Citation:
Fei Ye, Yan Liu, Qianru Lv, Boru Gao, Jingjing Xia, Xinyu Li, Mengmeng Dou, Kun Zhao, Munir Ahmad, Zhourong Xiao, Sufeng Wang, Shuaijie Wang, Qingrui Zhang. Unveiling the mechanism of efficient detoxification by Pd species in chlorinated pollutant degradation[J]. Chinese Chemical Letters,
2026, 37(2): 111136.
doi:
10.1016/j.cclet.2025.111136
Unveiling the mechanism of efficient detoxification by Pd species in chlorinated pollutant degradation
English
Unveiling the mechanism of efficient detoxification by Pd species in chlorinated pollutant degradation
State Key Laboratory of Metastable Materials Science and Technology, Hebei Key Laboratory of Heavy Metal Deep-Remediation in Water and Resource Reuse, School of Environmental and Chemical Engineering, Yanshan University, Qinhuangdao 066004, China
b.
State Environmental Protection Key Laboratory of Simulation and Control of Groundwater Pollution, Chinese Research Academy of Environmental Sciences, Beijing 100012, China
c.
Beijing Key Laboratory of Aqueous Typical Pollutants Control and Water Quality Safeguard, School of Environmental, Beijing Jiaotong University, Beijing 100044, China
d.
College of Environmental Science and Engineering, North China Electric Power University, Beijing 102206, China
e.
Institute of Carbon Neutrality, Zhejiang Wanli University, Ningbo 315100, China
zhangqr@ysu.edu.cn (Q. Zhang). 1 These authors contributed equally to this work.
Received Date:
29 November 2024 Accepted Date:
20 March 2025 Revised Date:
06 February 2025 Available Online:
15 February 2026
Abstract:
Successfully generating reactive oxygen species (ROS) in a targeted and efficient manner for the detoxification of chlorinated organic pollutants (CPs) is a significant and demanding challenge. Herein, we present an in-situ photoreduction strategy to fabricate a composite of palladium (Pd) nanoparticles anchored few-layer carbon nitride nanosheets (Pd-CN). This innovative Pd-CN is then leveraged to activate peroxymonosulfate (PMS) in pursuit of our objective. The incorporation of Pd nanoparticles enhances PMS absorption and targets its terminal oxygen, thereby aiding in the cleavage of the O-O bond. This process generates crucial intermediates, including adsorbed hydroxyl radicals (*OH) and adsorbed atomic oxygen (O*), which are essential for the production of 1O2. Consequently, the Pd-CN catalyst demonstrates strong preference for 1O2 generation during the PMS activation process, successfully degrading over 95% of pollutants such as 4-chlorophenol (4-CP), 2,4-dichlorophenol (2,4-DCP), and 2,4,6-trichlorophenol (2,4,6-TCP) within just 20 min. Additionally, the catalyst exhibits total organic carbon (TOC) removal rates ranging from 49.4% to 31.4%, while the rates for de-chlorination fall between 68.6% and 72.7%. A subsequent continuous-flow treatment experiment has confirmed the application potential of this system, demonstrating consistent catalytic activity for up to 8 h. This promising technique presents an efficient strategy for addressing the high toxicity of chlorinated organic pollutants in contaminated water.
Chlorinated organic compounds are a group of extremely hazardous persistent organic pollutants extensively employed in various industries [1,2]. When the concentration of these compounds, present in natural water bodies, reaches ppb (part per billion) levels, they can pose a substantial risk to the well-being of aquatic ecosystems and human health [3]. As a result, there is an urgent requirement to devise a highly effective technique to eliminate these contaminations from water environments. Among various physical [4,5], chemical [6–10], and biological remediation [11,12] technology, advanced oxidation processes (AOP) utilizing singlet oxygen (1O2) generated via peroxymonosulfate (PMS) activation have emerged as an especially promising solution. This is because 1O2 has the unique capability to dismantle C-Cl bonds and neutralize their toxicity by directly targeting the electronegative chlorinate atoms through electrophilic attacks [13–16]. However, the significant challenge remains in consistently and effectively generating 1O2 in a selective and efficient manner.
To achieve the goal of highly selective and efficient generation of 1O2 from activation of PMS, one key strategy involves the creation of highly effective single atom catalysts (SACs) using transition metals, with a carefully orchestrated coordination environment (CE) for the metal sites [16,17]. For example, it has been found that when a Co atom is coordinated with the -N3O1 unit, it is more efficient in enhancing the generation of 1O2 than when arranged with -N4 [18]. This difference in effectiveness can be attributed to the fact that the coordination environment of the former facilitates the absorption of PMS and the creation of the essential adsorbed atomic oxygen (O*) intermediate required for generating 1O2. Moreover, nearly perfect selectivity for generating 1O2 has also been achieved with both Co-N2+2 and Fe-N4 SACs [15,19]. Nevertheless, the harsh requirement for precisely regulating the coordination environment and the high surface free energy that leads to metal atom aggregation tendencies significantly restrict the widespread production and application of SACs [20]. To conquer this challenge, it is crucial to create substitute catalysts through a simple and scalable approach, imbued with a dependable structure for triggering PMS activation to generate 1O2.
In contrast to metal SACs, metal nanoparticles (NPs) originating from the dynamic transformation of SACs generally demonstrate superior stability and greater compatibility with coordination environments [21,22]. Especially, in particular scenarios involving distinct electronic and steric configurations, along with specific reaction pathways and environments, metal nanoparticles are even reported surpass single-atom catalysts in catalytic efficiency [23,24]. When it comes to triggering PMS activation, it is observed that transition metal nanoparticles often take a radical approach to create hydroxyl radicals (•OH) and persulfate radicals (SO4•−) instead of generating 1O2 as the primary reactive oxygen species (ROS) [25,26]. This behavior is attributed to the intense absorption and electron-donating properties between these metals and PMS. Opting for noble metal NPs with limited electron-donating capabilities rather than conventional transition metal NPs is believed to be an effective strategy for altering the PMS activation mechanism from radical to non-radical pathways leading to 1O2 [27]. Among the distinguished noble metals such as Au, Ag, Pd, Pt, Ir, and Rh, Pd stands out for its considerably high work function value of around 5.55 ± 0.1 eV [28], signifying its limited electron-donating capacity. This characteristic suggests that it should demonstrate excellent selectively triggering PMS to generate 1O2. However, research also suggests the potential for radical pathways to emerge when PMS is activated using Pd-based catalysts with various supports [29]. This underscores the importance of developing efficient Pd/support systems that enable highly selective PMS activation, ultimately resulting in the generation of 1O2 through a non-radical pathway.
Taking into account all the aforementioned analyses, we have selected a few-layer carbon nitride (abbreviated as CN) as a support material, and employed the in-situ photo-deposition method to anchor Pd nanoparticles to the CN surface. The morphology, microstructure, and composition of the prepared Pd-CN catalyst are thoroughly analyzed to demonstrate the successful creation of a specific catalyst structure. Subsequent measurements of chlorinated organic pollutants (CPs) degradation, TOC removal, and de-chlorination rates, in combination with qualitative quenching technology and electron paramagnetic resonance (EPR) capturing analysis, as well as quantitative measurement utilizing 9,10-anthracenediyl-bis(methylene)dimalonic acid (ABDA) probe, collectively show the exceptional catalytic performance of the Pd-CN catalyst in degrading target pollutants by activating PMS to selectively produce 1O2. Finally, the potential PMS activation mechanism over Pd-CN, and the application potential of this system are further evaluated through density functional theory (DFT) calculations, and a continuous-flow treatment experiment. This research offers a straightforward and effective approach to the design of efficient heterogeneous catalysts for activating PMS to degrade target CPs pollutants via a non-radical pathway dominated by 1O2.
The fabrication of the hybrid catalyst (Pd-CN) involving Pd NPs supported on few-layer CN is achieved using a straightforward photo-deposition method, as illustrated in Fig. 1a. The involved chemicals, and detailed synthesis, characterization as well as activity measurement methods are shown in Texts S1-S5 (Supporting information).
Figure 1
Figure 1.
(a) Schematic diagram illustrating the fabrication process of the Pdx-CN. (b) XRD patterns and (c) survey XPS spectra of CN and Pd3-CN. (d) High resolution spectrum of Pd 3d. (e) The STEM images of Pd3-CN, with the corresponding elemental mapping shown in (f-h), scale bar: 50 nm. The color of red, blue, and green represent the elements of C, N, and Pd, respectively. (i) TEM image of Pd3-CN and (j) enlargement selected region in 1i, and the inset depicts the HRTEM image of the selected region in 1j.
The successful integration of Pd into CN is initially examined through X-ray diffraction (XRD) analysis. Comparing the XRD patterns of Pd3-CN with those of CN in Fig. 1b, it is evident that although both materials display characteristic peaks corresponding to the (002) interlayer stacking of conjugated aromatic segments and the (100) in-plane repeating motif [30], there are slight differences in their relative intensities and positions. The comparatively weaker intensity of the (002) peak in CN, as opposed to Pd-CN, can be attributed to the fluffy structural nature of CN. However, this fluffy characteristic is anticipated to vanish after undergoing a sequence of treatments including ultrasonic dispersion, filtration, and vacuum drying as part of the Pd-CN preparation process (Fig. S1 in Supporting information). While, a lower shift in angle observed in the (002) peak for Pd-CN in comparison to CN indicates that the formation of Pd-CN mainly modifies the overall fluffy shape without causing significant reduce to the interlayer spacing of CN. Despite efforts to detect the successful introduction of Pd through XRD characterization, solid evidence is lacking as there are no distinctive peaks of Pd metal or oxide identified likely due to the low loading amount or small size of the Pd particles.
In order to identify the trail of Pd emerges in the Pd-CN composite, its survey X-ray photoelectron spectroscopy (XPS) spectrum is investigated. Compared with CN, apart from nearly similar signal intensity of C, N, and O elements, the enlarged survey XPS spectrum of Pd-CN also presents another distinct characteristic peaks at binding energies of 338.1 and 343.2 eV (Fig. 1c), corresponding to the Pd 3d orbit [31]. This suggests the successful incorporation of Pd species into the CN composite. Further analysis of the Pd 3d spectrum of the Pd3-CN catalyst in Fig. 1d reveals that the Pd species in Pd3-CN primarily existed in the form of Pd4+ and Pd2+ rather than Pd0. This is evident from the presence of dominant Pd 3d5/2 and Pd 3d3/2 peaks at 337.0 and 341.2 eV, which correspond to Pd4+, along with weaker peaks at 337.9 and 343.2 eV related to the valence state of Pd2+ species, while no characteristic peaks for Pd0 are detected. The main reason for the preservation of the majority of Pd4+ and the reduction of a minority of Pd4+ to Pd2+ instead of Pd0 may be attributed to the strong interaction between Pd4+ and the nitrogen groups from the CN support. Additionally, only a small number of photogenerated electrons participate in the in-situ photo reduction process in a pure water system without a photogenerated holes scavenger.
The subsequent TEM and elemental mapping images of Pd3-CN offer more direct evidence of the presence of Pd species. In Fig. 1e, the high angle angular dark field-scanning transmission electron microscopy (HAADF-STEM) image reveals the sporadic distribution of several ~3 nm bright spots on the support (CN), indicating that some Pd species may exist in the form of nanoparticles. Moreover, the observed uniform distribution of Pd elements corresponds well with the elemental mapping results of C and N elements (Figs. 1f-h), suggesting the presence of even smaller Pd species. To accurately understand the microstructure of the Pd-CN composite, another region where Pd NPs are also anchored to CN is selected and enlarged to identify the lattice pattern of the hetero-component. As shown in Figs. 1i and j, CN exhibits an amorphous stacked structure, while Pd NPs mainly consist of PdO, as evidenced by the clear lattice distance of 0.22 nm corresponding to the (111) plane of PdO [32]. Based on above comprehensive characterizations, the successful synthesis of Pd NPs anchored Pd-CN composite is verified.
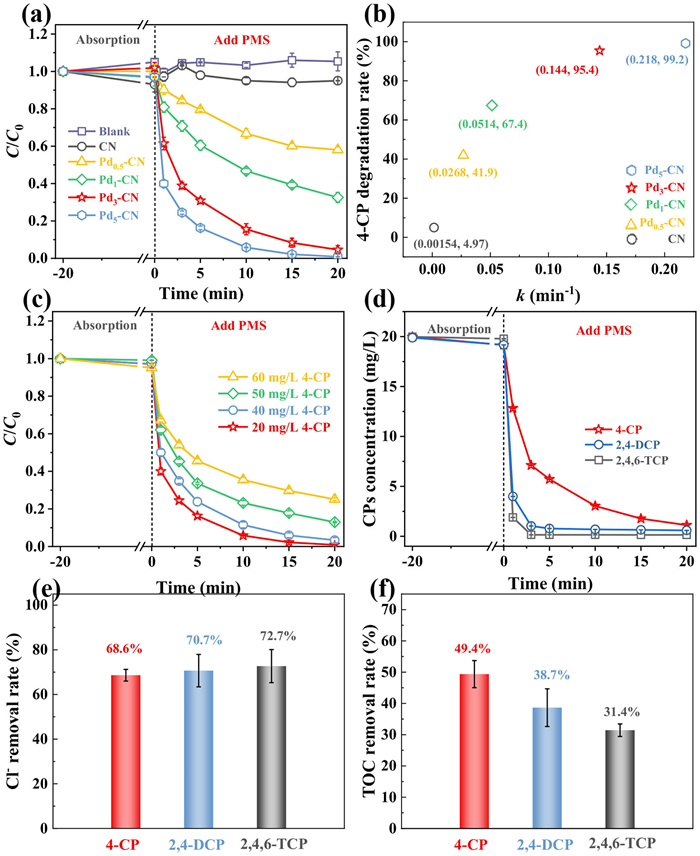
The catalytic degradation capability of the Pd-CN/PMS system is investigated using 4-chlorophenol (4-CP) as the model pollutant, and the pertinent results are presented in Fig. 2. As shown in Figs. 2a and b, the incorporation of Pd into CN significantly enhances the degradation performance of 4-CP compared to both the original CN/PMS and PMS systems. Specifically, by varying the weight percentage of Pd (in the K2PdCl6 precursor) to CN, the resulting Pdx-CN composites (x = 0.5, 1, 3, 5) demonstrate 8.4–19.9 times higher 4-CP degradation rates (%) and 4.7–38.1 times faster kinetic rates (min−1) than pristine CN. These findings underscore the pivotal role of Pd species in activating PMS.
Figure 2
Figure 2.
(a) 4-CP degradation performance among various catalysts/PMS coexisted system (catalyst dosage: 0.2 g/L, PMS dosage: 0.16 g/L, 4-CP concentration: 20 mg/L). (b) The comparison of degradation kinetics and degradation rates among various catalysts. The influence of (c) 4-CP concentration and (d) different CPs (4-CP, 2,4-DCP, 2,4,6-TCP) on the degradation performance of Pd3-CN/PMS system. (e) The de-chlorination and (f) TOC removal efficiencies of Pd3-CN/PMS system during degradation of CPs.
Among the series of Pdx-CN catalysts, Pd3-CN is chosen as the representative catalyst for subsequent investigations. This selection is primarily based on its kinetic rate (0.144 min−1), which is nearly half that of Pd5-CN (0.218 min−1), yet achieves an approximate 4-CP degradation rate (95.4% vs. 99.2%) comparable to the latter. Subsequent investigation into the impact of experimental parameters on the degradation performance of 4-CP over Pd3-CN/PMS system reveals that increasing the dosage of PMS and raising the environmental temperature both initially enhance the performance, before gradually stabilizing (Figs. S2a and b in Supporting information). As a result, we have determined that the ideal PMS dosage and operating temperature are 8 mg and 25 ℃, respectively, in the system where 10 mg of Pd3-CN is dispersed in a 50 mL solution containing 4-CP at a concentration of 20 mg/L.
In addition, the efficiency of catalytic degradation by the Pd3-CN/PMS system is assessed by varying the initial concentration of 4-CP and changing the target pollutant from 4-CP to 2,4-DCP and 2,4,6-TCP. As illustrated in Fig. 2c, an increase in the initial 4-CP concentration (ranging from 20 to 40, 50, and 60 mg/L) leads to a gradual decrease in degradation kinetics (from 0.218 to 0.156, 0.0905, and 0.0585 min−1) and degradation rates (from 99.2% to 96.7%, 74.9%, and 87.1%), suggesting that its capacity for treating pollutants tends to reach a saturation point. Hence, to boost the catalytic degradation performance of this system for CPs with elevated concentrations, prolonging the reaction duration and fine-tuning the proportion of catalyst to PMS may prove to be an effective strategy. In terms of the degradation performance towards CPs with varying degrees of substituted Cl atoms, an intriguing observation has been made. It appears that the degradation kinetics gradually accelerate as the number of Cl atom substitutes increases from 1 to 3 (4-CP, 2,4-DCP, to 2,4,6-TCP) (refer to Fig. 2d). This phenomenon can be attributed to the crucial role played by the breaking of C-Cl bonds in CPs molecules during their degradation. The de-chlorination process seems to occur more easily as the number of Cl atom substitutes in the phenol molecule increases. This hypothesis is supported by subsequent measurements of Cl− concentration and TOC removal rates in different CPs preservation systems (Figs. 2e and f). The de-chlorination rates gradually increase to 68.6%, 70.7%, and 72.7% during the degradation processes of 4-CP, 2,4-DCP, and 2,4,6-TCP, respectively. Conversely, there is a decreasing trend in TOC removal rates from 49.4% to 38.7% and 31.4%, which aligns with expectations.
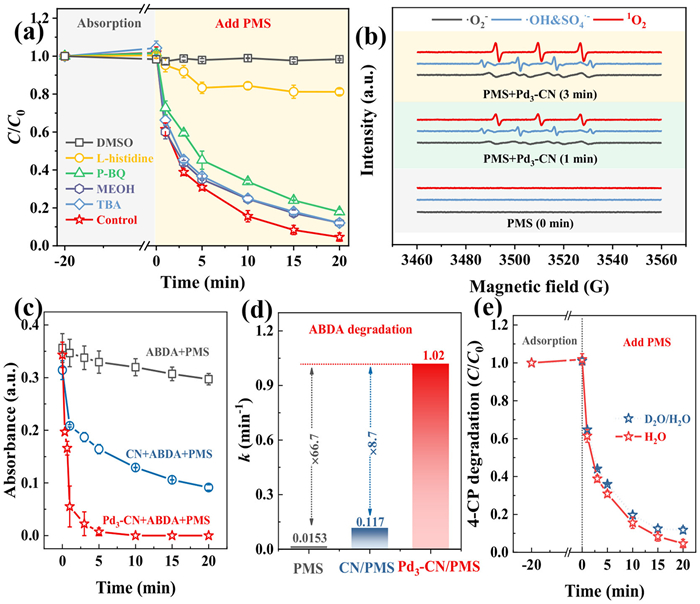
In order to reveal the primary active ingredients responsible for breaking down CPs, a series of quenching tests are initially performed. As illustrated in Fig. 3a, the introduction of Dimethyl sulfoxide (DMSO), intended to quench the bounded reactive oxygen species (ROS) on the catalyst's surface, greatly hinders the degradation of 4-CP. This suggests that the generation of surface-bounded ROS is likely a critical initial step in the activation of PMS, thereby playing a crucial role in triggering the catalytic degradation process [33].
Figure 3
Figure 3.
(a) The degradation of 4-CP over Pd3-CN/PMS system in the presence of various active specie scavengers (DMSO: 260 mmol/L, l-histidine: 26 mmol/L, p-BQ: 26 mmol/L, methanol: 260 mmol/L, and TBA: 260 mmol/L). (b) The comparison of EPR spectra detected over CN and Pd3-CN presented catalyst/PMS coexist system for the discrepancies on 1O2, •O2−, SO4•−/•OH generation. (c) Comparison of the variation on absorbance of ~21 mg/L of ABDA over PMS, CN + PMS, and Pd3-CN + PMS system, and (d) the corresponding ABDA decomposition kinetics. (e) Comparison the degradation efficiency of 4-CP in the Pd3-CN+PMS system, tested in both H2O and a D2O/H2O (50/50, v/v) mixture.
Similarly, when adding l-histidine, p-BQ, methanol, and TBA to scavenge 1O2, •O2−, SO4•−/•OH, •OH, respectively, each experimental group separately shows 80%, 14%, 5%, and 5% lower 4-CP degradation rates compared to the control group. This suggests that 1O2, •O2−, and •OH all contribute to 4-CP degradation, with 1O2 playing the dominant role. However, further increasing the dosage (from 26 mmol/L to 52 mmol/L) even achieves almost 100% quenched 4-CP degradation performance (Fig. S3 in Supporting information). This intriguing phenomenon directly challenges previous assumptions and raises important questions about the decline in performance. It suggests that the reduction may be linked to an overestimation of the active species' role, as the scavengers can also consume PMS during the quenching of the respective radicals. Consequently, there is a need for advanced technologies that can enhance the qualitative and quantitative assessment of radical species and their concentrations.
Subsequent EPR spectral analysis is performed for qualitatively identifying the presence of 1O2, •O2−, SO4•−/•OH radical signals. Additionally, radical probing techniques utilizing KI, blue tetrazolium chloride (NBT), and ortho-hydroxybenzoic acid (o-HBA) are employed for quantitatively assessments of PMS consumption, along with measurements of the generation levels of •O2− and •OH radicals. These findings reinforce the conclusion that 1O2, •O2−, and •OH radicals are all implicated in the Pd3-CN/PMS system, with 1O2 being the primary active species. This is evident from the gradually increasing characteristic signals of 1O2, •O2−, and •OH radicals with intensities of 1:1:1, 1:1:1:1, and 1:2:2:1 in the EPR spectra as time progresses (Fig. 3b). Additionally, only 4.73 and 0.19 µmol of •O2−, and •OH are quantitatively linked to the consumption of 37% and 0.7% of PMS, respectively, out of a total depletion of 25.48 µmol (as shown in Figs. S4a-c in Supporting information). This suggests that the majority of PMS, over 60%, continues to be actively converted into 1O2, while •O2− might merely act as an intermediate or byproduct during the formation of 1O2 [34].
To offer more compelling and direct evidence that 1O2 is indeed produced in the Pd3-CN/PMS system and plays a critical role in the degradation of 4-CP, we initially utilize the widely accepted probe, 9,10-anthracenediyl-bis(methylene)dimalonic acid (ABDA). This probe serves to specifically detect 1O2 by indicating its generation through the consumption of ABDA in a 1:1 reaction ratio. Consequently, the Pd3-CN/PMS system demonstrates the capability to decompose nearly 2.5 µmol of ABDA within just 3 min, as determined from the standard curve correlating ABDA concentration with its absorbance value illustrated in Fig. S5 (Supporting information). This rate is approximately 66.7 times and 8.7 times greater than that observed in systems using only PMS and in the CN/PMS system, respectively (Figs. 3c and d). This indicates a substantial production of 1O2 within the Pd3-CN/PMS system, considering that ABDA is generally understood to interact with merely around 2% of the 1O2 generated in aqueous environments [35].
Moreover, owing to the capability of physically quenching 1O2 is significantly reduced in D2O compared to H2O (with quenching rates of kd(D2O) = 1.6 × 104 s−1 and kd(H2O) = 2.5 × 105 s−1). This difference suggests that using D2O instead of H2O as a solvent in the degradation process of 4-CP could enhance the efficiency of a 1O2-dominated system. However, when half of the H2O is substituted with D2O in the degradation reaction of 4-CP using Pd3-CN and PMS, the degradation performance does not improve; in fact, it shows a slight decline, as illustrated in Fig. 3e. This appears to contradict earlier predictions that suggested the degradation performance would enhance due to the lifetime of 1O2 would be extended in a D2O solvent. Nevertheless, there is a rationale behind this outcome. Because we overlook the detrimental effects of the solvent isotope and the exchange interactions introduced by D2O on the generation of 1O2 in the Pd3-CN/PMS system. This phenomenon has been documented in previous studies [36] and is further corroborated by comparing the decomposition of PMS in both H2O and a D2O/H2O mixture (Fig. S4d in Supporting information). Overall, it is clear that 1O2 continues to play a crucial role in the degradation of 4-CP. This conclusion is supported by our observations that we can achieve comparable degradation efficiency in a D2O/H2O mixture with significantly lower amounts of PMS consumed than in a pure H2O system.
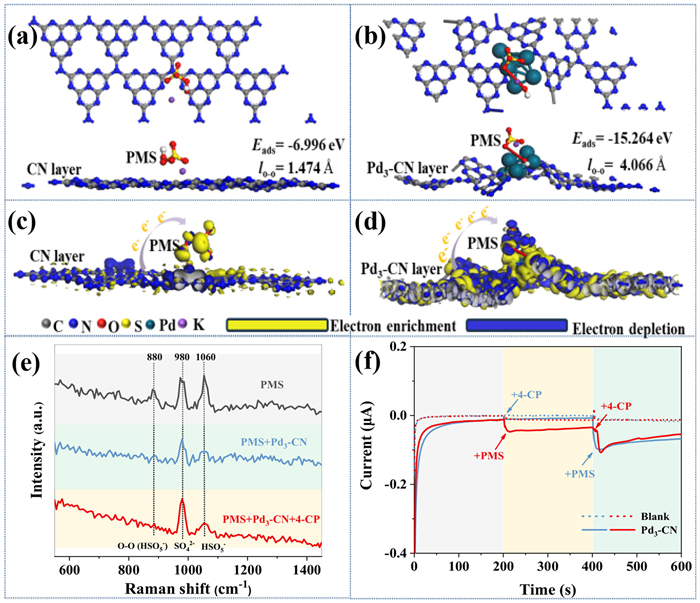
The improved catalytic efficiency of incorporating Pd species into the Pd3-CN/PMS system can be further explained by its ability to boost PMS activation and enhance the selectivity of products towards 1O2. In addition to the promotion of PMS exhaustion rate increasing from 4.6% to 87% by Pd species, as presented in Fig. S3, further electrochemical characterization reveals that Pd3-CN facilitates charge transfer more effectively than the original CN during PMS activation. This is evidenced by a smaller Nyquist semicircle radius, a steeper slope of the tangent line in both high and low frequency regions in electrochemical impedance spectroscopy (EIS) plots (Fig. S6a in Supporting information), and a higher reduction current in linear sweep voltammetry (LSV) curves (Fig. S6b in Supporting information) for Pd3-CN compared to CN. Moreover, to gain a deeper understanding of how Pd enhances PMS activation activity and improves 1O2 generation selectivity, DFT calculations have been conducted to model the interaction and electron transfer between Pd3-CN (or CN) and PMS (Figs. 4a-d). The results reveal that the adsorption energy (Eads) of PMS on Pd3-CN is significantly lower than that on CN (−15.264 eV vs. −6.996 eV). This is supported by the notably extended bond length of O-O in PMS from 1.474 Å for CN to 4.066 Å for Pd3-CN, as well as the observed charge transfer from Pd to PMS, as evidenced by the distinct charge distribution pattern. This implies that the terminal oxygen atom, characterized by its markedly elongated O-O bond, is more likely to attach to the Pd site's surface. As a result, it is anticipated that this interaction will facilitate the breaking of the O-O bond through electron acquisition, leading to the formation of a surface-bound *OH intermediate. This initial step is crucial for the subsequent creation of surface-bound O* and will ultimately promote the conversion into 1O2 [37].
Figure 4
Figure 4.
The most stable geometry, adsorption energy, the length of O-O bond of (a) CN/PMS system and (b) Pd3-CN/PMS system. The deformation charge density of (c) CN/PMS system and (d) Pd3-CN/PMS system. (e) In-situ Raman spectra of the Pd3-CN/PMS systems. (f) The i-t curves obtained at 0 V vs. Ag/AgCl (0.1 mol/L NaSO4) in the Pd3-CN/PMS system.
In order to provide more direct evidence for the specific site where PMS is attacked by Pd3-CN, in-situ Raman spectroscopy is employed. As can be seen from Fig. 4e, the Raman spectrum of pristine PMS exhibits three characteristic peaks at 880, 980, and 1060 cm−1, corresponding to the O-O bond, SO42−, and HSO5−, respectively [38]. After the addition of Pd3-CN, the consumption of HSO5− and the cleavage of the O-O bond occur, as both the signals at 1060 and 880 cm−1 decrease significantly. Further introduction of 4-CP into the PMS/Pd3-CN system leads to a more thorough elimination of PMS, with nearly complete disappearance of the peak of the O-O bond and an increase in the intensity of the SO42− peak. These results demonstrate that the introduction of Pd3-CN triggers the activation of the majority of PMS in the system by initially adsorbing HSO4− and then cleaving the O-O bond, while the addition of 4-CP further promotes the activation process to some extent to generate active species. A similar conclusion can also be drawn from the current-time curves of Pd3-CN recorded in Fig. 4f. It is worth noting that the successive injection of PMS and 4-CP at 200 s and 400 s both induce an instantaneous current response in the Pd3-CN electrode, which can be attributed to the electron transfer from Pd3-CN to PMS to initially form the Pd3-CN-PMS metastable reactive complex, and then further transferring electrons from 4-CP to the Pd3-CN-PMS complex. In contrast, the successive injection of 4-CP and PMS at 200 s and 400 s only causes a current jump at 400 s, indicating that the formation of the Pd3-CN-PMS complex is a prerequisite for the activation of PMS to degrade 4-CP.
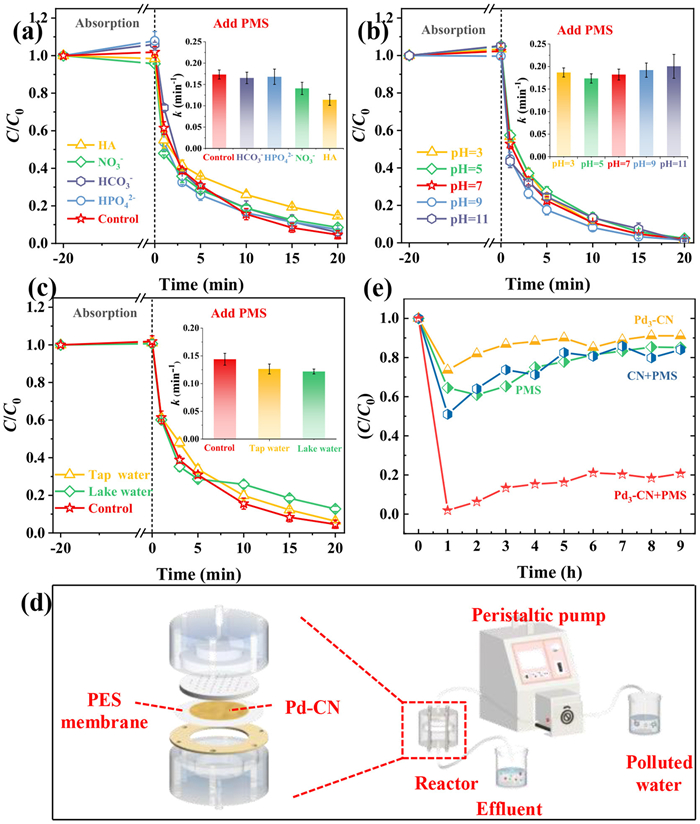
To conduct a preliminary exploration of the applicability of the Pd3-CN/PMS system in complex water quality environments, its anti-disturbance performance towards various anions and HA, as well as its activity preservation ability within a broad pH range or in various natural water bodies, is measured. As shown in Fig. 5a, the presence of NO3−, HPO42−, and HCO3− does not interfere, while the coexistence of HA slightly inhibits the 4-CP degradation kinetics over the Pd3-CN/PMS system, suggesting that HA has a slight influence on the degradation kinetics. This can be explained by the fact that active species such as •OH and •O2− are scavenged by HA, while the majority of active species that are not sensitive to HA, such as 1O2, still contribute to the degradation of 4-CP. This is also consistent with the aforementioned inference that 1O2 is the dominant active specie. In addition, the 4-CP degradation kinetics can further maintain stability when the solution pH value is switched within a wide range of 3–11, or when replaced with tap water or lake water (taken from Yanming lake of Yanshan University), complex water bodies (Figs. 5b and c), demonstrating its strong resistance to interference. Finally, the powder Pd3-CN catalyst is filtered on the PTFE membrane and together being installed on the filter device to evaluate its capacity for continuous water treatment (Fig. 5d). As a result, when a mixture solution containing 10 mg/L of 4-CP and 2.6 mmol/L PMS flows through this catalytic membrane system at a flow velocity of 40 L m−2 h−1, continuous over 80% removal of 4-CP is achieved with 50 mg of catalyst deposited on the membrane for > 8 h. In contrast, the systems utilizing a bare PTFE membrane with PMS, a bare PTFE membrane with Pd3-CN, or a PTFE membrane with CN and PMS can only manage to adsorb or degrade roughly 10%−15% of the pollutant (Fig. 5e). This result further verifies the good application potential of the Pd3-CN/PTFE/PMS system for sustainable water treatment.
Figure 5
Figure 5.
The influence of (a) various anions and HA, (b) solution pH value, and (c) different water bodies on 4-CP degradation performance of Pd3-CN/PMS system. (d) Continuous flow experiment of Pd3-CN/PMS system for 4-CP removal and (e) 4-CP removal efficiency, conditions: water flux = 40 L m−2 h−1, catalyst amount = 50 mg, PMS dosage = 2.6 mmol/L, 4-CP concentration = 10 mg/L.
In this work, we successfully anchor Pd species onto the surface of few-layer CN through a simple in-situ photo deposition strategy. The fabricated Pd-CN component is utilized for the activation of PMS to achieve efficient degradation of chlorinated pollutants. The introduction of Pd species notably enhances the degradation kinetics of the pristine CN/PMS system for 4-CP degradation from 0.00154 min−1 to 0.144 min−1. Additionally, the Pd-CN/PMS system also displays de-chlorination rates of 68.6%−72.7% and TOC removal rates of 31.4%−49.4% during CPs degradation. Subsequent radical quantitative analysis combined with radical scavenger results and DFT calculations reveals that the introduction of Pd promotes the absorption of PMS by binding with the terminal O atom of the PMS molecule, and subsequently improves the activation of PMS to selectively generate 1O2. Finally, the application potential of the Pd-CN/PMS is further verified through the stable and efficient removal of the target pollutant in over 8 h of continuous treatment of flowing water.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The support and resources from the Center for High Performance Computing at Beijing Jiaotong University (http://hpc.bjtu.edu.cn) are gratefully acknowledged. This work was supported by the Natural Science Foundation of Hebei Province (No. B2024203026), the Yanzhao Golden Platform Talent Project (Education Platform) of Hebei Province (No. HJYB202517). the National Natural Science Foundation of China (Nos. U22A20403, 22006128), and the Open Foundation of MOE Key Laboratory of Resources and Environmental System Optimization, College of Environmental Science and Engineering, North China Electric Power University (No. KLRE-KF202308).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111136.
[1]
K. Zhao, X. Quan, Y. Su, et al., Environ. Sci. Technol. 55 (2021) 14194–14203. doi: 10.1021/acs.est.1c04943
[2]
J.Y. Sun, S. Garg, T.D. Waite, Environ. Sci. Technol. 58 (2024) 13131–13144. doi: 10.1021/acs.est.4c03842
Figure 1
(a) Schematic diagram illustrating the fabrication process of the Pdx-CN. (b) XRD patterns and (c) survey XPS spectra of CN and Pd3-CN. (d) High resolution spectrum of Pd 3d. (e) The STEM images of Pd3-CN, with the corresponding elemental mapping shown in (f-h), scale bar: 50 nm. The color of red, blue, and green represent the elements of C, N, and Pd, respectively. (i) TEM image of Pd3-CN and (j) enlargement selected region in 1i, and the inset depicts the HRTEM image of the selected region in 1j.
Figure 2
(a) 4-CP degradation performance among various catalysts/PMS coexisted system (catalyst dosage: 0.2 g/L, PMS dosage: 0.16 g/L, 4-CP concentration: 20 mg/L). (b) The comparison of degradation kinetics and degradation rates among various catalysts. The influence of (c) 4-CP concentration and (d) different CPs (4-CP, 2,4-DCP, 2,4,6-TCP) on the degradation performance of Pd3-CN/PMS system. (e) The de-chlorination and (f) TOC removal efficiencies of Pd3-CN/PMS system during degradation of CPs.
Figure 3
(a) The degradation of 4-CP over Pd3-CN/PMS system in the presence of various active specie scavengers (DMSO: 260 mmol/L, l-histidine: 26 mmol/L, p-BQ: 26 mmol/L, methanol: 260 mmol/L, and TBA: 260 mmol/L). (b) The comparison of EPR spectra detected over CN and Pd3-CN presented catalyst/PMS coexist system for the discrepancies on 1O2, •O2−, SO4•−/•OH generation. (c) Comparison of the variation on absorbance of ~21 mg/L of ABDA over PMS, CN + PMS, and Pd3-CN + PMS system, and (d) the corresponding ABDA decomposition kinetics. (e) Comparison the degradation efficiency of 4-CP in the Pd3-CN+PMS system, tested in both H2O and a D2O/H2O (50/50, v/v) mixture.
Figure 4
The most stable geometry, adsorption energy, the length of O-O bond of (a) CN/PMS system and (b) Pd3-CN/PMS system. The deformation charge density of (c) CN/PMS system and (d) Pd3-CN/PMS system. (e) In-situ Raman spectra of the Pd3-CN/PMS systems. (f) The i-t curves obtained at 0 V vs. Ag/AgCl (0.1 mol/L NaSO4) in the Pd3-CN/PMS system.
Figure 5
The influence of (a) various anions and HA, (b) solution pH value, and (c) different water bodies on 4-CP degradation performance of Pd3-CN/PMS system. (d) Continuous flow experiment of Pd3-CN/PMS system for 4-CP removal and (e) 4-CP removal efficiency, conditions: water flux = 40 L m−2 h−1, catalyst amount = 50 mg, PMS dosage = 2.6 mmol/L, 4-CP concentration = 10 mg/L.


 DownLoad:
DownLoad:

 下载:
下载:






