Ma, F. Chen, F. Yao, D. Wang, Chem. Eng. J. 378 (2019) 122149. doi: 10.1016/j.cej.2019.122149
S. Yang, Y. Shi, X. Wang, et al., Water. Res. 242 (2023) 120317. doi: 10.1016/j.watres.2023.120317
C. Yu, Z. Wu, H. Shi, et al., Chin. Chem. Lett. 35 (2024) 109334. doi: 10.1016/j.cclet.2023.109334
L. Lai, H. Zhou, Y. Hong, et al., Chin. Chem. Lett. 34 (2023) 108580.
Y. Yuan, Z. Zhou, X. Zhang, et al., Chin. Chem. Lett. 34 (2023) 107932. doi: 10.1016/j.cclet.2022.107932
C. Zhou, P. Zhou, P. Zhang, Y. Sun, B. Lai, Res. Environ. Sci. 36 (2023) 1255–1264.
Z. Xiong, L. Gu, X. Wang, Energy Environ. Protect. 38 (2024) 52–64.
F. Qi, Z. Chen, B. Xu, et al., Appl. Catal B 84 (2008) 684–690. doi: 10.1016/j.apcatb.2008.05.027
B. Wang, H. Zhang, F. Wang, et al., Catalysts 9 (2019) 241. doi: 10.3390/catal9030241
H.L. Wang, L.S. Zhang, Z.G. Chen, et al., Chem. Soc. Rev. 43 (2014) 5234–5244. doi: 10.1039/c4cs00126e
Z. Wang, H. Ma, C. Zhang, et al., Chem. Eng. J. 354 (2018) 42–52. doi: 10.1016/j.cej.2018.07.177
A. Lv, C. Hu, Y. Nie, J. Qu, Appl. Catal. B 117-118 (2012) 246–252. doi: 10.1016/j.apcatb.2012.01.020
P. Yan, J. Shen, L. Yuan, et al., Sep. Purif. Technol. 228 (2019) 115766. doi: 10.1016/j.seppur.2019.115766
W. Xing, X. Xu, M. Zhang, et al., J. Hazard. Mater. 438 (2022) 129413. doi: 10.1016/j.jhazmat.2022.129413
J. Zhang, Q. Bai, X. Bi, et al., Nano Today 43 (2022) 101429. doi: 10.1016/j.nantod.2022.101429
S.Q. Tian, J.Y. Qi, Y. P, et al., Water. Res. 193 (2021) 116860. doi: 10.1016/j.watres.2021.116860
S.Q. Tian, J.Y. Qi, Y.P. Wang, Y.L. Liu, J. Ma, Water. Res. 193 (2021) 116860. doi: 10.1016/j.watres.2021.116860
D. Leybo, U.J. Etim, M. Monai, et al., Chem. Soc. Rev. 53 (2024) 10450–10490. doi: 10.1039/d4cs00527a
H.S. Bae, M.A. Mahadik, Y.S. Seo, et al., Chem. Eng. J. 408 (2021) 127260. doi: 10.1016/j.cej.2020.127260
Y. Yin, R. Lv, X. Li, L. Lv, W. Zhang, Appl. Catal. B 299 (2021) 120685. doi: 10.1016/j.apcatb.2021.120685
J. Low, J. Yu, M. Jaroniec, S. Wageh, A.A. Al-Ghamdi, Adv. Mater. 29 (2017) 1601694. doi: 10.1002/adma.201601694
K.Kaur Parul, R. Badru, P.P. Singh, S. Kaushal, J. Environ. Chem. Eng. 8 (2020) 103666. doi: 10.1016/j.jece.2020.103666
L. Zhang, Z. Wang, C. Hu, B. Shi, J. Colloid. Interface Sci. 553 (2019) 598–605. doi: 10.1016/j.jcis.2019.06.030
Y. Guo, S. Huang, Y. Guo, et al., Appl. Catal. B 312 (2022) 121388. doi: 10.1016/j.apcatb.2022.121388
R.R. Ding, W.Q. Li, C.S. He, et al., Appl. Catal. B 291 (2021) 120069. doi: 10.1016/j.apcatb.2021.120069
L. Wu, Z. Sun, Y. Zhen, et al., Environ. Sci. Technol. 55 (2021) 15400–15411. doi: 10.1021/acs.est.1c04600
S. Li, Y. Yang, H. Zheng, et al., Water Res. 225 (2022) 119176. doi: 10.1016/j.watres.2022.119176
Y.Y. Yang, H.P. Feng, X.G. Zhang, et al., Chem. Eng. J. 490 (2024) 151309. doi: 10.1016/j.cej.2024.151309
Z. He, J. Luo, G. Zhu, et al., J. Colloid. Interface Sci. 678 (2025) 186–200. doi: 10.1016/j.jcis.2024.08.155
F. Tompkins, Nature 186 (1960) 3–6. doi: 10.1038/186003a0
C. Yang, W. Zhong, K. Shen, et al., Adv. Energy Mater. 12 (2022) 2200077. doi: 10.1002/aenm.202200077
S. Guo, H. Wang, W. Yang, et al., Appl. Catal. B 262 (2020) 118250. doi: 10.1016/j.apcatb.2019.118250
N. Zhang, B. Zhang, A. He, et al., J. Environ. Chem. Eng. 11 (2023) 110717. doi: 10.1016/j.jece.2023.110717
X. Shi, X. Chen, L. Chen, et al., Appl. Surf. Sci. 601 (2022) 154237. doi: 10.1016/j.apsusc.2022.154237
X. Liang, L. Wang, T. Wen, et al., Sci. Total. Environ. 804 (2022) 150161. doi: 10.1016/j.scitotenv.2021.150161
Y.L.H.D.T. Pan, ACS. Appl. Mater. Interfaces 15 (2023) 9362–9372. doi: 10.1021/acsami.2c21120
R. Niu, C. Zhang, C. Li, P. Liu, J. Hazard. Mater. 476 (2024) 135010. doi: 10.1016/j.jhazmat.2024.135010
W. Yang, F. Qi, W. An, et al., ACS Catal. 14 (2024) 5936–5948. doi: 10.1021/acscatal.3c06234
S. Wang, H. Shi, S. Liang, et al., Small 20 (2024) 2311740. doi: 10.1002/smll.202311740
W.H.T.Z.Y. Sun, Environ. Sci. Technol. 53 (2019) 13332–13343. doi: 10.1021/acs.est.9b03689
J.A. Bolarin, Z. Zhang, H. Cao, et al., J. Magnesium Alloys 11 (2023) 2740–2749. doi: 10.1016/j.jma.2021.11.005
Q. Yang, Y. Wang, Q. Tian, et al., J. Mater. Chem. A 12 (2024) 7207–7214. doi: 10.1039/d3ta07981c
X. Zhang, J. Chen, C. Wang, et al., Nanotechnology 26 (2015) 175705. doi: 10.1088/0957-4484/26/17/175705
Z. Li, J.Chen L.Qian, et al., J. Alloys. Compd. 861 (2020) 157994.
F. Zhan, G. Wen, R. Li, et al., Phys. Chem. Chem. Phys. 26 (2024) 11182–11207. doi: 10.1039/d3cp06126d
A. Kumar, M. Kumar, V.N. Rao, et al., J. Mater. Chem. A 9 (2021) 17006–17018. doi: 10.1039/d1ta04180k
J. Cai, A. Cao, J. Huang, et al., Appl. Catal. B 267 (2020) 118378. doi: 10.1016/j.apcatb.2019.118378
T. Liu, S. Yang, J. Guan, et al., Small Methods 6 (2024) 101156.
N. Mouchani, A.H. Farahmand-Dashtarjandi, A. Yourdkhani, R. Poursalehi, N.B. Simhachalam, Surf. Interfaces 42 (2023) 103456. doi: 10.1016/j.surfin.2023.103456
L. Takacs, Prog. Mater. Sci. 18 (2009) 276–282.
F. Chang, C. Yang, X. Wang, et al., J. Clean. Prod. 380 (2022) 135167. doi: 10.1016/j.jclepro.2022.135167
L. Mao, Z. Song, J. Fan, et al., Sep. Purif. Technol. 334 (2024) 126035. doi: 10.1016/j.seppur.2023.126035
Y. Luo, Y. Wang, Y. Zhu, et al., Biochar. 4 (2022) s42773.
J. He, P. Wu, L. Lu, H. Li, H.M. Li, ACS. Appl. Mater. Interfaces 11 (2019) 36666–36675. doi: 10.1021/acsami.9b12063
T. Li, Y. Hu, J. Zhang, et al., Nano Energ. 126 (2024) 109690. doi: 10.1016/j.nanoen.2024.109690
J. Zheng, Y. Lyu, C. Xie, et al., Adv. Mater. 30 (2018) 1801773. doi: 10.1002/adma.201801773
F. Wang, J. Guo, L. Han, et al., Chem. Eng. J. 478 (2023) 147365. doi: 10.1016/j.cej.2023.147365
L. Yan, J. Qin, B. Liang, Q. Wang, M. Geng, Adv. Funct. Mater. 33 (2023) 2301886. doi: 10.1002/adfm.202301886
X. Xu, H. Yang, R. Tu, et al., Appl. Catal. B 342 (2024) 123358. doi: 10.1016/j.apcatb.2023.123358
F. Liu, J. Shen, D. Xu, et al., Chem. Eng. J. 334 (2018) 2283–2292. doi: 10.1016/j.cej.2017.11.114
H. Yuan, J. Li, W. Yang, et al., ACS Appl. Mater. Interfaces 10 (2018) 16410–16417. doi: 10.1021/acsami.8b01209
D. Liang, Y. Hu, C. Xiao, et al., Sci. Total. Environ. 834 (2022) 155278. doi: 10.1016/j.scitotenv.2022.155278
Y. Liu, Y. Chen, Y. Li, et al., Chem. Eng. J. 476 (2023) 146874. doi: 10.1016/j.cej.2023.146874
Z. Zhang, H. Ai, M.L. Fu, et al., J. Hazard. Mater. 436 (2022) 129000. doi: 10.1016/j.jhazmat.2022.129000
L. Sun, M. Feng, Y. Peng, et al., J. Mater. Chem. A 12 (2024) 8796–8804. doi: 10.1039/d4ta00655k
T. Jiang, W. Xie, S. Geng, et al., Chin. J. Catal. 43 (2022) 2434–2442. doi: 10.1016/S1872-2067(22)64137-8
S. Sun, C. Liu, S. Zhang, et al., Nano Energy 126 (2024) 109606. doi: 10.1016/j.nanoen.2024.109606
C. Dong, Z. Qu, X. Jiang, Y. Ren, J. Hazard. Mater. 391 (2020) 122181. doi: 10.1016/j.jhazmat.2020.122181
X. Zheng, Y. Li, W. You, et al., Chem. Eng. J. 430 (2022) 132917. doi: 10.1016/j.cej.2021.132917
L. Yan, G. Dong, X. Huang, Y. Zhang, Y. Bi, Appl. Catal. B 345 (2024) 123682. doi: 10.1016/j.apcatb.2023.123682
X. Lin, S. Li, H. He, et al., Appl. Catal. B: Environ. 223 (2018) 91–102. doi: 10.1016/j.apcatb.2017.06.071
L. Zhou, C. Wang, Y. Li, et al., Chin. Chem. Lett. 34 (2023) 107605. doi: 10.1016/j.cclet.2022.06.028
X. Xu, X. Ding, X. Yang, et al., J. Hazard. Mater. 364 (2019) 691–699. doi: 10.1016/j.jhazmat.2018.10.063
W.R. Chen, C. Liu, S.A. Boyd, B.J. Teppen, H. Li, Environ. Sci. Technol. 47 (2013) 1357–1364. doi: 10.1021/es303895w
F. Pan, S. Fu, Y. Wang, et al., Appl. Catal. B 356 (2024) 124185. doi: 10.1016/j.apcatb.2024.124185
Z.Wang L.Wu, J. Jia, et al., Appl. Catal. B 343 (2024) 123526. doi: 10.1016/j.apcatb.2023.123526
M. Xu, Y. Zhang, H. Yin, et al., Appl. Catal. B 322 (2023) 122085. doi: 10.1016/j.apcatb.2022.122085
T. Wu, X. Tang, Y. Lin, et al., Chem. Eng. J. 486 (2024) 150079. doi: 10.1016/j.cej.2024.150079
L. Liang, P. Cao, X. Qin, Appl. Catal. B 325 (2023) 122321. doi: 10.1016/j.apcatb.2022.122321
Q. Shao, Z. Cheng, L. Gao, et al., Appl. Catal. B 339 (2023) 123154. doi: 10.1016/j.apcatb.2023.123154
F. Liu, J. Shen, D. Xu, et al., Chem. Eng. J. 334 (2017) 2283–2292.
P. Yan, J. Shen, Y. Zhou, et al., Appl. Catal. B 277 (2020) 119055. doi: 10.1016/j.apcatb.2020.119055
Y. Cheng, J. Kang, P. Yan, et al., Appl. Catal B 341 (2024) 123325. doi: 10.1016/j.apcatb.2023.123325
Z.C. Yizhen Cheng, Pengwei Yan, ACS Catal. 14 (2024) 4040–4052. doi: 10.1021/acscatal.3c05554

Figure 2 (a) XRD patterns of high entropy alloy catalysts. Reprinted with permission [81]. Copyright 2021, Elsevier. (b) XRD patterns of doping of Pr6O11 by different metals. Reprinted with permission [59]. Copyright 2024, Elsevier. (c) XRD patterns of mesoporous SInO2 and NaInO2 compounds with different molar ratios. Reprinted with permission [82]. Copyright 2018, Elsevier. (d) XPS spectra of NiCo2O4/HCS-320-V. Reprinted with permission [83]. Copyright 2018, Elsevier. (e) XPS spectra of Au2Pd1/TiO2-xCA@HNTs. Reprinted with permission [85]. Copyright 2023, Elsevier. (f) XPS of Co 2p level for Mg-Co-Ce composites. Reprinted with permission [86]. Copyright 2022, Elsevier.

Figure 3 (a) EPR images of Fe-Co LDH at different calcination temperatures. Reprinted with permission [48]. Copyright 2021, Elsevier. (b) EPR spectra of MFO and CMFO-0.5. Reprinted with permission [49]. Copyright 2022, Elsevier. (c) HRTEM images of Fe/TiO2-x-S. Reprinted with permission [91]. Copyright 2022, Elsevier. (d) Raman spectra of MnOx(z)-CeO2. Reprinted with permission [93]. Copyright 2018, Elsevier. (e) Raman spectra of α-MnO2 with different reduction times. Reprinted with permission [94]. Copyright 2023, Elsevier. (f) Raman spectrum of BMO-0 and BMO-3. Reprinted with permission [95]. Copyright 2019, Elsevier.

Figure 4 (a) The Proposed mechanisms of MB degradation in CoFe-LDO-BC + O3 system. Reprinted with permission [55]. Copyright 2023, Elsevier. (b) PDOS of Fe 3d. Reprinted with permission [97]. Copyright 2024, Elsevier. (c) The charge density of α-MnO2-x and α-MnO2-x-2. Reprinted with permission [98]. Copyright 2024, Elsevier.

Figure 5 (a) Catalytic ozonation mechanism over Co-ZFO@Mn-CN. Reprinted with permission [99]. Copyright 2023, Elsevier. (b) The mechanism diagram of ROS production and ATZ degradation in CCA/O3 and CCAA/O3. Reprinted with permission [100]. Copyright 2024, Elsevier. (c) Possible interface catalytic mechanisms. Reprinted with permission [105]. Copyright 2024, Elsevier. (d) Relative energies of different coordinates and interfacial reaction pathways on zinc oxides. Reprinted with permission [106]. Copyright 2024, Elsevier.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:
