
Figure 1.
(a) Preparation procedure. (b) Fe K-edge XANES spectra. (c) FT-EXAFS spectra. (d) EXAFS fitting curves. (e, f) 57Fe Mössbauer spectra and (g) temperature-dependent magnetic susceptibilities of Fe1-NSC and Fe1-NC.
Local spin-state manipulation of iron single-atom sites induced by sulfur modification to boost Fenton-like reaction
Lina Zou , Dengke Wang , Shiqin Lai , Xunheng Jiang , Siqi Chen , Lanqing Deng , Dong Fan , Hengshuai Li , Zhigang Zhou , Denglong Chen , Xiangyang Yao , Jianping Zou

Single-atom catalysts (SACs) have garnered great attention in peroxymonosulfate (PMS)-based Fenton-like oxidation [1]. However, most developed SACs-based systems undergo unsatisfied PMS activation since the electron cloud density of the coordinated N atoms around metal site is uniformly dispersed and unsuitable for PMS approach and adsorption [2]. The general try-and-error methods of constructing single-atom sites lack efficiency in tuning the activity of SACs. In this scenario, it is important to develop an innovative strategy to regulate the SACs for enhancing the PMS activation efficiency.
PMS activation principally involves the adsorption and chemical bond cleavage at the material interface. The adsorption strength of PMS is viewed as a prominent indicator in regulating its activation kinetics, particularly in determining the final reactive species generation [3–6]. Theoretical studies reveal that the spin-linked occupancy of eg orbitals of transition metals has profound impacts on the adsorption behavior and binding energy of reactants [7]. Hence, rational design of the spin state of metal sites is crucial in manipulating the adsorption configurations to boost PMS activation. However, the spin state of reactive sites in conventional heterogeneous catalysts cannot be easily regulated due to their customized coordination structures and complicated surface components. Recent achievements indicate that the coordination structures of SACs can be modified via breaking the symmetry of planar metal-N4 by doping with alien atoms (e.g., S, P and B) [8]. The similar atomic radius to N atom but with lower electronegativity makes the heteroatoms tailor the electronic structure of metal-N4 via tuning the near-range coordinated interactions [9]. This may efficiently modulate the spin state of active centers and uplift their intrinsic PMS activation property.
To verify this assumption, Fe-based SACs modified with S (Fe1-NSC) is prepared. Experiments and theoretical calculations reveal that the spin state transition from high spin (HS) to medium spin (MS) Fe3+ sites is achieved after S introduction and facilitates the orbital overlap between Fe3+ 3d and O 2p, reinforcing the dissociation kinetics of PMS. Finally, Fe1-NSC shows excellent performance in PMS activation for pollutant degradation with specific activity reached 36.1 × 10–3 L min-1 m-2, 4.2-folds that of Fe1-NC (8.61 × 10–3 L min-1 m-2) and superior to the existed catalysts reported to date. Furthermore, the spin state modulation via S-modification can be extended to Mn, Co and Cu sites for highly efficient PMS activation.
Fig. 1a illustrates the synthesis of S-modified Fe1-NSC. The porphyrin-based skeletons wrapped on NH2-modified SiO2 (Si@Pyr) are first prepared by an acid-catalyzed polymerization of terephthalaldehyde and pyrrole (Fig. S1 in Supporting information). Fe/S co-doped products are then fabricated by immersing Si@Pyr into a solution containing thiophene and Fe3+. The resultant products are subsequently pyrolyzed and treated with acid to generate the targeted Fe1-NSC. For comparison, Fe1-NC without S-doping and S-modified carbon (NSC) are also synthesized.
Fe1-NSC and Fe1-NC exhibit hollow spherical morphology similar to the NSC host (Figs. S2-S5 in Supporting information). The bumpy surfaces pitted with crevices are observed on both Fe-based SACs, in contrast to the NSC pristine. No crystalline Fe is observed, in line with the result of X-ray diffraction patterns (Fig. S6 in Supporting information). The microstructures are imaged by high-angle annular dark-field scanning TEM (HAADF-STEM) (Figs. S2 and S7 in Supporting information). The individual bright dots reveal atomically dispersed Fe species. Energy dispersion X-ray spectroscopy (EDX) of Fe1-NSC signifies the homogeneous distribution of C, N, S and Fe (Fig. S2). These results suggest that the Fe single-atom sites are successfully obtained. Brunauer-Emmett-Teller analyses exhibit slightly higher surface areas in both Fe-based SACs relative to the pristine NSC (Fig. S8 and Table S1 in Supporting information). The Fe contents in Fe1-NSC and Fe1-NC are detected to be 0.40 and 0.38 wt%, respectively.
The coordination environment of Fe is probed by X-ray absorption spectroscopy and X-ray photoelectron spectroscopy (XPS) measurements. The Fe K-edge X-ray absorption near-edge structure (XANES) of Fe1-NSC shows a rising edge position close to that of Fe2O3, indicating the main Fe3+ species (Fig. 1b). This is also declared by the result of Fe 2p XPS analyses (Fig. S9 in Supporting information). The Fourier transform (FT)-extended X-ray absorption fine structure (EXAFS) spectrum displays no Fe-Fe and Fe-S bonds in Fe1-NSC, ruling out the generation of metallic Fe and FeS (Fig. 1c). Only one peak at 1.57 Å assigned to the scattering of Fe-N bond is observed. The N-bent Fe species are also witnessed by Fe K-edge wavelet transformed (WT)-EXAFS showing a maximum intensity at 5 Å-1 (Fig. S10 in Supporting information). The Fourier-transformed EXAFS of Fe1-NSC can be fitted using scattering path of Fe-N from FeN4 structure (Fig. 1d). The on-site Fe-N bonds of Fe1-NSC diverge into two sets of Fe-N bond lengths of 1.89 and 2.01 Å, differing from that of Fe1-NC with only one set of Fe-N length of 1.93 Å (Fig. S11 and Table S2 in Supporting information). The deviation of Fe-N length in Fe1-NSC is probably induced by asymmetric interaction of N with S, inferring that S interacts with Fe via second coordination. The subordinated coordination of S is also certified by the distinct pre-edge peak. Compared with FePc with square-planar FeN4, Fe1-NSC shows a left-shifted pre-edge peak, delivering a broken D4h symmetry of FeN4 (Fig. S12 in Supporting information) [10]. These, coupled with C-S-N(C) groups monitored from S 2p XPS spectra (Fig. S13 in Supporting information), demonstrate that the Fe configuration in Fe1-NSC is built up from four N atoms in the first shell and two S atoms in the second shell. Accordingly, the FeN4S2 model dominated in the Fe1-NSC is theoretically constructed (Fig. S14 in Supporting information). The most stable one with the lowest free energy is found to show close structure parameters of experimental characterizations (Table S3 in Supporting information).
The spin states of Fe are investigated and the 57Fe Mössbauer spectra for both Fe-based SACs can be only fitted with MS Fe3+, HS Fe3+ and HS Fe2+ (Figs. 1e and f). HS Fe3+ is the dominant Fe species in Fe1-NC, while MS Fe3+ is the main Fe species for Fe1-NSC according to the quantitative analyses (Table S4 in Supporting information). The content of MS Fe3+ reaches 61% in Fe1-NSC at the expense of the original HS Fe3+, suggesting that the Fe1-NC experiences a spin crossover from HS to MS configuration after S introduction (Fig. S15 in Supporting information). Furthermore, the zero-field cooling temperature-dependent magnetization tests are conducted. The magnetic moment (μeff) for Fe1-NC and Fe1-NSC is simulated to be ca. 6.16 and 3.43 μB, respectively (Fig. 1g). The unpaired electrons (n) are calculated to be 5.2 for Fe1-NC and 2.6 for Fe1-NSC. It indicates that a specific electron spin transition from HS to MS Fe3+ occurs after S-modification. The spin conversion may permit Fe species of Fe1-NSC to achieve an ideal d-electron configuration and have strong adsorption toward PMS [11].
The Fe-based SACs for PMS activation are assessed by p-nitrophenol (4-NP) degradation. PMS and catalysts alone cannot degrade 4-NP (Fig. 2a and Fig. S16 in Supporting information). Notably, complete 4-NP degradation is achieved within 10 min in Fe1-NSC/PMS system (Fig. 2a). However, low 4-NP removal is observed in NSC/PMS and Fe1-NC/PMS systems. The Fe1-NSC/PMS delivers an even higher removal rate and kinetic constant (k4-NP) of 4-NP than the landmark Fe2+/PMS, Co2+/PMS, Co3O4/PMS and Fe2O3/PMS (Fig. S17 in Supporting information). Besides, Fe1-NSC can maintain stable and excellent catalytic ability not only in the presence of wide pH and various anions but also for the 4-NP degradation in tap water and natural water relative to the Fe1-NC (Figs. S18-S21 in Supporting information). The used Fe1-NSC shows XRD, TEM and HAADF-STEM images similar to the fresh one and without metal leaching, inferring its high stability (Fig. S22 in Supporting information). Compared with Fe1-NC/PMS, Fe1-NSC/PMS system also exhibits remarkable degradation performance toward broad emerging pollutants with removal ratios > 98% and high reaction kinetics (Figs. S23 and S24 in Supporting information). These results highlight the admirable regulatory capability of S on Fe single sites for boosting PMS activation and wastewater treatment.
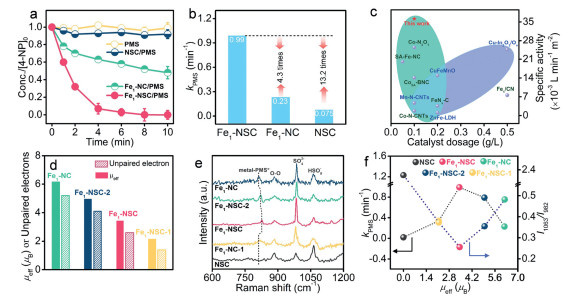
The major reactive species during the PMS activation process are identified. Both TEMP-1O2 and DMPO-•O2- are recorded over Fe1-NSC/PMS, differing from that of Fe1-NC/PMS with only TEMP-1O2 detected (Fig. S25 in Supporting information). Notably, the intensity of DMPO-•O2- and TEMP-1O2 are markedly reduced when p-BQ is added. This suggests that the part of 1O2 in Fe1-NSC/PMS is derived from the dismutation of •O2-. No signals for DMPO-•SO4- and DMPO-•OH or FeⅣ species are detected (Fig. S26 in Supporting information). These results clarify the predominant role of •O2- in the Fe1-NSC/PMS system for pollutant degradation.
PMS concentration in both single-Fe-atom systems declines along with the pollutant degradation, validating the surface chemical evolution of PMS rather than adsorption (Fig. S27 in Supporting information). The kinetic constant (kPMS) of PMS decomposition over Fe1-NSC is calculated to be 0.99 min-1, 4.3 and 13.2 times that of Fe1-NC (0.23 min-1) and NSC (0.075 min-1) (Fig. 2b). Besides, Fe1-NSC shows improved specific activity (36.1 × 10–3 L min-1 m-2) [12], 4.2 and 12.6 times of that Fe1-NC (8.61 × 10–3 L min-1 m-2) and NSC (2.87 × 10–3 L min-1 m-2), respectively, highlighting the superiority of S modification on Fe sites for PMS activation. Remarkably, the specific activity of Fe1-NSC surpasses that of the cutting-edge catalysts reported to date [13–20], corroborating that Fe1-NSC is among the ranks of excellent PMS activators (Fig. 2c).
To explore the origin of the enhanced PMS activation over Fe1-NSC, the effect of spin state on PMS activation is analyzed. The degradation experiments indicate that Fe3+ rather than Fe2+ governs the PMS activation since Fe1-NSC and Fe1-NC have comparable amounts of Fe2+, yet the former shows better PMS activation efficiency than the latter. To further identify the dominant role of Fe3+, other S-modified Fe-based SACs (Fe1-NSC-1 and Fe1-NSC-2) with varied μeff are prepared by altering the Fe/S molar ratio (Fig. 2d and Fig. S28 in Supporting information). A volcano curve of PMS activation kinetics is observed with the increase of μeff and both Fe1-NC and Fe1-NSC-1 display low PMS activation rates (Fig. S29 in Supporting information). This may be explained that Fe species with too high or too low eg orbital occupation are unfavorable for PMS adsorption. Previous studies infer that the semi-filled antibonding orbitals are easy to approach substrates, and the single electron in antibonding orbitals can penetrate the Π* and σ orbitals of oxygenated species [21]. These deduce that partly occupied eg orbitals may be conducive to PMS adsorption. To ascertain it, in situ Raman spectra are conducted to analyze the PMS adsorption (Fig. 2e). Three peaks at 1063, 982 and 884 cm-1 are observed in PMS solution and assigned to free HSO5-, SO42- and O—O groups, respectively [22]. A new peak at 829 cm-1 belonging to metal-PMS* is detected when Fe-based SACs are added to the PMS solution [23]. Note that the metal-PMS* peaks for S-modified SACs systems show evident red-shift from 812 cm-1 to 826 cm-1 as the decrease of μeff, suggesting the reinforced interaction between PMS and Fe sites. When the μeff further decreased, the red-shift phenomenon gradually disappeared. Moreover, the ratio of I1063/I982 presents an inverted volcanic shape with the reduction of μeff (Fig. 2f). The minimum value of I1063/I982 is gained for the Fe1-NSC/PMS system, confirming that the Fe1-NSC facilitates optimal PMS adsorption and rapid decomposition.
Furthermore, the orbital electronic occupancy of Fe1-NSC is theoretically investigated. The density of state (DOS) plots for Fe 3d orbital display that the bonding t2g orbital of Fe1-NSC is occupied by more spin-down electrons than that of Fe1-NC (Fig. S30 in Supporting information). The partial DOS plots of Fe1-NSC exhibit a pronounced disparity in electron density within its Fe t2g orbital, particularly in the dyz and dxy orbitals, relative to that of Fe1-NC (Fig. S31 in Supporting information). These results infer that the spin polarization of Fe 3d orbital electrons in the Fe1-NSC has been weakened. To gain insight into it, the centers of Fe 3d and N 2p are determined by integrating the partial DOS (Fig. 3a). The energy difference between the N 2p and Fe 3d centers in Fe1-NSC is larger than that in Fe1-NC, suggesting a reduction of Fe-N covalency in Fe1-NSC. These infer that the Fe sites with more positive charges in Fe1-NSC are more capable of exchanging electrons with PMS. Additionally, we estimate the maximum valence band energies (EVB) of Fe1-NSC and Fe1-NC to be around 1.43 and 1.71 eV, respectively (Fig. 3a), indicating that the valence band of Fe1-NSC is closer to the Fermi level (Ef) than that of Fe1-NC. Since the valence electrons are contributed from d orbital, it suggests Fe1-NSC has lower d-band energy than Fe1-NC, thus beneficial for better PMS adsorption and boosted activation efficiency.
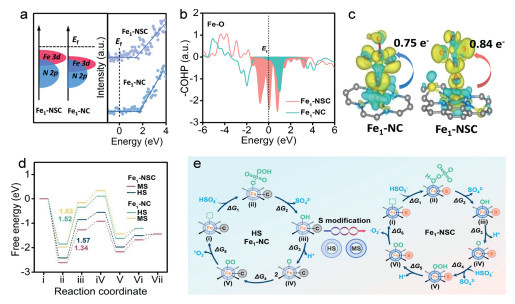
The PMS adsorption models guided by the Fe spin states have been established and optimized. Although PMS is adsorbed on Fe sites through the formation of Fe-O bonds in all models, PMS is preferentially chemisorbed on the Fe site in the Fe1-NSC system with the O atom (O1) at the -OH side, in contrast to the Fe1-NC system where the PMS has a higher affinity for adsorption on the Fe site with the O atom (O2) at the -SO4 side (Fig. S32 and Table S5 in Supporting information). The observed variation in adsorption energy (Ead.PMS) also supports the dissimilar adsorption configurations of the optimized models and Ead.PMS follows the order of Fe1-NSC (545.0 kJ/mol) > Fe1-NC (255.2 kJ/mol) > NSC (69.3 kJ/mol). The structural distortion of the bound PMS on catalysts increases with the increase of Ead.PMS, and the O—O bond is lengthened from 1.49 Å on Fe1-NC to 1.62 Å on Fe1-NSC system, which will be more conducive to PMS activation. Besides, the length of the Fe-O bond formed on the Fe1-NC catalyst is measured to be 1.96 Å, while it is reduced to 1.91 Å on the Fe1-NSC system. Previous studies reveal that the Fe-O bond can exhibit mixed ionic-covalent properties that play a key role in determining catalytic activity [24]. To validate this, the crystal orbital Hamiltonian (COHP) is calculated to explore the bonding characteristics between catalyst and PMS (Fig. 3b). Unlike the bonding contribution of HS Fe3+ in Fe1-NC, a stronger antibonding state appears at the Fermi level in MS Fe3+ of Fe1-NSC, confirming that electrons in the O 2p orbital in PMS are more easily transferred to MS Fe3+ 3d orbital. Besides, the Bader charge analyses exhibit a charge transfer of 0.84 e– from the adsorbed PMS to the Fe species in the Fe1-NSC system, while it is only 0.75 e– for the Fe1-NC system (Fig. 3c). These results indicate that the spin crossover effect indeed manipulates the activation of PMS molecules by single-Fe-atom catalysts.
To thermodynamically elucidate the effect of spin state on PMS activation, the key intermediates on Fe1-NC and Fe1-NSC are proposed (Fig. S33 in Supporting information) [25]. The distinction between these two catalysts can be attributed to the reversed spin configurations of Fe sites after S modification. The free energy differences of the isomeric reactants and rate-limiting barrier are modulated owing to the spin-dependent orbital hybridization (Fig. 3d). The PMS activation occurs through the high-spin pathway in Fe1-NC system because the O—O bond fracture of PMS on the HS Fe site (step ii) is the rate-determining step (1.52 eV), which has slightly lower energy than that of the MS intermediates (1.82 eV). After S modification, the determining activation energy at MS Fe sites in Fe1-NSC (1.34 eV) is lower than that of the Fe1-NC. Importantly, the differences in relative stability between HS and MS configuration at the rate-limiting step are enlarged due to a stronger spin-orbital interaction. This selectivity of the reaction pathway in MS configuration plays a critical role in determining PMS activation performance (Fig. 3e).
The S-modification strategy to regulate the local spin state is also applicable to other SACs (M1-NSC, M = Mn, Co, Cu). M1-NSC are prepared following procedures similar to Fe1-NSC and well characterized (Fig. S34 in Supporting information). The μeff and the unpaired electrons (n) for M1-NSC are calculated and lower than those of counterparts (Fig. S35 in Supporting information). The resultant M1-NSC exhibits high 4-NP degradation rates (Fig. S36 in Supporting information). Especially, the kPMS over M1-NSC show > 3 times higher than those counterparts. These results demonstrate the versatility of S-modification in modulating the spin state for enhancing PMS activation.
In summary, an S-modified Fe-based SACs is prepared. The S atom with a similar atomic radius to N but with lower electronegativity makes the spin state of the nearby Fe site reconfigured to form MS Fe3+. Due to the unique features of the unoccupied eg orbitals in MS Fe3+, the dissociation kinetics of PMS are reinforced via the enhancement of Fe 3d and O 2p, thus boosting the PMS activation and organic pollutant degradation performance. This work provides a universal scheme for the spin-state regulation of SACs toward Fenton-like applications and highlights the great potential of heteroatom doping in the rational design of highly efficient heterogeneous catalysts.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Lina Zou: Investigation, Formal analysis, Data curation. Dengke Wang: Writing – review & editing, Supervision, Project administration, Funding acquisition, Conceptualization. Shiqin Lai: Methodology, Investigation, Formal analysis. Xunheng Jiang: Software, Methodology. Siqi Chen: Software, Methodology, Investigation. Lanqing Deng: Methodology, Investigation, Data curation. Dong Fan: Software, Investigation. Hengshuai Li: Supervision, Software, Methodology. Zhigang Zhou: Visualization, Investigation. Denglong Chen: Resources, Methodology. Xiangyang Yao: Visualization, Investigation. Jianping Zou: Supervision, Funding acquisition, Conceptualization.
We gratefully acknowledge the financial support of the National Natural Science Foundation of China (Nos. 52260006, 52000097, 52170082 and 51938007), and the Natural Science Foundation of Jiangxi Province (Nos. 20242BAB23049, 20243BCE51075 and 20212ACB203008).
Supplementary material associated with this article can be found, in the online version, at doi:
X.J. Hu, D.X. Zhou, H. Wang, et al., Chin. Chem. Lett. 34 (2023) 108050. doi: 10.1016/j.cclet.2022.108050
Y.J. Zhang, S.Y. Liu, D.D. Chen, X. Xu, Chin. Chem. Lett. 35 (2024) 108666. doi: 10.1016/j.cclet.2023.108666
H.C. Yan, C. Lai, S.Y. Liu, et al., Water Res. 234 (2023) 119808. doi: 10.1016/j.watres.2023.119808
Y. Wu, P.F. Wang, H.N. Che, et al., Angew. Chem. Int. Ed. 136 (2024) e202316410. doi: 10.1002/ange.202316410
L.F. Cui, P.F. Wang, H.N. Che, et al., Water Res. 244 (2023) 120514. doi: 10.1016/j.watres.2023.120514
C. Xue, P.F. Wang, H.N. Che, et al., Appl. Catal. B: Environ. 340 (2024) 123259. doi: 10.1016/j.apcatb.2023.123259
K.X. Song, B.B. Yang, X. Zou, W. Zhang, W.T. Zheng, Energy Environ. Sci. 17 (2024) 27–48. doi: 10.1039/d3ee02644b
Y. Wu, Z.W. Zhuang, C. Chen, et al., Chem Catal. 3 (2023) 100586.
Y. Gao, B.Z. Liu, D.S. Wang, Adv. Mater. 35 (2023) 2209654. doi: 10.1002/adma.202209654
Y.F. Li, D.L. Fang, X.L. Li, et al., Energy Environ. Mater. 6 (2023) e12449. doi: 10.1002/eem2.12449
V.H. Do, J.M. Lee, ACS Nano 16 (2022) 17847–17890. doi: 10.1021/acsnano.2c08919
D.K. Wang, M.J. Suo, S.Q. Lai, et al., Appl. Catal. B: Environ. 321 (2023) 122054. doi: 10.1016/j.apcatb.2022.122054
Z.W. Wang, E. Almatrafi, H. Wang, et al., Angew. Chem. Int. Ed. 61 (2022) e202202338. doi: 10.1002/anie.202202338
Y.W. Gao, Y. Zhu, T. Li, et al., Environ. Sci. Technol. 55 (2021) 8318–8328. doi: 10.1021/acs.est.1c01131
B.F. Zhang, X.Q. Li, K. Akiyama, P.A. Bingham, S. Kubuki, Environ. Sci. Technol. 56 (2022) 1321–1330. doi: 10.1021/acs.est.1c05980
J. Miao, Y. Zhu, J.Y. Lang, et al., ACS Catal. 11 (2021) 9569–9577. doi: 10.1021/acscatal.1c02031
M.J. Huang, Y.S. Li, C.Q. Zhang, et al., Proc. Natl. Acad. Sci. U. S. A. 119 (2022) e2202682119. doi: 10.1073/pnas.2202682119
Y. Bao, C. Lian, K. Huang, et al., Angew. Chem. Int. Ed. 61 (2022) e202209542. doi: 10.1002/anie.202209542
Z.Y. Zhao, P.F. Wang, C.L. Song, et al., Chem. Int. Ed. 62 (2023) e202216403. doi: 10.1002/anie.202216403
J.S. Song, N.N. Hou, X.C. Liu, et al., Adv. Mater. 35 (2023) 2209552. doi: 10.1002/adma.202209552
Q.Y. Wu, Z.W. Yang, Z.W. Wang, W.L. Wang, Proc. Natl. Acad. Sci. U. S. A. 120 (2023) e2219923120. doi: 10.1073/pnas.2219923120
J.J. Yang, J. Pignatello, C. Yang, et al., Appl. Catal. B: Environ. Energy 347 (2024) 123793. doi: 10.1016/j.apcatb.2024.123793
S. Li, X.R. Liu, Y.J. Zheng et al., Chin. Chem. Lett. 35 (2024) 108971. doi: 10.1016/j.cclet.2023.108971
G. Zhou, P.F. Wang, H. Li, et al., Nat. Commun. 12 (2021) 4827. doi: 10.1038/s41467-021-25095-4
D.K. Wang, S.Q. Chen, S.Q. Lai, et al., Chin. Chem. Lett. 34 (2023) 107861. doi: 10.1016/j.cclet.2022.107861

Figure 1 (a) Preparation procedure. (b) Fe K-edge XANES spectra. (c) FT-EXAFS spectra. (d) EXAFS fitting curves. (e, f) 57Fe Mössbauer spectra and (g) temperature-dependent magnetic susceptibilities of Fe1-NSC and Fe1-NC.

Figure 2 (a) Degradation performance of 4-NP. (b) The kinetic constant of PMS decomposition. (c) The comparisons of specific activity. (d) The magnetic moments. (e) In-situ Raman spectra of PMS adsorption. (f) The kinetics of PMS consumption and the ratio of I1063/I982 in Raman spectra over different systems.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
