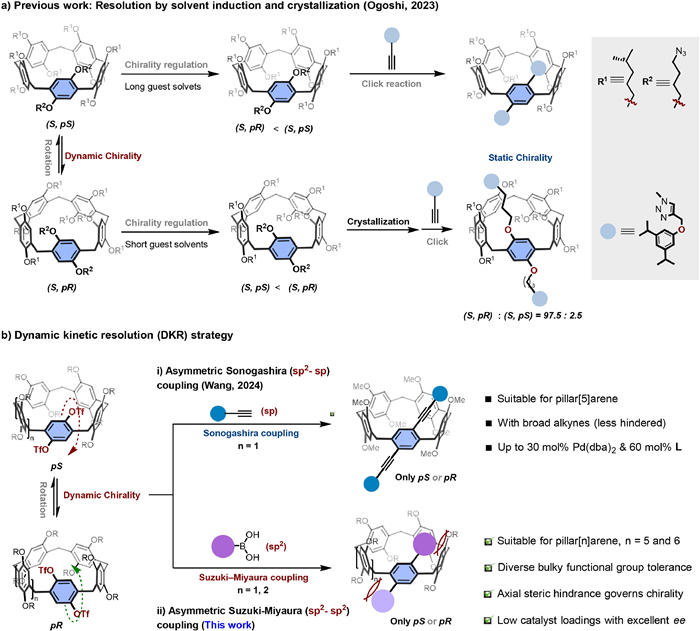
Figure 1.
Strategies for the construction of static chirality PAs (a: resolution by solvent induction and crystallization; b: DKR approaches).
Enantioselective synthesis of bulky planar-chiral pillar[n]arenes through dynamic kinetic resolution
Yan-E Zhang , Yingtao Jiang , Yun Zhang , Hu Wang , Zitong Wu , Rui Li , Yumiao Ma , Tao Tu

Planar chiral macrocyclic arenes, including paracyclophanes [1,2], pillar[n]arenes [3–10], prism[n]arenes [11,12] and pagoda[n]arenes [13], have attracted great attention due to their unique structures and extensive applications in chiral recognition [14–22], self-assembly [23–25], chiral solvating agents [26–29], asymmetric catalysis [30], and circularly polarized luminescence (CPL) materials [31–37]. Among them, pillar[n]arenes (PAs), as potential chiral macrocyclic arenes, exhibit planar chirality due to the specific arrangement of rim substituents on their π-rings. Each repeating unit within the structure contributes to this unique chiral property, making PAs a key subject of study in the field of chiral supramolecular chemistry [39–43]. In the case of pillar[5]arenes, eight stereoisomers exist, consisting of four diastereomeric pairs of enantiomers. Typically, pillar[n]arenes favor chirality-aligned conformations, like all-pS or all-pR arrangements with reduced steric hindrance. However, the interconversion between pS and pR stereoisomers readily occurs via facile flipping of the π-panel rims [34–36]. This stereo-dynamic nature presents significant challenges in the asymmetric synthesis of configurationally stable macrocyclic molecules, especially in a catalytic manner.
Generally, upon the introduction of sterically demanding substituents, such as cyclohexylmethoxy groups, at each position, or the incorporation of two macrocyclic moieties onto any phenyl ring within the single pillar[5]arene unit, flipping can be effectively prevented, allowing enantiomer separation via chiral column chromatography or diastereomer formation with other chiral reagents [37–41]. However, theoretically, the maximum isolated yield of a single enantiomer via racemic resolution is 50%. In 2023, Ogoshi and coworkers synthesized pillar[5]arene diastereomers via click chemistry with stereogenic carbons at both rims. By employing solvent induction and crystallization amplification approaches, they introduced bulky groups to achieve stable chiral PAs in high enantiomeric excess (ee, Fig. 1a) [38]. Despite these advances, the synthesis of chiral PAs remains hindered by complex starting material preparation, limited substrate scope, and susceptibility to racemization at moderate temperatures. These challenges underscore the need for more efficient, robust, and versatile strategies for the synthesis chiral PAs.
The dynamic kinetic resolution (DKR) strategy, pioneered by Bäckvall and colleagues [44], constitutes an efficient approach for the synthesis of enantiopure compounds by converting racemic mixtures into a single enantiomer, offering superior atom economy compared to traditional resolution protocols. We conceived that DKR may be regarded as an ideal approach to access desired planar chiral PAs, fully utilizing racemic starting materials and enhancing enantiomer production efficiency [45–48]. The asymmetric Suzuki–Miyaura cross-coupling reactions provide a versatile approach for synthesizing chiral biaryls via direct C(sp2)–C(sp2) bond formation due to its operational simplicity, non-toxicity, high stability and availability of substrates [49,50]. Herein, we demonstrate the enantioselective synthesis of chiral pillar[n]arenes (n = 5, 6) through Pd-catalyzed Suzuki–Miyaura coupling reactions using DKR strategy (Fig. 1b-ⅱ). During the preparation of this manuscript, Wang's research group reported a study on the construction of C(sp2)-C(sp) bonds via the asymmetric Sonogashira coupling reactions [51], leading to the formation of planar-chiral pillar[5]arene (Fig. 1b-ⅰ). Compared to less hindered C(sp2)-C(sp) bonds, the connection between C(sp2)-C(sp2) bonds presents much greater steric hindrance, making their construction more challenging. Moreover, planar-chiral PAs formed by C(sp2)-C(sp2) bonds exhibit larger conjugated systems and enhanced rigidity, which may confer superior performance in the field of circularly polarized luminescence. The DKR strategy using Suzuki–Miyaura coupling effectively achieves enantiopure PAs and may provide a robust platform for their applications in chiral recognition, supramolecular assembly, and chiral luminescent materials [11–28]. It is noteworthy that during the preparation and revision of this manuscript, two independent studies have reported similar findings on the enantioselective construction of chiral pillar[5]arenes [52,53], further validating the feasibility of the proposed strategy.
We commenced our investigation with the model coupling between prochiral pillar[5]arene triflates 1 and benzyl boric acid 2a. We hypothesized that the flipping of substituents could be suppressed by introducing two bulky and rigid aryl rims into a single aromatic unit of the pillar[5]arene, resulting in the configurationally stable planar chiral pillar[5]arene 3a. Initially, Pd(OAc)2 and Pd2(dba)3 were selected to investigate their catalytic efficiency [49] and we found Pd(OAc)2 was preferred for the coupling process. Since the base is critical [54–56] various bases, including K3PO4, Na3PO4, K2CO3, Na2CO3, CH3COOK, TMSOK, KF and organic triethylamine were screened, in which Na3PO4 delivered the best outcomes [57]. Although the target disubstituted product 3a was obtained in a moderate yield of 37%, accompanied by the formation of monosubstituted derivative 4 as the major side product (57% yield, entry 1, Table S1 in Supporting information), optimization of the catalytic conditions revealed that increasing the catalyst loading to 20 mol% substantially enhanced the reaction efficiency. This modification resulted in a remarkable improvement in the production of 3a, achieving an isolated yield of 86% (Table S1, entry 9). Additionally, to selectively access monosubstituted 4 rather than 3a, 10 mol% Pd(OAc)2 was involved with THF as the solvent at a decreased temperature of 50 ℃. Under these conditions, up to 87% yield of monosubstituted 4 was observed after 24 h (Table S1, entry 10). Notably, the unreacted triflate rim retained its rotational flexibility at room temperature, enabling further synthesis of planar chiral PAs with two distinct substituents on each rim.
With established protocol in hand, its asymmetric reaction conditions were then investigated. Given the profound impact of chiral ligands on both enantiomeric excess (ee) and overall yields of the desired chiral products [58,59], a series of chiral ligands were tested for their ability to facilitate dynamic kinetic resolution.
Extensive experiments revealed that classic bisphosphine ligands, such as BINAP (L1), MeOBIPHEP (L4) efficiently catalyzed the reaction, albeit with relatively low ee values (<45%). PHOX (L2) and BIDIME (L3) resulted in low production and poor enantioselectivity. Surprisingly, phosphoramidites (L5-L11) significantly enhanced enantioselectivity and up to 82% ee could be achieved, although the yields were still low (Scheme 1, in aqueous toluene). Further investigation indicated the inferior yields could be attributed to the partial hydrolysis of the OTf rims of 2a, likely due to the relatively high-water content in the toluene-water mixture. After further screening other solvents, 1,4-dioxane effectively suppressed the hydrolysis, increasing the yield to 86% with a slightly inferior ee (78%, Table S2 in Supporting information). With these promising results, other chiral phosphoramidites were further involved (Scheme 1, in dioxane). Finally, in the presence of 40 mol% L19, the desired product 3a was obtained with an impressive 93% yield and 98% ee value. The absolute configuration of 3a is unambiguously determined as all-pS conformation by single-crystal X-ray diffraction analysis (Scheme 2). Notably, once the configuration of one of the five units in the resultant pillar[5]arenes is fixed as pS by the attachment of two bulky rims, the other four units adopt the same planar chiral configuration, resulting in the chirality-aligned all-pS conformer as the most energetically favored enantiomer.


After establishing the optimal asymmetric reaction conditions, the substrate scope and limitations were further explored. As shown in Scheme 2, a wide range of aryl boronic acids, bearing various electron-donating (-OMe, -alkyls and -OBn) or electron-withdrawing (-F, -CN, -NO2, -CF3) groups at the para-position of the phenyl ring, were well compatible. The corresponding homo-rimmed planar chiral pillar[5]arenes 3a-3j were obtained in moderate good to excellent yields (65%−93% yields) and impressive enantioselectivities (96%−99.5% ee). The reaction proceeded smoothly even with boronic acids containing sterically bulky conjugated systems, including [1,1′-biphenyl]−4-ylboronic acid, (4-(pyridine-4-yl)phenyl)boronic acid, naphthalen-2-ylboronic acid and anthracene-2-ylboronic acid, yielding the desired planar chiral pillar[5]arenes 3k-3n in good yields (69%−87%) with excellent enantioselectivity (96%−98% ee). Along with increasing the conjugation and steric hindrance of the selected boronic acid skeletons, pillar[5]arenes 3o-3r were still obtained in good yields (60%−80%) by slightly raising the reaction temperature to 90 ℃ within 48 h. Delightedly, the enantioselectivity remained unaffected by the elevated reaction temperature, exceeding 96% ee was still observed. Remarkably, pillar[5]arenes 3o and 3p, can be used to construct CPL-emitting assemblies, while 3q serves as an excellent AIE-active CPL materials, and 3r can be employed in the construction of chiral supramolecular polymers.
Unexpectedly, when meta-methoxyphenylboronic acid, (3,5-dimethoxyphenyl)boronic acid or anthracen-9-ylphenylboronic acid was involved, the desired pillar[5]arenes 3s, 3t or 3u could be obtained in good yields but as racemic mixtures. In contrast to the conventionally less-hindered para-methoxyphenyl and 2-anthracenyl analogues, the meta-methoxyphenyl, 3,5-dimethoxyphenyl and 9-anthracenyl groups may exhibit shorter axial length within pillar[5]arene framework, potentially reducing the energy barrier for the molecular flipping. Therefore, we conceived that these three compounds may adopt restricted flipping at room temperature, but undergo rapid interconversion at elevated temperature, thereby enabling racemization. To test this hypothesis, two enantiomers of 3s, 3t and 3u were separated by chiral HPLC. Regardless of which enantiomer was isolated, subsequent dissolution in 1,4-dioxane followed by heating at 80 ℃ for 2 h resulted in the observation of two distinct peaks with equivalent peak areas (50:50 ratio, as evidenced by HPLC analysis, see Supporting information), demonstrating the occurrence of thermal racemization. Density functional theory (DFT) calculations revealed the free energy barriers for the flipping of 3s and 3t at 80 ℃ were 17.6 kcal/mol and 17.1 kcal/mol, respectively. These values are significantly lower than those of the para-substituted counterparts (e.g., >35 kcal/mol for 3a), highlighting the crucial role of axial length in conformational stability. The sizes of the 3,5-dimethoxyphenyl (4.30 Å) and 9-anthracenyl (4.32 Å) groups are significantly smaller than the cavity size of pillar[5]arenes (~8.5 Å, Fig. S7 in Supporting information). Transition state analysis indicated that the flipping aryl ring must align with the pillar[n]arene's equatorial plane. Critically, para-substituents project axially and directly clash with the macrocyclic skeleton (Fig. S9 in Supporting information), elevating the flipping barrier. Conversely, meta-substituents orient radially into the open space above and below the macrocycle (Fig. S8 in Supporting information), minimizing steric interactions. These results constitute the first experimental evidence that axial steric hindrance, rather than radial substitution, governs conformational locking in pillar[n]arenes. These findings suggest that sufficient axial-steric hindrance is crucial for successful asymmetric induction in this catalytic reaction. Notably, even when using the monosubstituted intermediate 4 under optimized conditions with meta-methoxyphenylboronic acid, the resulting product 5a remained racemic, implying that dual axial bulky substituents are required to fully suppress racemization at the elevated reaction temperature. Collectively, these findings establish that sufficient axially steric bulk is essential for effective asymmetric induction in this PA system.
Strikingly, when bulkier aryl, including 4-(F, CF3, NO2, CHO, tBu, or Si(CH3)3)-phenyl, [1,1′-biphenyl]−4-yl, naphthalen-2-yl, and anthracen-2-yl boronic acids were coupled with monosubstituted intermediate 4 under optimized conditions, hetero-rimmed planar chiral pillar[5]arenes (5b-5j) bearing diverse functional groups were synthesized with excellent enantioselectivities (>95% ee, Scheme 3). This success highlights the generality of the strategy for installing sterically demanding axial substituents to achieve stereochemical control.

Compared to pillar[5]arenes, pillar[6]arenes possess a larger cavity, which facilitates the interconversion between different conformations. Moreover, ethoxy groups exhibit greater steric hindrance than methoxy groups, making the catalytic asymmetric synthesis of planar chiral pillar[6]arenes more arduous. After successfully establishing the optimal catalytic system for planar chiral pillar[5]arenes, we investigated the newly developed asymmetric protocol for the enantioselective synthesis of planar chiral pillar[6]arenes. Using para-methoxyphenylboronic acid and 2-naphthylboronic acid as the coupling partners, the reaction was carried out under optimal conditions to obtain target pillar[6]arenes 7a and 7c in good yields (Scheme 4). However, chiral HPLC analysis at room temperature revealed only a single peak for both products, suggesting that the axial length of the introduced bulky macrocyclic groups (para-methoxyphenyl and 2-naphthyl) is insufficient to inhibit the flipping of the pillar[6]arenes. DFT-optimized geometries (Fig. S9) further confirm that the size of para-methoxyphenyl (7.50 Å) and 2-naphthy (7.62 Å) is significantly smaller than that of the pillar[6]arenes (~11.2 Å). When a slightly larger amount of dibenzo[b,d]furan-2-ylboronic acid is used, the resulting product 7b exhibits two peaks at room temperature. However, racemization still occurs at the reaction temperature (80 ℃, See Supporting information for details). To our excitement, further increasing the bulkiness of the boronic acids, the resulting planar chiral pillar[6]arenes 7d-7j exhibited stable conformations and underwent chiral HPLC separation into two distinct peaks. Additionally, both the yield (61%−90%) and ee (87%−99%) were good under optimal conditions (Scheme 4). This is the first successful example in the catalytic asymmetric synthesis of planar chiral pillar[6]arenes to date and further demonstrates the broad feasibility of our newly developed protocol. Therefore, an efficient and convenient approach for the synthesis of such planar chiral pillar[n]arenes (n = 5, 6) may hold unique and potential feasibility in chiral recognition and the construction of chiral luminescent materials [25–29].

The extraordinarily high induction of planar chirality by a chiral phosphoramidite-ligated palladium complex was unprecedented and intriguing. To gain deep insight into the chirality transfer from a chiral ligand to planar chiral macrocycles, we conducted DFT calculations at the state-of-the-art ωB97M-V/TZVP/def2-TZVP//PBE0-D3BJ/def2-SV(P) level [60,61].
Initially, we examined the configurational stability of PAs with different substitution patterns. As two arylation process occur sequentially, we calculated the Gibbs free energy barrier for flipping the key aryl ring of the substance 1, mono-arylated intermediate 4, and di-arylated product 3a (Fig. S6 in Supporting information). The flipping barriers for 1 and 4 are 8.0 and 10.2 kcal/mol (Fig. 2a), respectively, which are easily racemized even at room temperature. In contrast, the di-arylated product 3a shows a higher flipping barrier (>35 kcal/mol, Fig. 2b), which is hardly racemized, reflecting its configurational stability. In addition, the cavity size of pillar[5]arene macrocycle is 8.46 Å, which is larger than the size of OTf group (4.83 Å) but incompatible to a para-substituted aryl group (for instance, 7.50 Å for para-methoxyphenyl group, Fig. S8). For monosubstituted 4, racemization could occur through in-cavity rotation of the OTf group. However, for bis-substituted analogue 3, flipping is blocked by hinderance of axial di-substitutes (Fig. 2b).
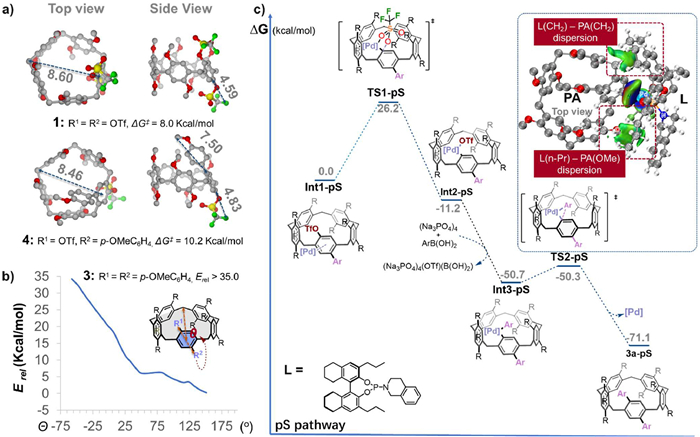
To understand the origin of enantioselectivity, we further investigated the reaction mechanism. Due to 4′s configurational flexibility, enantioselectivity is determined by the second arylation. The Suzuki-Miyaura coupling likely proceeds through sequential oxidative addition (OA), transmetalation, and reductive elimination (RE). The Pd(0) catalyst first coordinates to the substrate, which can undergo rapid interconversion, leading to the formation of arene-Pd complex Int1-pR or Int1-pS, in which Int1-pS is favored by 1.9 kcal/mol. After the coordination, OA occurs via either TS1-pS or TS1-pR, resulting in a Pd(II) intermediate Int2-pS or Int2-pR with an exergonicity of −10.6 or −11.2 kcal/mol. The OA barrier is 28.2 kcal/mol for the pR-pathway and 26.2 kcal/mol for the pS-pathway, while the reverse reaction barrier (~38 kcal/mol) is inaccessible (Fig. 2c and Fig. S10 in Supporting information), indicating an irreversible OA step. Notably, the huge size of the ligand-Pd moiety (11.41 Å, Fig. S8) prevents Int2-pS or Int2-pR from racemization. Thus, overall enantioselectivity is determined by the OA step, favoring the pS-pathway by 2.0 kcal/mol. Once the OA step is completed, transmetalation and RE proceed with negligible barrier (0.3 kcal/mol via TS2-pS), completing the overall DKR process.
The energetic preference for TS2-pS can be attributed to the dispersion interaction between the substrate fragment and the chiral ligand, illustrated by the Independent Gradient Model (IGM [62–64]) isosurface (Fig. 2c and Fig. S11 in Supporting information). Green isosurfaces represent attractive non-covalent interactions, while red isosurfaces (if present) indicate repulsive steric interactions. Two key dispersion interactions stabilize TS2-pS: One between the PA's methylene (CH2) group and the ligand's tetrahydronaphthalene skeleton, and another between the ligand's propyl group and the PA's methoxy group. In contrast, TS2-pR exhibits weaker dispersion interactions as visualized by the significantly sparser isosurfaces, primarily between the two propyl groups and the substrate's triflate and methoxy groups (Fig. 2c and Fig. S11). This significantly emphasizes that the attractive dispersion in TS2-pS leads to the preferential formation of the pS product.
In conclusion, we have successfully established an efficient, practical, and versatile protocol for the precise synthesis of diverse chirality-aligned pillar[n]arenes (PAs, n = 5,6) using asymmetric Pd-catalyzed Suzuki–Miyaura (sp2-sp2) cross-coupling via dynamic kinetic resolution (DKR) process. A series of robust and chirality-aligned homo- and hetero-diaryl PAs with structural diversity were achieved with excellent enantioselectivity (>95% ee) by introducing sterically bulky aryl substituents at the rims of PA skeleton. These outcomes constitute the first experimental evidence that axial steric hindrance, rather than radial substitution, governs conformational locking in pillar[n]arenes, which is supported by DFT calculation and control experiments. This work not only offers a promising approach for synthesizing planar chiral macrocyclic arenes, but also provides diverse candidates for potential applications of chiral molecular machines, enantioselective sensors, and chiral luminescent materials.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Yan-E Zhang: Writing – review & editing, Writing – original draft, Supervision, Data curation. Yingtao Jiang: Writing – original draft, Investigation, Formal analysis, Data curation. Yun Zhang: Investigation. Hu Wang: Investigation. Zitong Wu: Writing – review & editing, Formal analysis. Rui Li: Investigation. Yumiao Ma: Writing – review & editing, Formal analysis. Tao Tu: Writing – review & editing, Supervision.
We thank the financial support from the Natural Science Foundation of Ningxia Province (Nos. 2024AAC03023 and 2024AAC03024). We would like to thank Analysis and Testing Center of Ningxia University for their assistance. We would also like to thank Dr. Yuze Li for his help in mass spectrometry testing.
Supplementary material associated with this article can be found, in the online version, at doi:
Z. Hassan, E. Spuling, D.M. Knoll, S. Bräse, Angew. Chem. Int. Ed. 59 (2020) 2156–2170. doi: 10.1002/anie.201904863
Z. Hassan, E. Spuling, D.M. Knoll, S. Bräse, Chem. Soc. Rev. 47 (2018) 6947–6963. doi: 10.1039/C7CS00803A
T. Ogoshi, T. Yamagishi, Y. Nakamoto, Chem. Rev. 116 (2016) 7937–8002. doi: 10.1021/acs.chemrev.5b00765
J. Zhou, L. Rao, G. Yu, et al., Chem. Soc. Rev. 50 (2021) 2839–2891. doi: 10.1039/D0CS00011F
P.P. Jia, L. Xu, Y.X. Hu, et al., J. Am. Chem. Soc. 143 (2021) 399–408. doi: 10.1021/jacs.0c11370
J. Yao, W. Wu, C. Xiao, et al., Nat. Commun. 12 (2021) 2600–2608. doi: 10.1038/s41467-021-22880-z
W.J. Li, Q. Gu, X.Q. Wang, et al., Angew. Chem. Int. Ed. 60 (2021) 9507–9515. doi: 10.1002/anie.202100934
P.D. Sala, R.D. Regno, C. Talotta, et al., J. Am. Chem. Soc. 142 (2020) 1752–1756. doi: 10.1021/jacs.9b12216
X. Liang, Y. Shen, D. Zhou, et al., Chem. Commun. 58 (2022) 13584–13587. doi: 10.1039/D2CC05690A
X.N. Han, Y. Han, C.F. Chen, J. Am. Chem. Soc. 142 (2020) 8262–8269. doi: 10.1021/jacs.0c00624
Z. Liu, Y. Liu, Chem. Soc. Rev. 51 (2022) 4786–4827. doi: 10.1039/D1CS00821H
H. Nie, Z. Wei, X.L. Ni, Y. Liu, Chem. Rev. 122 (2022) 9032–9077. doi: 10.1021/acs.chemrev.1c01050
H.T. Zhu, Q. Li, Z.C. Gao, et al., Angew. Chem. Int. Ed. 59 (2020) 10868–10872. doi: 10.1002/anie.202001680
Y. Chen, L.L. Fu, B.B. Sun, et al., Chem. Eur. J. 27 (2021) 5890–5896. doi: 10.1002/chem.202004003
Z.Y. Zhang, C. Li, Acc. Chem. Res. 55 (2022) 916–929. doi: 10.1021/acs.accounts.2c00043
G. Sun, X. Zhang, Z. Zheng, et al., J. Am. Chem. Soc. 146 (2024) 26233–26242. doi: 10.1021/jacs.4c07924
R.L. Huang, X.Q. Wei, P.Y. Wang, et al., Org. Lett. 26 (2024) 1405–1409. doi: 10.1021/acs.orglett.3c04367
H. Zhu, Q. Li, B.B. Shi, et al., J. Am. Chem. Soc. 142 (2020) 17340–17345. doi: 10.1021/jacs.0c09598
J. He, Y.Q. Fan, Z. Lian, et al., Adv. Opt. Mater. 12 (2023) 302221.
N.L. Strutt, H. Zhang, J.F. Stoddart, Chem. Commun. 50 (2014) 7455–7458. doi: 10.1039/c4cc02559h
Y. Lv, C. Xiao, C. Yang, Chem. Commun. 56 (2020) 6197–6200. doi: 10.1039/D0CC02055A
J.F. Chen, T.B. Wei, Q.X. Gao, H. Yao, Q. Lin, Chem. Commun. 60 (2024) 6728–6740. doi: 10.1039/D4CC01698J
J.Q. Wang, X.N. Han, Y. Han, C.F. Chen, Chem. Commun. 59 (2023) 13089–13106. doi: 10.1039/D3CC04187E
P. Li, Y. Chen, Yu. Liu, Chin. Chem. Lett. 30 (2019) 1190–1197. doi: 10.1016/j.cclet.2019.03.035
X. Zhao, A. Wang, S. Wang, et al., Chin. Chem. Lett. 36 (2025) 110205. doi: 10.1016/j.cclet.2024.110205
J.F. Chen, X. Yin, B. Wang, et al., Angew. Chem. Int. Ed. 59 (2020) 11267–11272. doi: 10.1002/anie.202001145
J.F. Chen, X. Yin, K. Zhang, et al., J. Org. Chem. 86 (2021) 12654–12663. doi: 10.1021/acs.joc.1c01175
J.F. Chen, G. Tian, K. Liu, et al., Org. Lett. 24 (2022) 1935–1940. doi: 10.1021/acs.orglett.2c00313
F.Y. Chen, W.C. Geng, K. Cai, D.S. Guo, Chin. Chem. Lett. 35 (2024) 109161. doi: 10.1016/j.cclet.2023.109161
K. Wang, X. Tian, J.H. Jordan, et al., Chin. Chem. Lett. 33 (2022) 89–96. doi: 10.1016/j.cclet.2021.06.026
M. Xue, Y. Yang, X.D. Chi, Z.B. Zhang, F. Huang, Acc. Chem. Res. 45 (2012) 1294–1308. doi: 10.1021/ar2003418
T. Ogoshi, T. Yamagishi, Y. Nakamoto, Chem. Rev. 116 (2016) 7937–8002. doi: 10.1021/acs.chemrev.5b00765
N.L. Strutt, H.C. Zhang, S.T. Schneebeli, J.F. Stoddart, Acc. Chem. Res. 47 (2014) 2631–2642. doi: 10.1021/ar500177d
K. Kato, S. Fa, S. Ohtani, et al., Chem. Soc. Rev. 51 (2022) 3648–3687. doi: 10.1039/D2CS00169A
S. Fa, T. Kakuta, T. Yamagishi, T. Ogoshi, Chem. Lett. 48 (2019) 1278–1287. doi: 10.1246/cl.190544
T. Ogoshi, K. Kitajima, T. Aoki, T. Yamagishi, Y. Nakamoto, J. Phys. Chem. Lett. 1 (2010) 817–821. doi: 10.1021/jz900437r
J. Jiao, J. Yu, X. Tian, X.Y. Hu, Chin. Chem. Lett. 36 (2025) 111026. doi: 10.1016/j.cclet.2025.111026
T. Ogoshi, K. Masaki, R. Shiga, K. Kitajima, T. Yamagishi, Org. Lett. 13 (2011) 1264–1266. doi: 10.1021/ol200062j
T. Ogoshi, K. Kitajima, T. Aoki, et al., J. Org. Chem. 75 (2010) 3268–3273. doi: 10.1021/jo100273n
J. Ji, W. Wu, X. Wei, et al., Chem. Commun. 56 (2020) 6197–6200. doi: 10.1039/D0CC02055A
Y. Lv, C. Xiao, C. Yang, New J. Chem. 42 (2018) 19357–19359. doi: 10.1039/C8NJ04802A
K. Wada, M. Suzuki, T. Kakuta, et al., Angew. Chem. Int. Ed. 62 (2023) e202217971. doi: 10.1002/anie.202217971
O. Verho, J.E. Bäckvall, J. Am. Chem. Soc. 137 (2015) 3996–4009. doi: 10.1021/jacs.5b01031
C. Gagnon, É. Godin, C. Minozzi, et al., Science 367 (2020) 917–921. doi: 10.1126/science.aaz7381
J. Guerrero-Morales, M. Scaglia, E. Fauran, G. Lepage, S.K. Collins, Nat. Synth. 3 (2024) 1275–1282. doi: 10.1038/s44160-024-00591-9
Q.L. Lu, X.D. Wang, S. Tong, J. Zhu, M.X. Wang, ACS Catal. 14 (2024) 5140–5146. doi: 10.1021/acscatal.4c00598
K. Wang, W. Wang, D. Lou, et al., ACS Cent. Sci. 10 (2024) 2099. doi: 10.1021/acscentsci.4c01370
J. Zhang, K. Wang, C. Zhu, JACS Au 4 (2024) 502–511. doi: 10.1021/jacsau.3c00623
H. Yang, J.W. Sun, W. Gu, W.J. Tang, J. Am. Chem. Soc. 142 (2020) 8036–8043. doi: 10.1021/jacs.0c02686
R. Pearce-Higgins, L.N. Hogenhout, P.J. Dochert, et al., J. Am. Chem. Soc. 144 (2022) 15026–15032. doi: 10.1021/jacs.2c06529
X.H. Zhou, X. Zhang, Y.R. Song, et al., Angew. Chem. Int. Ed. (2024) e202415190.
T.R. Luan, C. Sun, Y.L. Tian, et al., Nat. Commun. 16 (2025) 2370. doi: 10.1038/s41467-025-57461-x
X.H. Zhou, X. Song, Y.M. Lin, et al., Sci. China Chem. 68 (2025) 4940–4947. doi: 10.1007/s11426-025-2636-1
A.A. Thomas, S.E. Denmark, Science 352 (2016) 329–332. doi: 10.1126/science.aad6981
C.C.C. Johansson Seechurn, M.O. Kitching, T.J. Colacot, V. Snieckus, Angew. Chem. Int. Ed. 51 (2012) 5062–5085. doi: 10.1002/anie.201107017
C.P. Delaney, V.M. Kassel, S.E. Denmark, ACS Catal. 10 (2020) 73–80. doi: 10.1021/acscatal.9b04353
J.P.G. Rygus, C.M. Crudden, J. Am. Chem. Soc. 139 (2017) 18124–18137. doi: 10.1021/jacs.7b08326
S. Zhao, T. Gensch, B. Murray, et al., Science 362 (2018) 670–674. doi: 10.1126/science.aat2299
C. Adamo, V. Barone, J. Chem. Phys. 110 (1999) 6158–6170. doi: 10.1063/1.478522
S. Grimme, J. Antony, S. Ehrlich, H. Krieg, J. Chem. Phys. 132 (2010) 154104. doi: 10.1063/1.3382344
A.V. Marenich, C.J. Cramer, D.G. Truhlar, J. Phys. Chem. B 113 (2009) 6378–6396. doi: 10.1021/jp810292n
F. Weigend, R. Ahlrichs, Phys. Chem. Chem. Phys. 7 (2005) 3297–3302. doi: 10.1039/b508541a
A. Schaefer, C. Huber, R. Ahlrichs, J. Chem. Phys. 100 (1994) 5829–5835. doi: 10.1063/1.467146
C. Lefebvre, G. Rubez, H. Khartabil, et al., Phys. Chem. Chem. Phys. 19 (2017) 17928–17936. doi: 10.1039/C7CP02110K

Figure 1 Strategies for the construction of static chirality PAs (a: resolution by solvent induction and crystallization; b: DKR approaches).

Scheme 1 Optimization of reaction conditions and screening of ligands. Reaction conditions: 1 (0.01 mmol), 2a (0.05 mmol), Pd(OAc)2 (20 mol%), Na3PO4 (5.0 equiv.) and ligand L (40 mol%) in (a) aqueous toluene (2.0 mL, toluene:H2O = 19:1) or (b) 1,4-dioxane (2.0 mL) at 80 ℃ under N2 for 48 h. Isolated yields. The ee value was determined by chiral-phase HPLC.

Scheme 2 Scope of homo-rimmed planar chiral pillar[5]arenes. Reaction conditions: 1 (0.01 mmol), 2 (0.05 mmol), Pd(OAc)2 (20 mol%), Na3PO4 (5.0 equiv.) and L19 (40 mol%) in 1,4-dioxane (2.0 mL) at 80 ℃ under N2 for 48 h. Isolated yields. The ee value was determined by chiral-phase HPLC. a At 90 ℃ for 48 h under otherwise identical conditions.

Scheme 3 Scope of hetero-rimmed planar chiral pillar[5]arenes. Reaction conditions: 4 (0.01 mmol), 2 (0.025 mmol), Pd(OAc)2 (20 mol%), Na3PO4 (5.0 equiv.) and L19 (40 mol%) in 1,4-dioxane (2.0 mL) at 80 ℃ under N2 for 48 h. Isolated yields. The ee value was determined by chiral-phase HPLC.

Scheme 4 Scope of homo-rimmed planar chiral pillar[6]arenes. Reaction conditions: 6 (0.01 mmol), 2 (0.025 mmol), Pd(OAc)2 (20 mol%), Na3PO4 (5.0 equiv.) and L19 (40 mol%) in 1,4-dioxane (2.0 mL) at 80 ℃ under N2 for 48 h. Isolated yield. The ee value was determined by chiral-phase HPLC. a 90 ℃, 48 h, under otherwise identical conditions. b With K3PO4 (5.0 equiv.) under otherwise identical conditions.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
