
Figure 1.
Photoproduction of Fe(Ⅱ) in the systems of Fe(Ⅲ) alone (a), Fe(Ⅲ) complexes with AL (b), BH (c) and PA (d) at pH 3.0. The total Fe(Ⅲ) (Fetot) was 100 µmol/L and PROC concentration was 12.5 mg/L.
Photoreduction of Fe(Ⅲ) mediated by structurally different plant-related organic compounds: An EPR study
Qingchao Li , Fengmin Ma , Pu Wang , Yu Fu , Jinhui Cao , Chunxiao Xu , Jialin Chen , Lingli Wang , Zhaohui Wang

Natural organic matter (NOM) is prevalent in soil and water, playing a crucial role in sustaining soil fertility and active organic carbon levels [1,2]. The NOM typically comprises diverse organic constituents, including carboxyl, alcoholic/phenolic hydroxyl, ketone, aldehyde, quinone, and additional chemical groups. These particular functional groups govern the reactivity of the NOM, particularly in photochemical reactions [3]. Light, particularly within the 280–400 nm spectrum, can facilitate the degradation of larger NOM molecules, yielding various low-molecular-weight organic or inorganic compounds [4]. Furthermore, NOM can produce various reactive oxygen species (ROS) upon light exposure, including hydroxyl radical (•OH), superoxide anion radical (O2•−), singlet oxygen (1O2) and carbon dioxide radical (CO2•-) [3,5]. Iron (Fe), the predominant transition metal in the environment, regulates the global biogeochemical cycles of various elements, including carbon, oxygen, nitrogen, and sulfur [6]. Almost entirely dissolved iron is organically complexed in organic matter-rich environment, thereby significantly influencing its speciation, solubility, and mobility [7]. The direct photoreaction through ligand-metal charge transfer (LMCT) is the primary mechanism for generating bioavailable Fe(Ⅱ) in surface water [1]. In addition to the photoinduced inner-sphere LMCT process, another photochemical coupling pathway between Fe(Ⅲ) and NOM is the outer-sphere charge transfer from photo-produced ROS to Fe(Ⅲ)-organic complexes [4,8]. The photochemical reactions of Fe(Ⅲ)-carboxylate complexes have been thoroughly investigated because of their high photosensitivity and natural abundance [9,10], such as oxalic acid, malonic acid, malic acid, citric acid and tartaric acid [11]. For instance, photolysis of Fe(Ⅲ)-oxalate complex may lead to a superfast conversion of Fe(Ⅲ) to Fe(Ⅱ) and production of C2O4• and CO2•- [12].
Plant-related organic compound (PROC) serves as the primary source of NOM [13]. Its composition is intricate, encompassing both readily leachable organic constituents (e.g., water-soluble components, unprotected cellulose, oligosaccharides, and hemicellulose) and other refractory components [14,15]. The relative abundance of these organic constituents can vary depending on the plant species. For instance, contents of carbon and nitrogen in PROC derived from broadleaved tree species are generally greater than those from coniferous species. Alkaline lignin (AL) and its derivatives, as significant components of PROC, accumulate in the soil due to their strong retention and stability [16]. Furthermore, betaine hydrochloride (BH) is crucial in amino acid synthesis as a methyl transfer agent and can form crystalline complexes with various acids [17]. Phytic acid (PA) represents a primary form of phosphate storage in food products, constituting approximately 60%−80% of the total phosphorus in seeds [18]. These PROCs are eventually introduced into the soil and aquatic systems through the decomposition of litter, where they interact with metal elements in the environment to form metal-organic complexes. While most studies examined the variations in photochemical reactions of Fe(Ⅲ) complexes with structurally similar carboxylates [10,19], the photochemical behaviors of iron-PROC complexes featuring structurally diverse organic functional groups have yet to be thoroughly investigated.
This study employs three representative types of PROCs, namely AL, BH, and PA (Fig. S1 in Supporting information), to examine the differences in the photochemical behavior of Fe(Ⅲ)-PROC complexes. To achieve this, the concentrations of Fe(Ⅱ), levels of organic carbon, and the generation of reactive oxygen species (ROS) were assessed using various analytical techniques, including UV–vis spectroscopy, total organic carbon analyzer (TOC) and electron paramagnetic resonance (EPR). A comprehensive list of materials used in this research is provided in Text S1 (Supporting information), while the experimental protocols and analytical methodologies are detailed in Texts S2 and S3 (Supporting information). The primary objectives of this investigation are: (ⅰ) To explore the PROC-mediated reduction of Fe(Ⅲ) under both illuminated and non-illuminated conditions, and (ⅱ) to evaluate the photoreactivity of different PROC-associated Fe(Ⅲ) species and their contributions to free radical production. The findings may advance our understanding of iron redox cycling and radical production mediated by the typical low-molecular-weight PROC.
Without any organic ligands, UV irradiation facilitated the generation of Fe(Ⅱ) and the dynamic equilibrium between the photoreduction of Fe(Ⅲ) and the photooxidation of Fe(Ⅱ) by dioxygen culminated in a photo steady state, while Fe(Ⅲ)-OH complexes were the dominant light-absorbing species in the Fe2(SO4)3 solution [8,10]. At pH 3.0, FeOH2+ reaches its maximum concentration (Fig. S2 in Supporting information), making it the most significant iron species in the process of photoreduction. With a further increase in pH, the species become predominantly Fe(OH)2+. Then, a quantitative analysis was performed to compare the concentrations of Fe(Ⅱ) produced at a pH of 3.0 across various complexes of Fe(Ⅲ) bound to PROCs (Fig. 1). In the dark, the yield of Fe(Ⅱ) from the Fe(Ⅲ)-AL complex rapidly increased to 66 µmol/L within the first 10 min, but this was not the case for Fe(Ⅲ)-BH and Fe(Ⅲ)-PA complexes. Under UV illumination, the maximum yield of Fe(Ⅱ) from the Fe(Ⅲ)-AL and Fe(Ⅲ)-BH complexes surpassed that observed in the dark. Notably, the Fe(Ⅱ) yield from the Fe(Ⅲ)-BH complex was greater than that of Fe(Ⅲ) sulfate under identical conditions. Interestingly, no Fe(Ⅱ) formation was detected for Fe(Ⅲ)-PA complex either in the dark or under UV irradiation. The photoreduction of Fe(Ⅲ) was significantly impeded as the pH increased from 4.0 to 6.0 (Fig. S3 in Supporting information).
The observed variability in the Fe(Ⅱ) yield with different types of PROCs can be attributed to the distinct functional groups present within these PROCs. The macromolecular structure of AL is characterized by a multitude of active functional groups and bonds, such as Ar-CHO, Ar-COOH, Ar-OH, Ar-CO-R, and Ar-CHCH-Ar. The reduction potential of these groups is significantly more negative than that of iron species, (E0Fe(Ⅲ)/Fe(Ⅱ) = 0.771 V vs. NHE) [20,21]. Consequently, these functional groups facilitate the reduction of Fe(Ⅲ) to Fe(Ⅱ) as described in Eq. 1.
|
|
(1) |
In addition to direct reduction by organic ligands, the photoreduction of Fe(Ⅲ) primarily occurs through two mechanisms: Photoinduced LMCT process and indirect pathways involving photochemically generated free radicals [7]. In the case of the Fe(Ⅲ)-AL complex, the photo formation of Fe(Ⅱ) predominantly resulted from LMCT pathways. The yield of Fe(Ⅱ) in the Fe(Ⅲ)-BH solution was observed to be 1.5 times higher than that in the Fe(Ⅲ) sulfate system. The BH, with a positively charged trimethylammonium group and a negatively charged carboxyl group, may be weakly complexed with Fe(Ⅲ), akin to acetic acid [8]. The observed shift in the redox equilibrium towards Fe(Ⅱ) could be ascribed to the decreased Fe(Ⅱ) re-oxidation by •OH, with BH as a radical scavenger [22]. Phytic acid (PA) is capable of forming stable complexes with various metal ions in its deprotonated form. Within the pH range of our experimental conditions, 6–8 protons may detach from PA myo-inositol [23]. The deprotonated PA subsequently coordinated with Fe(Ⅲ) in an octahedral configuration, involving the phosphoinositol positions P2, P1, P3, and P5, as well as adjacent phosphate groups P4 and P6. The stabilization constants (log K) for the complexes [FePA]4-, [FePA]5- and [FePA]6- are reported to be 8.99, 12.70 and 18.20, respectively [24]. The strong chelation of deprotonated PA with Fe(Ⅲ) may effectively inhibit the photoreduction of Fe(Ⅲ), similar to the phenomenon observed for Fe(Ⅲ) phosphate complexes [22].
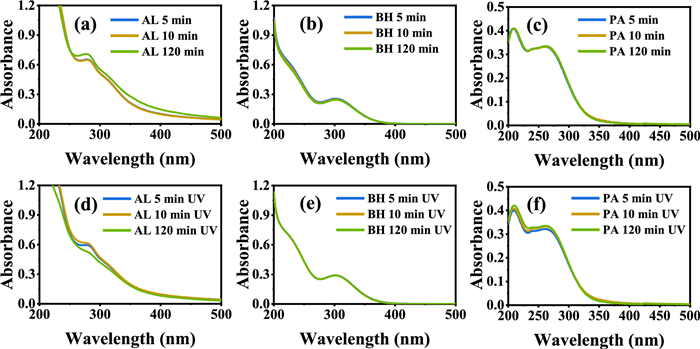
The changes in absorbance of Fe(Ⅲ) and PROCs were analyzed using a UV-vis spectrophotometer, as illustrated in Fig. S4 (Supporting information). Fig. S4a demonstrates that the hydrolysis of Fe(Ⅲ) occurred with increasing pH, accompanied by a blue shift in the UV-visible spectrum at 274 nm. The characteristic peak associated with AL was observed at 281 nm and remained stable despite the increase in pH. Conversely, no significant changes in absorbance were noted for the other two PROCs, as depicted in Figs. S4b–d. The Fe(Ⅲ)-PROC complexes exhibited distinct peaks at 281 nm (AL), 305 nm (BH), and 216/265 nm (PA) in the dark (Figs. 2a–c). The intensities of all characteristic peaks remained constant over time. Notably, the characteristic peak at 281 nm for the Fe(Ⅲ)-AL complex diminished after 120 min of UV light exposure, implying organic ligands decomposition under UV irradiation (Fig. 2d). The absorption peak at 281 nm corresponds to the n→π* transition of aromatic rings or non-conjugated phenolic groups [25]. Its disappearance can be attributed to UV-induced mineralization [26,27]. In the case of the Fe(Ⅲ)-BH solution, only the characteristic peak of Fe(Ⅲ) aquo complex was observed, indicating no formation of chromophoric Fe(Ⅲ)-BH complex (Fig. 2e). For the Fe(Ⅲ)-PA complex, two new characteristic peaks were observed at 214 nm and 265 nm [28], which suggest that the newly formed complex is stable and unaffected by UV light (Fig. 2f). In conclusion, the results indicate that among the Fe(Ⅲ)-PROC complexes studied, only the AL complex exhibited changes in its UV-vis spectrum upon exposure to UV light. Fig. S5 (Supporting information) illustrates the variation in TOC concentration of the three Fe(Ⅲ)-PROC complexes when exposed to UV light. Notably, a decrease in TOC concentration was observed solely in the Fe(Ⅲ)-AL complex, suggesting that certain carbon-containing organic moieties within the AL complex were mineralized. In contrast, the TOC values of the other two complexes were unchanged, indicating an absence of organic matter mineralization in the solution.
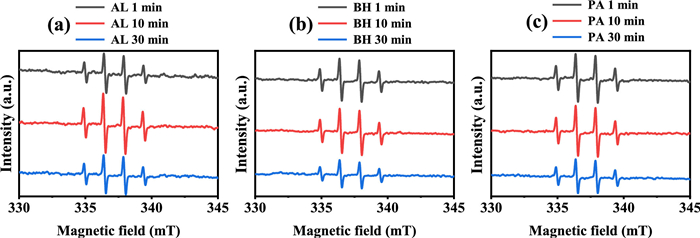
Spin-trapping EPR technique was utilized to identify the possible short-lived radicals present in the different Fe(Ⅲ)-PROCs systems (Fig. 3). All recorded spectra exhibited a characteristic 1:2:2:1 quartet signal, which is assigned to DMPO-OH (aH = aN = 1.49 mT) [10]. In the Fe(Ⅲ)-AL system, the intensity of the DMPO-OH signal peaked at 10 min (Fig. 3a), whereas the signals associated with the Fe(Ⅲ)-BH and Fe(Ⅲ)-PA complexes exhibited a gradual decline over time (Figs. 3b and c). This observation indicates that the mechanisms underlying the formation of the DMPO-OH signal may differ among the various Fe(Ⅲ)-PROCs complexes. It should be noted that the detection of the DMPO-OH signal via EPR does not necessarily confirm the formation of authentic •OH. In an aqueous solution of DMPO containing Fe(Ⅲ), the DMPO-OH signal can arise from the oxidation of DMPO by Fe(Ⅲ), followed by a nucleophilic attack from water [29]. As illustrated in Fig. S6 (Supporting information), the generation of DMPO-OH was observed in solutions containing only Fe(Ⅲ), as well as in those with Fe(Ⅲ)-AL and Fe(Ⅲ)-BH; however, no such signal was detected in the Fe(Ⅲ)-PA system. This absence of signal may be attributed to the strong binding of PA to Fe(Ⅲ), which likely obstructs the formation of a chelate between DMPO and Fe(Ⅲ), a critical step in the oxidation of DMPO [29]. Interestingly, in the UV/Fe(Ⅲ)-PA system, a discernible DMPO-OH adduct was observed (Fig. 3c), suggesting that the Fe(Ⅲ)-PA system underwent a photochemical reaction that results in the generation of relatively loosely bound Fe(Ⅲ), thereby facilitating the formation of the Fe(Ⅲ)-DMPO complex. Given that no Fe(Ⅱ) was produced in the photolyzed Fe(Ⅲ)-PA system (Fig. 1), it is plausible that the photoreaction involving the Fe(Ⅲ)-PA complex proceeds via a photodissociation mechanism, wherein only the Fe(Ⅲ)-O bond was cleaved upon photoexcitation.
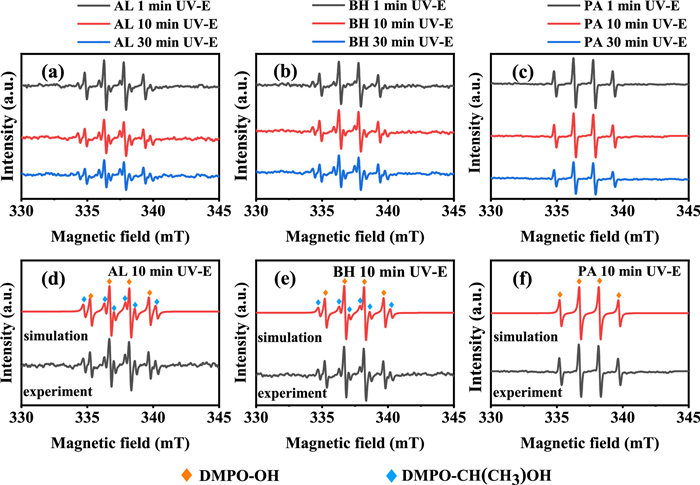
To evaluate whether the observed DMPO-OH adduct is attributable to the presence of trapped •OH radicals, amounts of ethanol was added into those Fe(Ⅲ)-PROC systems. Fig. 4 illustrates that a sextet signal with a 1:1:1:1:1:1 ratio was recorded for the Fe(Ⅲ)-AL and Fe(Ⅲ)-BH systems, which is identified as DMPO-CH(CH3)OH adduct (aN = 1.57 mT, aH = 2.27 mT) [30]. This finding indicates that the photolysis of the Fe(Ⅲ)-AL and Fe(Ⅲ)-BH systems indeed produced •OH, which abstracts H atom from ethanol to yield CH(CH3)OH carbon-centred radical. In contrast, no such sextet signal was detected in the photolyzed Fe(Ⅲ)-PA system, providing direct evidence for the absence of photoactive Fe(Ⅲ)-OH complexes capable of producing •OH. In addition to Fe(Ⅲ)-OH complexes, some NOM itself may generate •OH under solar irradiation, with a reduction of molecular weight and aromaticity, as well as an increase of low molecular weight components [26,31]. However, the possibility of formation of •OH by photolyzed AL, BH or PA can be ruled out (Fig. S7 in Supporting information). Therefore, it can be concluded that the detected DMPO-OH adduct is likely a result of the direct oxidation of DMPO by Fe(Ⅲ) species or the trapped •OH generated from the photoreaction of Fe(Ⅲ)-OH complexes.
In summary, this study examined the influence of three structurally distinct PROCs on the photoreduction of Fe(Ⅲ) under acidic conditions. The reductive agent AL demonstrated the ability to directly convert Fe(Ⅲ) to Fe(Ⅱ). In contrast, BH, while unable to form strong complexes with iron, facilitated the generation of Fe(Ⅱ) by scavenging •OH and mitigating the reoxidation of Fe(Ⅱ). PA exhibited a strong binding affinity for Fe(Ⅲ), effectively inhibiting its photoreduction. The EPR analysis revealed that the DMPO-OH signal in the photolyzed Fe(Ⅲ)-PROC solutions originated from different pathways (Fig. S8 in Supporting information). Uncomplexed Fe(Ⅲ) was found to oxidize DMPO directly, resulting in the formation of a false DMPO-OH adduct. The introduction of ethanol into the photolyzed Fe(Ⅲ)-AL and Fe(Ⅲ)-BH systems resulted in the production of the DMPO-CH(CH3)OH adduct, confirming the presence of genuine •OH in these systems. The photolysis of the Fe(Ⅲ)-PA complex may yield loosely bound Fe(Ⅲ), which can oxidize DMPO, followed by a nucleophilic attack from water. This research provides compelling evidence of the varied roles that PROCs play in regulating iron redox cycling within geochemical processes.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Qingchao Li: Writing – original draft, Methodology, Investigation, Formal analysis, Data curation. Fengmin Ma: Investigation, Formal analysis, Data curation. Pu Wang: Validation, Software, Methodology. Yu Fu: Visualization, Validation. Jinhui Cao: Validation, Software. Chunxiao Xu: Visualization, Methodology. Jialin Chen: Validation, Investigation. Lingli Wang: Writing – review & editing, Supervision, Formal analysis. Zhaohui Wang: Writing – review & editing, Supervision, Resources, Funding acquisition, Conceptualization.
This work was supported by the Natural Science Foundation of Shanghai (No. 24ZR1419600).
Supplementary material associated with this article can be found, in the online version, at doi:
K. Barbeau, Photochem. Photobiol. 82 (2006) 1505–1516. doi: 10.1111/j.1751-1097.2006.tb09806.x
Q. Li, L. Wang, Y. Fu, et al., J. Soils Sediments 23 (2023) 1485–1500. doi: 10.1007/s11368-022-03381-y
Y. Xu, Y. Zhang, L. Qiu, et al., Eco-Environment Health 3 (2024) 529–542. doi: 10.1016/j.eehl.2024.06.002
U. Lueder, B.B. Jørgensen, A. Kappler, C. Schmidt, Environ. Sci. -Process. Impacts 22 (2020) 12–24. doi: 10.1039/c9em00415g
Y. Dong, W. Peng, Y. Liu, Z. Wang, J. Hazard. Mater. 401 (2021) 123884. doi: 10.1016/j.jhazmat.2020.123884
J. Huang, A. Jones, T.D. Waite, et al., Chem. Rev. 121 (2021) 8161–8233. doi: 10.1021/acs.chemrev.0c01286
A. Kappler, C. Bryce, M. Mansor, et al., Nat. Rev. Microbiol. 19 (2021) 360–374. doi: 10.1038/s41579-020-00502-7
Z. Wang, C. Chen, W. Ma, J. Zhao, J. Phys. Chem. Lett. 3 (2012) 2044–2051. doi: 10.1021/jz3005333
F. Wu, N. Deng, Chemosphere 41 (2000) 1137–1147. doi: 10.1016/S0045-6535(00)00024-2
Q. Li, Y. Fu, L. Wang, et al., Sci. Total. Environ. 927 (2024) 172333. doi: 10.1016/j.scitotenv.2024.172333
F. Li, J. Chen, C. Liu, J. Dong, T. Liu, Biol. Fertil. Soils 42 (2006) 409–417. doi: 10.1007/s00374-006-0084-7
C. Zhang, C. Yang, X. Liu, F. Cao, Y. Zhang, Sci. Total. Environ. 719 (2020) 137416. doi: 10.1016/j.scitotenv.2020.137416
Y. Zhong, W. Yan, R. Wang, Z. Shangguan, Biol. Fertil. Soils 53 (2017) 939–949. doi: 10.1007/s00374-017-1242-9
J. Voříšková, P. Baldrian, ISME J 7 (2013) 477–486. doi: 10.1038/ismej.2012.116
D.P. Bebber, S.C. Watkinson, L. Boddy, P.R. Darrah, Oecologia 167 (2011) 1177–1184. doi: 10.1007/s00442-011-2057-2
L. Liu, Z. Yang, W. Yang, et al., J. Environ. Sci. 139 (2024) 23–33. doi: 10.1016/j.jes.2023.05.016
M. Szafran, A. Katrusiak, Z. Dega-Szafran, I. Kowalczyk, J. Mol. Struct. 1031 (2013) 49–55. doi: 10.1016/j.molstruc.2012.07.030
K. Dost, G. Karaca, Food Anal. Method. 9 (2016) 1391–1397. doi: 10.1007/s12161-015-0319-z
D. Xiao, Y. Guo, X. Lou, et al., Chemosphere 103 (2014) 354–358. doi: 10.1016/j.chemosphere.2013.11.069
E. Evstigneyev, S. Shevchenko, H. Mayorova, A. Platonov, J. Wood Chem. Technol. 24 (2005) 263–278. doi: 10.1081/WCT-200038182
S. Yi, J. Cui, S. Li, et al., Appl. Surf. Sci. 319 (2014) 230–236. doi: 10.1016/j.apsusc.2014.06.151
W. Song, W. Ma, J. Ma, et al., Environ. Sci. Technol. 39 (2005) 3121–3127. doi: 10.1021/es0483701
G. Marolt, E. Gričar, B. Pihlar, M. Kolar, Front. Chem. 8 (2020) 582746. doi: 10.3389/fchem.2020.582746
J. Torres, S. Domínguez, M.F. Cerdá, et al., J. Inorg. Biochem. 99 (2005) 828–840. doi: 10.1016/j.jinorgbio.2004.12.011
O.Y. Abdelaziz, C.P. Hulteberg, Waste Biomass Valori 8 (2017) 859–869. doi: 10.1007/s12649-016-9643-9
A. Paul, C. Dziallas, E. Zwirnmann, E.T. Gjessing, H. Grossart, Aquat. Sci. 74 (2012) 443–454. doi: 10.1007/s00027-011-0239-y
Z. Gao, Y. Lin, B. Xu, et al., Sci. Total. Environ. 702 (2020) 134942. doi: 10.1016/j.scitotenv.2019.134942
Y. Zhong, D. Zhou, B. Zhang, M. Nishi, A. Murakami, ACS Appl. Polym. Mater. 4 (2022) 546–555. doi: 10.1021/acsapm.1c01446
K. Makino, T. Hagiwara, A. Hagi, et al., Biochem. Biophys. Res. Commun. 172 (1990) 1073–1080. doi: 10.1016/0006-291X(90)91556-8
L. Wang, J. Cao, P. Wang, et al., Environ. Health 3 (2025) 143–153. doi: 10.1021/envhealth.4c00142
X. Qiu, S. Ma, J. Zhang, et al., Environ. Sci. Technol. 56 (2022) 10149–10160. doi: 10.1021/acs.est.2c03309

Figure 1 Photoproduction of Fe(Ⅱ) in the systems of Fe(Ⅲ) alone (a), Fe(Ⅲ) complexes with AL (b), BH (c) and PA (d) at pH 3.0. The total Fe(Ⅲ) (Fetot) was 100 µmol/L and PROC concentration was 12.5 mg/L.

Figure 2 The UV-vis spectrum of solutions containing Fe(Ⅲ) with AL, BH and PA at pH 2.2 in the dark (a-c) and under UV irradiation (d-f), respectively. The Fetot concentration was 100 µmol/L and the PROC concentration was 25 mg/L.

Figure 3 The EPR spectrum of Fe(Ⅲ) complexes with (a) AL, (b) BH and (c) PA at pH 2.2 under UV irradiation. The Fetot concentration was 100 µmol/L and the PROC concentration was 25 mg/L.

Figure 4 The EPR spectrum of Fe(Ⅲ)-PROC complexes using ethanol (E) as a scavenger at pH 2.2 under UV irradiation. (a–c) are experimental data, and (d–f) are simulated data. The Fetot concentration was 100 µmol/L and the PROC concentration was 25 mg/L. Ethanol concentration was 0.5 mol/L.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载: