
Scheme 1.
Representative bioactive molecules, reported the synthesis of tricyclo[3.2.1.02,7]octane derivatives and our divergent synthesis strategy.
Diverse synthesis of bridged bicyclo[3.2.1]octa-2,6-diene and tricyclo[3.2.1.02,7]oct-3-ene frameworks via stepwise cascade reactions
Ze-Hong Zheng , Mu-Qiu Chen , Jin Zhou , Jie Wang , Yan-Rong Wei , Cheng Peng , Gu Zhan , Qian-Qian Yang , Bo Han

Since the 1990s, fragment-based drug discovery (FBDD) strategies have received widespread attention [1–6]. This approach involves identifying and optimizing small molecular fragments to develop bioactive lead compounds. With continuous advancements in medicinal chemistry, the emphasis of FBDD development has evolved from two-dimensional molecular structures to the intricacies of three-dimensional molecular frameworks [7–14]. Bridged-ring compounds, central to the structures of numerous natural products and pharmaceuticals, serve as pivotal intermediates in the synthesis of many crucial natural products and exhibit distinctive biological activities, exemplifying the essence of 3D structures. The quest for green and efficient synthesis, along with the investigation of their activities, has piqued the interest of organic chemists and pharmacologists [15–27]. Nonetheless, the inherent rigidity of the bridged-ring frameworks and the associated high ring strain present significant challenges in the construction of these frameworks and the attainment of stereoselective control. Consequently, the development of high-efficiency and eco-friendly synthetic approaches to construct intricate natural bridged-ring skeletons has emerged as a critical area of focus within the realms of organic and medicinal chemistry research.
The bridged-ring structures bicyclo[3.2.1]octane and tricyclo[3.2.1.02,7]octane serve as crucial cores and active centers in these frameworks. Natural products mitrephorone A and mitrephorone B [28–30], which belong to the ent–trachylobane-type diterpenoids, contain the tricyclo[3.2.1.02,7]octane nucleus. Salvileucalin B [14,31–33], extracted from Salvia leucantha, has shown cytotoxicity against A549 (human lung adenocarcinoma cells) and HT-29 (human colon adenocarcinoma cells) (Scheme 1a). Despite the exceptional bioactivity displayed by many natural molecules, constructing the bridged-ring skeletons of these complex compounds remains a core challenge in synthesis. Due to the complexity of bridged-ring frameworks, they are typically constructed in the later stages of synthesis [13,17,21,25,26,34–37], which can lead to prolonged synthetic routes and overly specific target molecules. Compared to the construction of the bicyclo[3.2.1]octane bridged-ring frameworks [38–46], strategies for building the more complex tricyclo[3.2.1.02,7]octane core are more limited due to the presence of the highest ring strain cyclopropane fragment in the bridged-ring skeleton [14,47–49]. Consequently, constructing the strained tricyclo[3.2.1.02,7]octane derivatives, which form a polycyclic system with fused three-membered, five-membered, and six-membered rings, has become a particularly challenging research endeavor.
Currently, one of the most effective methods for the total synthesis of the fascinating and complex tricyclo[3.2.1.02,7]octane structure involves base-induced or metal-catalyzed skeletal rearrangement. A frequently utilized strategy employs a tetraene fragment for 6π electrocyclization, followed by a [4 + 2] Diels-Alder cyclization to generate these derivatives (Scheme 1b, top) [31,32]. However, the reaction is highly sensitive to substrate specificity, particularly to the electronic effects and steric hindrance of substituents. An alternative approach uses carbene precursors to construct tricyclic tricyclo[3.2.1.02,7]octane derivatives through cyclopropanation [14,33] or rearrangement of the bicyclo[2.2.2]octane framework (Scheme 1b, below) [65]. Despite this, the synthetic preparation of these raw materials is complex, and the use of diazo carbene precursors entails certain safety hazards. Additionally, employing the di-π-methane rearrangement reaction of 1,4-diene fragments is a common strategy for synthesizing cyclopropanes, and it is also applied in constructing the tricyclo[3.2.1.02,7]octane structure. However, this strategy often faces challenges related to reaction multiplicity and stereoselectivity, frequently resulting in the formation of multiple rearrangement products. Furthermore, the reaction is significantly constrained by the substrate framework. Overall, significant breakthroughs in constructing tricyclo[3.2.1.02,7]octane bridged-ring derivatives remain elusive. Thus, efficiently and stereoselectively constructing these highly strained polycyclic frameworks remains one of the most critical challenges in synthesizing these complex molecules.
Allenoates, as crucial synthetic building blocks for molecular fragments [50–56], exhibit excellent reactivity and perform remarkably well in constructing complex bridged-ring frameworks [57–59]. Building on our previous work in synthesizing diverse pharmacophore architectures [60–62], we envisioned employing tandem reactions of allenoates to construct complex bridged-ring molecules. Herein, we report a multi-component tandem reaction involving allenoates and innovatively utilize photocatalysis to achieve highly stereoselective rearrangement reactions, efficiently constructing tricyclo[3.2.1.02,7]octane bridged-ring derivatives (Scheme 1c). Firstly, we employed allenoates to undergo γ-elimination [63] with α-cyano cinnamaldehyde via Lu's [3 + 2] cyclization [64], followed by a series of reactions including [5 + 2] cyclization involving the aldehyde group, acyl transfer, and formic acid elimination with another molecule of α-cyano cinnamaldehyde, leading to the formation of bicyclo[3.2.1]octa-2,6-diene bridged-ring derivatives. The success of this reaction relies on the orderly progression of multiple tandem reactions.
Notably, under visible light irradiation in the presence of a photosensitizer, the bicyclo[3.2.1]octa-2,6-diene bridged-ring derivatives undergo efficient skeletal rearrangement to form the more strained tricyclo[3.2.1.02,7]oct–3-ene framework. This strategy offers an efficient and diverse construction of bridged-ring scaffolds under mild conditions using simple raw materials, not only highlighting significant advances in one-pot, multi-step, highly stereoselective construction of multiple C—C bonds through organocatalysis but also developing a photocatalytic strategy for efficient transformation between complex bridged rings.
We initiated our study by investigating the model reaction between allenoate 1a and (E)-2-formyl-3-phenylacrylonitrile 2a using PPh3 as the catalyst in ethyl acetate (EtOAc) at 50 ℃, and selected results are summarized in Table 1. Fortunately, the reaction can be monitored to produce the bridged bicyclo[3.2.1]octa-2,6-diene product 3a. Additionally, the [3 + 2] cyclization product 3a', formed from the γ-elimination reaction of allenoates 1a and 2a, can be detected in situ by NMR, achieving a yield of 33% (entry 1). However, due to its almost similar polarity to product 3a, it cannot be cleanly separated. These intermediates provide evidence supporting the proposed reaction mechanism. To address this, we screened various co-catalysts, including different Brønsted acids, for synergistic effects. Unfortunately, apart from C3, which managed to yield 3a at medium to low levels and control the formation of 3a', other conditions resulted in poor conversion (entries 2–5). We then explored both organic and inorganic bases as co-catalysts (entries 6–10). Triethylamine (Et3N) and sodium p-toluenesulfonate C5 were able to facilitate the complete conversion to 3a, achieving yields over 60%. Inorganic bases such as K2CO3, Cs2CO3, and organic base DIPEA were slightly less effective. While using triethylamine as a co-catalyst, we tested various solvents, including dichloromethane (DCM), toluene, tetrahydrofuran (THF), N, N-dimethylformamide (DMF), acetonitrile (MeCN), and 1,2-dichloroethane (DCE)—but none significantly improved the yields. The addition of Na2SO4 and 3 Å molecular sieves as dehydrating agents also showed no significant benefit. Interestingly, when we switched to sodium p-toluenesulfonate C5, we observed a significant increase in yield compared to triethylamine, achieving an optimal yield of 78% for the bridged product 3a, while also controlling the [3 + 2] cyclization product 3a'.
 DownLoad:
CSV
DownLoad:
CSV
 |
||||
| Entry | Co-catalyst | Solvent | Yield (3a)b | Yield (3a')c |
| 1 | – | EtOAc | 40 | 33 |
| 2 | C1 | EtOAc | 55 | 23 |
| 3 | C2 | EtOAc | – | – |
| 4 | C3 | EtOAc | 22 | 10 |
| 5 | C4 | EtOAc | – | – |
| 6 | C5 | EtOAc | 60 | 22 |
| 7 | K2CO3 | EtOAc | 57 | 20 |
| 8 | Et3N | EtOAc | 42 | 26 |
| 9 | DIPEA | EtOAc | 44 | 23 |
| 10 | Cs2CO3 | EtOAc | 44 | 27 |
| 11 | Et3N | DCM | 44 | 31 |
| 12 | Et3N | Toluene | 24 | 17 |
| 13 | Et3N | THF | 58 | 21 |
| 14 | Et3N | DMF | mess | – |
| 15 | Et3N | MeCN | 31 | 12 |
| 16 | Et3N | DCE | 30 | 12 |
| 17e | Et3N | EtOAc | 64 | 7 |
| 18d | Et3N | EtOAc | 62 | 10 |
| 19e | C5 | EtOAc | 78 | 6 |
| a Reactions conditions: 1a (0.2 mmol), 2a (0.2 mmol), PPh 3 (20 mol%) and co-catalyst (20 mol%) in 2.0 mL of solvent at 50 ℃ for 12 h; > 19:1 dr determined by 1H NMR. b Yield of isolated product 3a. c Determined by in situ 1H NMR. d With Na 2SO 4 (50 mg) as additive. e With 3 Å molecular sieve (50 mg) as additives. |
||||
Having established the optimal reaction conditions, we then turned our attention toward the substrate scope of this three-component tandem reaction. As summarized in Scheme 2, electron-withdrawing substituted α-cyano cinnamaldehydes 2 exhibit good reactivity, allowing the construction of bridged bicyclo[3.2.1]octa-2,6-diene products 3b–3f with moderate yields. The relative configuration of 3 was identified by X-ray diffraction analysis of a single crystal with product 3a. Electron-donating substituted enals regardless of substitution at ortho-, meta-, or para-positions, display poor reactivity; the reaction is quite sensitive to the electronic effects of the enals, resulting in low yields for products 3g–3k. Nonetheless, both electron-withdrawing and electron-donating naphthalene-substituted enals yield bridged products 3l–3n with moderate yields. Additionally, we examined the adaptability of allenoates, finding that γ-phenyl substitution in allenoate is crucial for the reaction. When using γ-alkyl-substituted or unsubstituted allenoates, no target product was formed in the reaction. This might be because γ-aryl-substituted allenoates can generate stable γ-elimination [3 + 2] cyclization products, which then undergo a three-component cascade reaction. Allenoates with diverse substituents on the γ-aryl ring, such as 4-fluoro, 4-trifluoromethyl and 4-phenyl, demonstrate good reactivity adaptability.
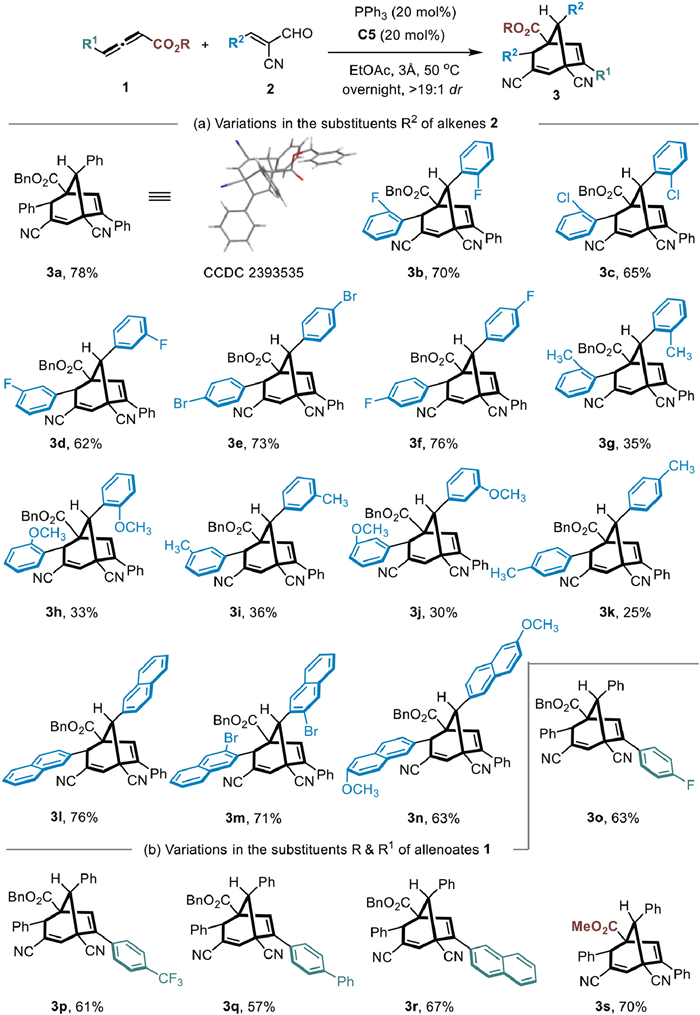
This versatility facilitates the formation of bridged products 3o–3q, achieving moderate yields of 57% to 63%. Substituting the benzene ring with a naphthalene ring and replacing the benzyl ester with a methyl ester in the allenoate both demonstrate strong reactivity. These modifications successfully construct the bicyclo[3.2.1]octa-2,6-diene frameworks 3r and 3s, achieving yields of 67% and 70%, respectively.
Meanwhile, we found compounds containing 1,4-diene fragments can transform into alkenyl cyclopropane frameworks when exposed to light, enabling the synthesis of various cyclopropane-fused complex skeletons [65–68]. However, there has been limited progress in de novo synthesis of this di-π-methane rearrangement reaction. We investigated the rearrangement process under light conditions using 1,4-diene fragments from de novo synthesized bicyclo[3.2.1]octa-2,6-diene frameworks to construct more complex tricyclo[3.2.1.02,7]octane bridged-ring derivatives. Initially, we attempted the conversion of bridged product 3a under light exposure, but it did not result in the expected skeletal rearrangement. By introducing iridium as a photosensitizer, we were thrilled to observe that 3a indeed underwent skeletal rearrangement, yielding the strained tricyclo[3.2.1.02,7]oct–3-ene framework 4a with comparable conversion and yield. Control experiments confirmed that both the photosensitizer and 460 nm blue light are essential for the reaction (more details please see Supporting information). As illustrated in Scheme 3, we speculated on the reaction process: under light, the double bonds of C2–C3 and C6–C7 undergo a π-π transition, forming a cyclopropane diradical intermediate Ⅰ. Subsequently, the C1–C7 bond of the three-membered ring opens to produce a new 1,3-diradical intermediate Ⅱ, which then rearranges into the cyclopropane product 4a. Compared to most established radical translocations, the development of di-π-methane rearrangement is limited. We subsequently assessed the compatibility of various substituents in the di-π-methane rearrangement and found that modifying the aromatic ring R1 of the allenoates to either a substituted phenyl ring (such as 4-fluoro or 4-phenyl) or a naphthyl ring consistently yielded polycyclic fused bridged products 4b–4d with high yields and excellent stereocontrol. The methoxy–substituted bridged skeleton 3 demonstrated exceptional reactivity, seamlessly rearranging to form the fused bridged cyclopropane skeleton 4e with 93% yield. Additionally, the presence of an electron-donating group as the R2 substituent on bridged skeleton 3 did not hinder the reaction yield, resulting in products 4f–4i with yields ranging from 85% to 99%.
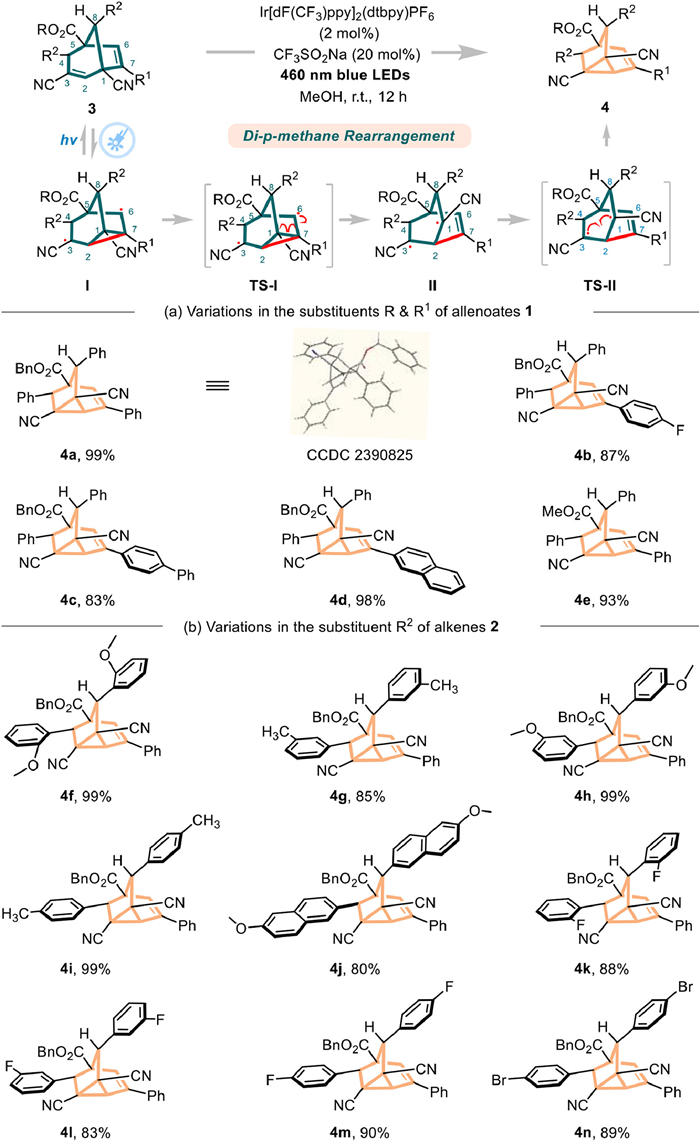
When bulkier substituted naphthyl rings were used, a slight decrease in rearrangement efficiency was observed, producing product 4j with 80% yield. Similarly, the effect of ortho-, meta-, or para-directing electron-withdrawing groups on the reaction was minimal, allowing for high yields of products 4k–4n. These findings demonstrate that this visible light-driven rearrangement reaction exhibits outstanding reactivity and chemoselectivity between bridged products 3 and 4.
To demonstrate the utility of this method, we conducted a scale-up experiment of the phosphine-catalysed reaction under standard conditions. The bridged framework 3a was successfully constructed with a moderate yield, although scaling up resulted in a slight decrease in yield (Scheme 4a). To streamline the preparation process for the bridged scaffold 4a, we employed a one-pot distributed synthetic strategy. Under standard phosphine catalysis conditions, allenoates 1a and 2a were reacted to yield 3a under phosphine catalysis. The reaction mixture was then directly concentrated, and a photosensitizer along with CF3SO2Na was added to the crude mixture. This was followed by subjecting it to photoreaction conditions at 460 nm. We were pleased to find that the bridged product 4a could be efficiently constructed without the need for intermediate separation (Scheme 4b). Subsequently, we further derivatized product 3a and discovered that NaBH4 could reduce the polar double bond to yield product 5a (Scheme 4c, ⅰ), whose relative configuration was determined via NOESY analysis. Interestingly, under photocatalytic conditions, 5a underwent photoinduced α-isomerization of the cyano group to produce 6a (Scheme 4c, ⅱ). Furthermore, selective reduction of the benzyl ester of 3a was achieved under Pd/H2 conditions, introducing a carboxylic acid functional group at the bridgehead carbon (7a) without affecting the double bond in the skeleton (Scheme 4c, ⅲ). Notably, carboxylic acids with bridged ring skeletons exhibit significant activity in many drug molecules [69], yet their synthesis remains highly challenging. Additionally, product 3a can selectively hydrolyze the CN- groups to amide under oxidative conditions (please see Supporting information for details), while the presence of H2O2 enables epoxidation of the double bond, constructing a bridged ring skeleton with an epoxypropane moiety (8a) (Scheme 4c, iv). This series of intriguing transformations stems from the rich functional groups present in the bridged ring skeleton of 3a, offering numerous possibilities for further research and applications.

We speculated on the multicomponent reaction process involving allenoates and α-cyano-cinnamaldehyde. As depicted in Scheme 5, the reaction begins with PPh3 attacking the allenoate, forming a zwitterionic intermediate Ⅲ. Then, nucleophilic attack at the α-position of Ⅲ constructs a cyclopentane intermediate Ⅴ. Finally, through the pathway of γ-H elimination, PPh3 is removed, entering the catalytic cycle. The [3 + 2] cyclization product 3a', due to the acidic hydrogen at the α-position of the ester group, can continue to undergo 1,4-addition with a second molecule of α-cyano-cinnamaldehyde 2. Subsequently, the carbanion of Ⅶ attacks the aldehyde carbonyl group to form Ⅷ, completing the [5 + 2] cyclization. Due to the presence of an adjacent aldehyde group, the alkoxide ion attacks the aldehyde group again, generating an oxetane hemiacetal. Through electron transfer, a series of reactions including acyl migration and formic acid removal are realized, ultimately yielding the bridged-ring product 3a. This coherent reaction sequence illustrates the complexity and precision in synthesizing bridged-ring compounds.
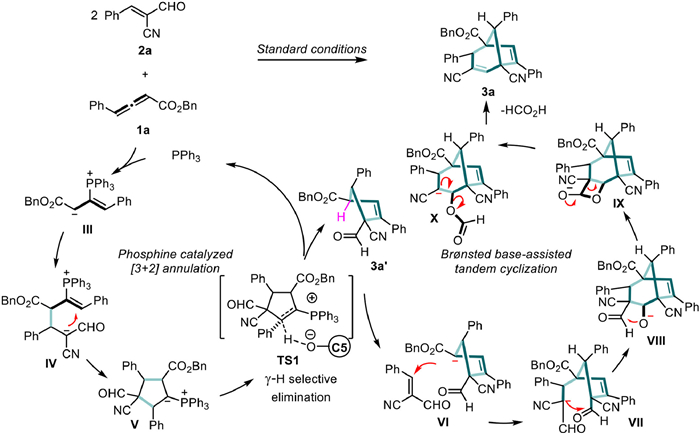
In summary, we present an innovative synthetic strategy that, for the first time, combines organocatalysis with photocatalysis to facilitate a multi-component cascade reaction between allenoates and α-cyano-cinnamaldehydes. This robust method paves the way for the construction of diverse strained bridged skeletons. In the phosphine-catalysed synthesis, the reaction showcases a one-pot, multi-step cascade process sensitive to electronic effects, allowing for the efficient and orderly formation of multiple C—C bonds to construct bicyclo[3.2.1]octa-2,6-diene frameworks (up to 5 C—C bonds formation). The generated bridged bicyclo[3.2.1]octa-2,6-diene skeletons can subsequently undergo light-driven, highly reactive, and stereoselective rearrangement reactions, facilitating the production of strained tricyclo[3.2.1.02,7]oct–3-ene pharmacophore scaffolds with high to excellent yields and exceptional chemoselectivity. Additionally, the richly functionalized 3a can participate in various selective reduction and oxidation reactions, enabling the synthesis of a diverse range of interesting, bridged ring skeletons. Further biological studies on these promising bridged cyclic compounds are currently underway in our laboratory.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Ze-Hong Zheng: Investigation, Formal analysis, Data curation. Mu-Qiu Chen: Methodology, Investigation, Data curation. Jin Zhou: Methodology, Formal analysis. Jie Wang: Investigation, Formal analysis. Yan-Rong Wei: Formal analysis, Data curation. Cheng Peng: Resources. Gu Zhan: Validation, Supervision. Qian-Qian Yang: Writing – original draft, Validation, Supervision, Conceptualization. Bo Han: Writing – review & editing, Validation, Supervision, Resources, Project administration, Conceptualization.
We are grateful for financial support from National Natural Science Foundation of China (Nos. 82374020 and 22401025), Science & Technology Department of Sichuan Province (No. 2024NSFTD0023), and Xinglin Scholar Research Promotion Project of Chengdu University of TCM.
Supplementary material associated with this article can be found, in the online version, at doi:
C.W. Murray, D.C. Rees, Nat. Chem. 1 (2009) 187–192. doi: 10.1038/nchem.217
Y. Wang, P. Tang, W. Tu, et al., Chin. Chem. Lett. 36 (2025) 109955. doi: 10.1016/j.cclet.2024.109955
J. Du, J. Guo, D. Kang, et al., Chin. Chem. Lett. 31 (2020) 1695–1708. doi: 10.1016/j.cclet.2020.03.028
A. Dai, Z. Zheng, L. Duan, et al., Chin. Chem. Lett. 35 (2024) 110462.
E.A. Crane, K. Gademann, Angew. Chem. Int. Ed. 55 (2016) 3882–3902. doi: 10.1002/anie.201505863
J. Shearer, J.L. Castro, A.D. G Lawson, et al., J. Med. Chem. 65 (2022) 8699–8712. doi: 10.1021/acs.jmedchem.2c00473
M. Wu, K. Ren, C. Zou, et al., Chin. Chem. Lett. 35 (2024) 110213.
Q. Zhang, Y. Kuang, L. Chang, et al., Chin. Chem. Lett. 35 (2024) 108338. doi: 10.1016/j.cclet.2023.108338
T. Mori, R. Zhai, R. Ushimaru, et al., Nat. Commun. 12 (2021) 4417. doi: 10.1038/s41467-021-24685-6
W.Y. Liu, X.X. Lei, W.J. Wang, et al., Chin. Chem. Lett. 35 (2024) 110478.
F. Zhang, Y. Wang, Z. Tan, et al., Chin. Chem. Lett. 35 (2024) 110581.
S. Levin, R.R. Nani, S.E. Reisman, Org. Lett. 12 (2010) 780–783. doi: 10.1021/ol902848k
Y.J. Hu, C.C. Gu, X.F. Wang, et al., J. Am. Chem. Soc. 143 (2021) 17862–17870. doi: 10.1021/jacs.1c09637
P. Chen, L. Liang, Y. Zhu, et al., Chin. Chem. Lett. 35 (2024) 109229. doi: 10.1016/j.cclet.2023.109229
L.X. Li, L. Min, T.B. Yao, et al., J. Am. Chem. Soc. 144 (2022) 18823–18828. doi: 10.1021/jacs.2c09548
K.R. Owens, S.V. McCowen, K.A. Blackford, et al., J. Am. Chem. Soc. 141 (2019) 13713–13717. doi: 10.1021/jacs.9b05815
S.J. Davidson, D. Barker, Angew. Chem. Int. Ed. 56 (2017) 9483–9486. doi: 10.1002/anie.201705575
L. Min, X. Liu, C.C. Li, Acc. Chem. Res. 53 (2020) 703–718. doi: 10.1021/acs.accounts.9b00640
J. Liu, X. Liu, J. Wu, C.C. Li, Chem 6 (2020) 579–615. doi: 10.1016/j.chempr.2019.12.027
K. Nagaraju, D. Ni, D. Ma, Angew. Chem. Int. Ed. 59 (2020) 22039–22042. doi: 10.1002/anie.202011093
Z. Lu, H. Li, M. Bian, A. Li, J. Am. Chem. Soc. 137 (2015) 13764–13767. doi: 10.1021/jacs.5b09198
Z. Zhou, A.X. Gao, S.A. Snyder, J. Am. Chem. Soc. 141 (2019) 7715–7720. doi: 10.1021/jacs.9b03248
L. Li, Z.L. Li, F.L. Wang, et al., Nat. Commun. 7 (2016) 13852. doi: 10.1038/ncomms13852
T.C. Coombs, Y. Zhang, E.C. Garnier-Amblard, L.S. Liebeskind, J. Am. Chem. Soc. 131 (2009) 876–877. doi: 10.1021/ja808533z
J.E. DeLorbe, D. Horne, R. Jove, et al., J. Am. Chem. Soc. 135 (2013) 4117–4128. doi: 10.1021/ja400315y
J.E. DeLorbe, S.Y. Jabri, S.M. Mennen, et al., J. Am. Chem. Soc. 133 (2011) 6549–6552. doi: 10.1021/ja201789v
Q. Tan, Z. Yang, D. Jiang, et al., Angew. Chem. Int. Ed. 58 (2019) 6420–6424. doi: 10.1002/anie.201902155
M.J.R. Richter, M. Schneider, M. Brandstätter, et al., J. Am. Chem. Soc. 140 (2018) 16704–16710. doi: 10.1021/jacs.8b09685
L.A. Wein, K. Wurst, P. Angyal, et al., J. Am. Chem. Soc. 141 (2019) 19589–19593. doi: 10.1021/jacs.9b11646
C. Li, D. Lee, T.N. Graf, et al., Org. Lett. 7 (2005) 5709–5712. doi: 10.1021/ol052498l
Y. Aoyagi, A. Yamazaki, C. Nakatsugawa, et al., Org. Lett. 10 (2008) 4429–4432. doi: 10.1021/ol801620u
C.C. Tseng, H. Ding, A. Li, et al., Org. Lett. 13 (2011) 4410–4413. doi: 10.1021/ol201748x
S. Levin, R.R. Nani, S.E. Reisman, J. Am. Chem. Soc. 133 (2010) 774–776.
T.T. Song, Y.K. Mei, Y. Liu, et al., Angew. Chem. Int. Ed. 63 (2024) e202314304. doi: 10.1002/anie.202314304
J.M. Anderson, N.D. Measom, J.A. Murphy, D.L. Poole, Angew. Chem. Int. Ed. 60 (2021) 24754–24769. doi: 10.1002/anie.202106352
X. Liu, J. Liu, J. Wu, et al., J. Am. Chem. Soc. 141 (2019) 2872–2877. doi: 10.1021/jacs.8b12647
W. Zhang, L. Li, C.C. Li, Chem. Soc. Rev. 50 (2021) 9430–9442. doi: 10.1039/d0cs01471k
K. Gao, J. Hu, H. Ding, Acc. Chem. Res. 54 (2021) 875–889. doi: 10.1021/acs.accounts.0c00798
M.H. Filippini, J. Rodriguez, Chem. Rev. 99 (1999) 27–76. doi: 10.1021/cr970029u
M. Presset, Y. Coquerel, J. Rodriguez, Chem. Rev. 113 (2012) 525–595.
J. Zhuo, C. Zhu, J. Wu, et al., J. Am. Chem. Soc. 144 (2021) 99–105.
W. Adam, O. De Lucchi, K. Peters, et al., J. Am. Chem. Soc. 104 (1982) 161–166. doi: 10.1021/ja00365a029
Z. Yuan, Z. Feng, Y. Zeng, et al., Angew. Chem. Int. Ed. 58 (2019) 2884–2888. doi: 10.1002/anie.201900059
B. Chen, Y. Zhang, R. Wu, et al., ACS Catal. 9 (2019) 11788–11793. doi: 10.1021/acscatal.9b04183
Z. Yuan, Y. Zeng, Z. Feng, et al., Nat. Commun. 11 (2020) 2544. doi: 10.1038/s41467-020-16221-9
I. Škorić, M. Šmehil, Ž. Marinić, et al, J. Chem. Phys. 207 (2009) 190–196.
D. Skropeta, R.W. Rickards, Tetrahedron Lett. 48 (2007) 3281–3284. doi: 10.1016/j.tetlet.2007.03.011
M. Ramirez, V. Vece, S. Hanessian, K.N. Houk, J. Org. Chem. 86 (2021) 17955–17964. doi: 10.1021/acs.joc.1c02296
S. Zhu, Z. Guo, Z. Huang, H. Jiang, Chem. Eur. J. 20 (2014) 2425–2430. doi: 10.1002/chem.201304839
X. Wang, Z. Han, Z. Wang, K. Ding, Acc. Chem. Res. 54 (2021) 668–684. doi: 10.1021/acs.accounts.0c00697
X. Fan, Y. He, X. Zhang, Chem. Rec. 16 (2016) 1635–1646. doi: 10.1002/tcr.201500301
X. Lu, C. Zhang, Z. Xu, Acc. Chem. Res. 34 (2001) 535–544. doi: 10.1021/ar000253x
L.W. Ye, J. Zhou, Y. Tang, Chem. Soc. Rev. 37 (2008) 1140–1152. doi: 10.1039/b717758e
H. Ni, W.L. Chan, Y. Lu, Chem. Rev. 118 (2018) 9344–9411. doi: 10.1021/acs.chemrev.8b00261
H. Guo, Y.C. Fan, Z. Sun, et al., Chem. Rev. 118 (2018) 10049–10293. doi: 10.1021/acs.chemrev.8b00081
X. Li, J. Liao, X. Zhuo, et al., Green Synth. Catal. 4 (2023) 54–57.
Y. Li, M. Dai, Angew. Chem. Int. Ed. 56 (2017) 11624–11627. doi: 10.1002/anie.201706845
L.Z. Li, Y.R. Huang, Z.X. Xu, et al., J. Am. Chem. Soc. 146 (2024) 24782–24787. doi: 10.1021/jacs.4c09384
C.H. Liu, Z.X. Yu, Angew. Chem. Int. Ed. 56 (2017) 8667–8671. doi: 10.1002/anie.201702288
Q.Q. Yang, C. Chen, D. Yao, et al., Angew. Chem. Int. Ed. 63 (2023) e202312663.
C. Chen, J. Zhou, J. Jiang, et al., Chin. Chem. Lett. 35 (2024) 108295. doi: 10.1016/j.cclet.2023.108295
Q.Q. Yang, S.J. Liu, W. Huang, et al., Research 7 (2024) 0410. doi: 10.34133/research.0410
C. Yuan, H. Tan, X.F. Jiang, et al., Asian J. Org. Chem. 8 (2019) 1893–1902. doi: 10.1002/ajoc.201900393
Z. Xu, X.J. Lu, Org. Chem. 63 (1998) 5031–5041. doi: 10.1021/jo9723063
X. Creary, M.A. Butchko, J. Am. Chem. Soc. 13 (2001) 1569–1578.
H.E. Zimmerman, R.W. Binkley, R.S. Givens, M. Sherwin, J. Am. Chem. Soc. 89 (1967) 3932–3933. doi: 10.1021/ja00991a064
Y. Zheng, Q.X. Dong, S.Y. Wen, et al., J. Am. Chem. Soc. 146 (2024) 18210– 18217. doi: 10.1021/jacs.4c04370
Z.C. He, S.K. Mellerup, L. Liu, et al., Angew. Chem. Int. Ed. 58 (2019) 6683–6687. doi: 10.1002/anie.201902231
P. Milbeo, J. Martinez, M. Amblard, et al., Acc. Chem. Res. 54 (2021) 685–696. doi: 10.1021/acs.accounts.0c00680

Scheme 1 Representative bioactive molecules, reported the synthesis of tricyclo[3.2.1.02,7]octane derivatives and our divergent synthesis strategy.

Scheme 2 Substrate scope of multicomponent reactions for the synthesis of bridged bicyclo[3.2.1]octa-2,6-diene products 3.

Scheme 3 Substrate scope of visible light-driven di-π-methane rearrangement reaction to construct tricyclo[3.2.1.02,7]oct–3-ene frameworks 4.

Scheme 4 Scale-up synthesis of 3a, one-pot stepwise strategy for the synthesis of tricyclo[3.2.1.02,7]oct–3-ene frameworks 4a and the subsequent derivatization of the product 3a.
Table 1. Screening conditions for three-component tandem reaction of allenoate 1a and (E)-2-formyl-3-phenylacrylonitrile 2a.a
|
||||
| Entry | Co-catalyst | Solvent | Yield (3a)b | Yield (3a')c |
| 1 | – | EtOAc | 40 | 33 |
| 2 | C1 | EtOAc | 55 | 23 |
| 3 | C2 | EtOAc | – | – |
| 4 | C3 | EtOAc | 22 | 10 |
| 5 | C4 | EtOAc | – | – |
| 6 | C5 | EtOAc | 60 | 22 |
| 7 | K2CO3 | EtOAc | 57 | 20 |
| 8 | Et3N | EtOAc | 42 | 26 |
| 9 | DIPEA | EtOAc | 44 | 23 |
| 10 | Cs2CO3 | EtOAc | 44 | 27 |
| 11 | Et3N | DCM | 44 | 31 |
| 12 | Et3N | Toluene | 24 | 17 |
| 13 | Et3N | THF | 58 | 21 |
| 14 | Et3N | DMF | mess | – |
| 15 | Et3N | MeCN | 31 | 12 |
| 16 | Et3N | DCE | 30 | 12 |
| 17e | Et3N | EtOAc | 64 | 7 |
| 18d | Et3N | EtOAc | 62 | 10 |
| 19e | C5 | EtOAc | 78 | 6 |
| a Reactions conditions: 1a (0.2 mmol), 2a (0.2 mmol), PPh 3 (20 mol%) and co-catalyst (20 mol%) in 2.0 mL of solvent at 50 ℃ for 12 h; > 19:1 dr determined by 1H NMR. b Yield of isolated product 3a. c Determined by in situ 1H NMR. d With Na 2SO 4 (50 mg) as additive. e With 3 Å molecular sieve (50 mg) as additives. |
||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
