Scheme 1.
Alkenyl chlorides and unsaturated 1, n-dihalides.
Deoxygenative hydrohalogenation of propargyl alcohols: Regio- and stereoselective synthesis of unsaturated distal dihalides
Zhihua Wang , Xiang-Zhao Zhu , Xinglei He , Chen-Xu Gong , Wang-Fu Liang , Wenfeng Wang , Yuqi Lin , Ke-Yin Ye

Organohalides comprise the key ingredients of numerous pharmaceuticals on the market [1,2], more than five thousand natural products [3], and are also widely used as intermediates in organic synthesis [4]. Compared with the C(sp3)-hybridized alkyl halides, the C(sp2)-hybridized alkenyl halides are less reactive but still serve as competent building blocks in diverse organic transformations [5–7]. For instance, the most widely use of vinyl chloride is its polymerization for the production of polyvinyl chloride (PVC) in vast quantities, which is second only to that of polyethylene [8]. The recent development of transition-metal catalysis renders the previously challenging alkenyl chlorides comparable partners in various types of cross-coupling reactions, along with their bromo and iodo derivatives [9–11]. Among them, polychlorinated compounds with one or more alkenyl halide scaffolds are also found in various biologically active natural products (Scheme 1A) [12,13].
There already exist well-established synthetic approaches for the facile preparation of diverse monoalkenyl chlorides in literature (Scheme 1B). These approaches include the chlorination of carbonyls or alkenyl precursors with toxic and difficult-to-handle phosphorous chlorides [14], the addition of corrosive HCl [15] or its surrogate to alkynes [16,17], olefin metathesis [18], and so on [19–21]. Unlike the well-defined stereochemistry of cyclic alkenyl chlorides, the E/Z isomers of linear alkenyl chlorides are occasionally obtained with unsatisfactory stereoselectivity [22]. In addition, there still lacks a general approach to access the analogous bisalkenyl halides of synthetic significance, likely owing to the challenges in the control of regioselectivity, E/Z stereochemistry, and nonselective formation of either conjugated or distal bisalkenyl halides.
We herein report a deoxygenative hydrohalogenation of the versatile and readily available propargyl alcohols [23,24] for the regio- and stereoselective preparation of alkenyl halides (Scheme 1C). This approach features high synthetic fidelity toward not only the stereospecifically Z-configurated alkenyl halides but also the synthetically challenging homologous 1,3- [25], 1,4- [26], and 1,5-dichlorides [27], as well as their dibromides and diiodides derivatives. The commercially available halodimethylsilane (HSiMe2X), or a surrogate combination of hydrosilane (Et3SiH) and halosilane (Me3SiX), readily serves both as the hydrogen and halogen sources. Preliminary mechanistic investigations suggest the regio- and stereoselective stepwise hydrogenation of in situ generated chloroallenes is the key step [28].
In the presence of 5 mol% of Co(salen) A catalyst and 3 equiv. chlorodimethylsilane (HSiMe2Cl) [29], propargyl alcohol (1a) was transformed into the target alkenyl chloride (2a) in 90% isolated yield (Table 1, entry 1). Notably, the chlorine atom was regioselectively installed at the remote carbon of the triple bond likely via an SN2′ manner. The stereoselectivity is also remarkable as only the Z-configurated alkenyl chloride was formed (Z/E > 20:1). This reaction was non-sensitive to air and moisture and thus inert atmosphere protection was unnecessary. Without a catalyst, it still proceeded with the same reactivity but at a much slower rate and with a lower conversion (entry 2). When Co(OAc)2 or CoCl2 was used as the catalysts, the conversion decreased (entry 3). Variations on the catalysts, including the electronic substituents and backbones of the supporting salen ligand (entry 4), various transition metals (Co, Ni, Cu, and Mn) in their different valences (+2 and +3), did not lead to higher yields (entries 5–8). By contrast, phosphine ligands were ineffective in this transformation (see Supporting information). Regarding the hydrochlorinating agents, both chlorodiphenylsilane (HSiPh2Cl) and trichlorosilane (HSiCl3) were not as effective as HSiMe2Cl (entries 9 and 10). Other inorganic chlorinating agents, such as KCl and NaCl, were not applicable (see Supporting information). Interestingly, the combination of Et3SiH and Me3SiX also afforded the target Z-alkenyl chloride but in a slightly lower yield (entry 11). Finally, this reaction was found to be sensitive to solvents as only dichloromethane, dichloroethane, and acetonitrile were tolerated among many solvents examined (entries 12–14).
 DownLoad:
CSV
DownLoad:
CSV
 |
||
| Entry | Deviation from standard conditions | Yield (%)b |
| 1 | — | 95 (90)c |
| 2 | Without Co(salen) | 50 |
| 3 | Co(OAc)2 or CoCl2 | 68, 57 |
| 4 | Co(salen) B-D | 52–92 |
| 5 | Ni(salen) E | 21 |
| 6 | Cu(salen) F | 81 |
| 7 | Co(salen)Cl G | 89 |
| 8 | Mn(salen)Cl H | 93 |
| 9 | HSiPh2Cl | 21 |
| 10 | HSiCl3 | n.d. |
| 11 | Et3SiH + Me3SiCl | 90 |
| 12 | MeCN | 87 |
| 13 | DCM | 85 |
| 14 | DMSO, DMF, THF, Dioxane, Toluene | < 5 |
| a Reaction conditions: 1a (0.2 mmol, 1.0 equiv.), HSiMe 2Cl (0.6 mmol, 3.0 equiv.), Co(salen) A (0.01 mmol, 5 mol%), DCE (0.1 mol/L), 50 ℃, 3 h. b Yield determined by 1H NMR analysis using CH 2Br 2 as the internal standard. c Isolated yield. n.d. = not detected. |
||
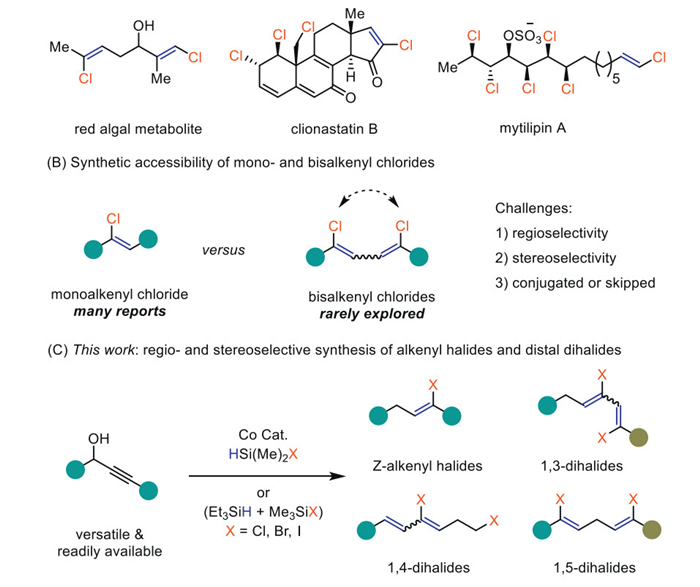
The substrate scope of this deoxygenative hydrochlorination of propargyl alcohols was then investigated (Scheme 2). Triphenyl propargyl alcohol afforded the target Z-alkenyl chloride (2b) in 98% yield whose stereochemistry was confirmed by its X-ray diffraction analysis. The high synthetic fidelity of this protocol provided easy access to diverse Z-alkenyl chlorides with an installment of a particular substituent in a specific position, as exemplified by the facile preparation of two thiophene derivatives (2c, 2d). Note that tert–butyl could be survived (2e). Propargyl alcohols containing parts of natural products and pharmaceuticals, such as DL-menthol ester (2f), ibuprofenamide (2g), epiandrosterone (2h), and icaridin ester (2i) were all readily transformed into the anticipated Z-alkenyl chlorides in good to excellent yields (Scheme 2A).
Despite the recent advances in the vicinal dichlorination of alkenes [30–34], straightforward and modular access to the analogously remote dichlorinated scaffold remains elusive. To further explore the synthetic potentials, we then investigated the possibility of applying the current approach for the ready preparation of diverse unsaturated remote dichlorides. Interestingly, penta-2,4-diyn-1-ols (3), propargyl alcohols with an extra conjugated alkyne moiety were readily converted into bisalkenyl chlorides s-cis-(1Z, 3Z)−4 in the presence of 5 mol% of Co(salen) D (Scheme 2B). Remarkably, the 1,3-diene scaffolds were selectively generated in an s-cis form (up to 13:1), as substantiated both by the crude nuclear magnetic resonance spectra and X-ray diffraction analysis of compound 4k. Density-function-theory (DFT) calculations suggested that two chlorine atoms located on the opposite side are more stable owing to the intermolecular and intramolecular H···Cl hydrogen bonds (Fig. S14 in Supporting information). This finding is remarkable because dienes in the Dies-Alder reaction need to adopt an s-cis configuration before an effective [4 + 2] cycloaddition [35]. Unfortunately, attempts to utilize these bisalkenyl chlorides in the Dies-Alder reaction were not successful yet.
The homologous bisalkenyl chlorides 1Z, 4Z-6 were also readily accessed by using penta-1,4-diyn-3-ols (5) as the substrates featuring two skipped alkyne moieties (Scheme 2C). Accordingly, a wide array of symmetric (6a-6h and 6m-6o) and nonsymmetric (6i-6l and 6p) bisalkenyl chlorides were formed in good to excellent yields in the presence of 1 mol% of Co(salen) A. Consistent with the formation of monoalkenyl chlorides (2), both the alkene moieties of 6 were generated in high Z-selectivity.
In addition, the X-ray diffraction analysis of the naphthalene derivative (6r) confirmed the formation of a skipped diene moiety. Surprisingly, cyclohexyl-substituted penta-1,4-diyn-3-ol only afforded the anticipated 6s as the minor product. Instead, its conjugated triene derivative (6t) was formed as the major product in 62% yield.
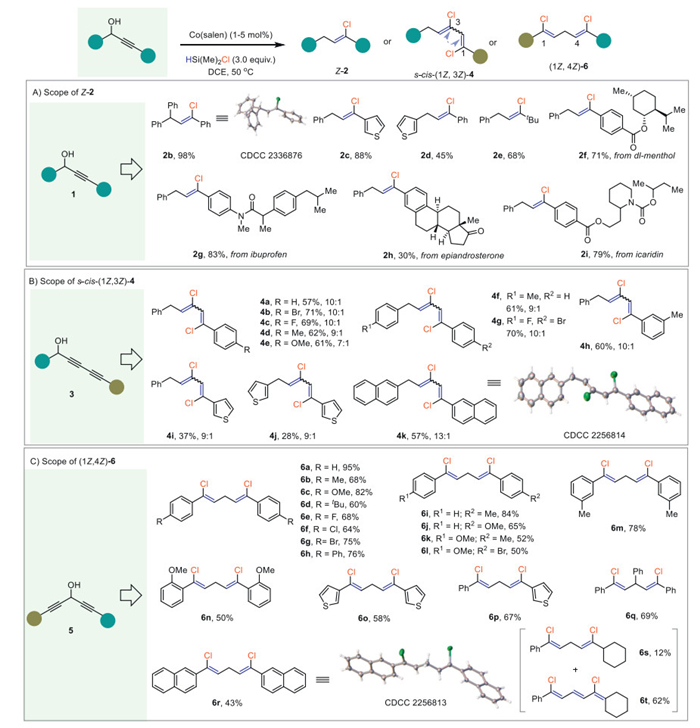
Interestingly, 3-cyclopropyl–prop-2-yn-1-ols (7) exhibited a distinct reaction pathway to give 1E, 3Z-bisalkenyl chlorides (8) where one chloride links to an sp2 hybridized carbon and the other to an sp3 hybridized terminal carbon (Scheme 3). This reaction proceeded smoothly even without a catalyst and the more economical TMSCl can also be used instead of HSiMe2Cl albeit in slightly lower yields. By contrast to s-cis-(1Z, 3Z)−4, the conjugated diene moiety in these bisalkenyl chlorides is in an s-trans form (up to 10:1), in which the Z-alkenyl chloride was still maintained. The mild reaction conditions further allowed the survival of conjugated alkene (8f) and alkyne (8g) moieties. In addition, linear- (8h) and cyclic aliphatic chains (8i-8k), as well as thiophene (8l) substituted derivatives were all well tolerated. The rapid access to the molecular complexity of this protocol was further demonstrated by the convenient installation of four C‒Cl bonds (8m) in one simple operation. A gram-scale preparation of the 1,4-dichloride (8a, 76%, 1.04 g) was also applicable (see Supporting information).
We continued to explore the preparation of the heavy congeners of unsaturated distal dichlorides, i.e., dibromides and diiodides (Scheme 4). Though HMe2SiBr and HMe2SiI are not commercially available, the adoption of their surrogate combination of Et3SiH and Me3SiX proved to be successful.

Compared with 1Z, 3Z-bisalkenyl chlorides (4), the s-cis selectivity for dibrominated (9a) and diiodinated (9b) reactions were relatively lower, i.e., 5:1 and 3:1, respectively. While the dibrominated (1Z, 3Z)−10a was formed as expected, the diiodination reaction further proceeded to partial hydrogenation to afford the Z-configurated alkenyl iodide (10b). Finally, (1Z, 4Z)-bisalkenyl bromide (11a) and iodide (11b) were readily accessible under the established reaction conditions. Unfortunately, the current protocol is not applicable for the preparation of alkenyl fluorides [36,37] because it requires the use of Me3SiF as the reagent whose strong Si‒F bond (bond energy = 565 kJ/mol) [38] hampers its reactivity in the current transformation.
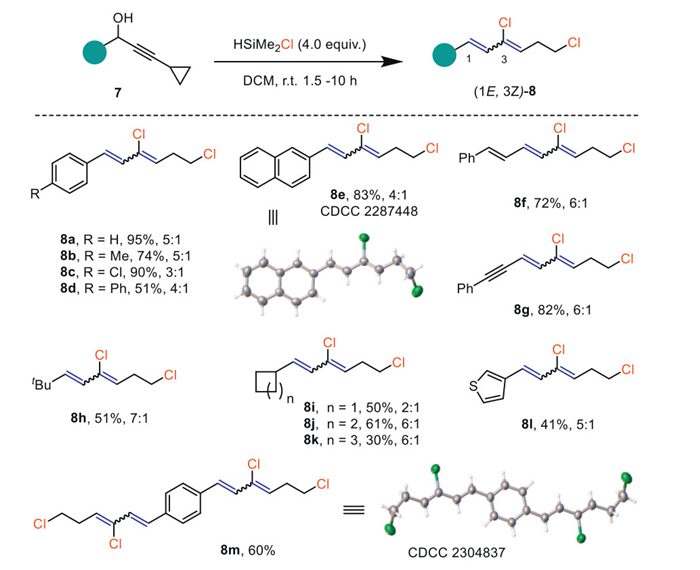
The synthetic utility of the obtained unsaturated distal 1, n-dichlorides was further investigated (Scheme 5). For instance, compound 4a underwent a facile oxidative Cl-migration with the Koser reagent (hydroxy(tosyloxy)iodobenzene, HTIB) [39] to afford the α-chlorinated enone (12), which was readily transformed into an isoxazole (13) upon treatment with NH2OH·HCl/KOH (Scheme 5A). For compound 6a, the superstoichiometric Koser reagent enabled both the skipped alkenyl halide moieties to proceed with double oxidative Cl-migrations (14), whose structure was unambiguously confirmed by X-ray diffraction analysis. The dichlorinated 1,5–dione (14) underwent reduction with NaBH4 into α-chlorohydrins (15) or reacted with thioacetamide to form a bisthiazole (16, Scheme 5B). Bithiazole heterocyclic derivatives are scaffolds of research interest with diverse therapeutic properties such as analgesic and antimicrobial [40,41]. In addition, the bisalkenyl chloride (6a) can also be employed in the Pd-catalyzed C‒C cross-coupling reaction (17) [42]. Remarkably, starting from propargyl alcohol (18) with three skipped phenylacetylenes, the corresponding skipped trialkenyl halides derivative (19) was conveniently prepared in 17% yield that is otherwise very tedious and challenging to achieve by other means (Scheme 5C).
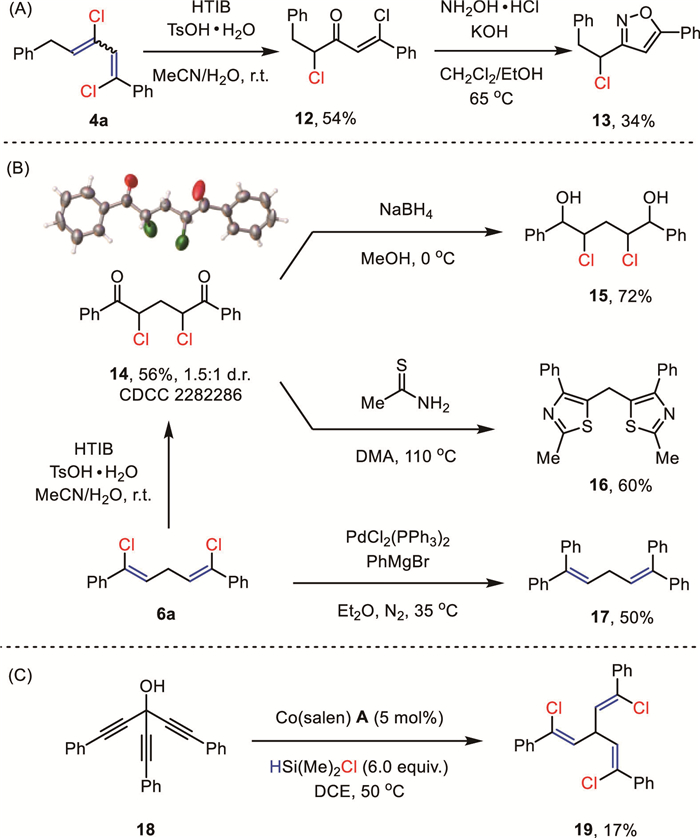
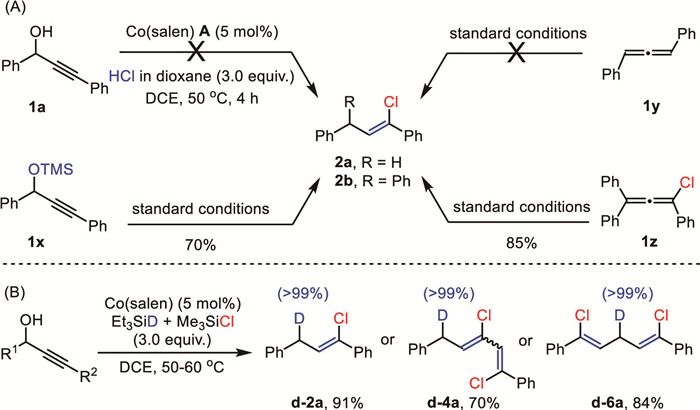
A couple of mechanistic experiments were carried out to shed light on the reaction mechanism (Scheme 6). Replacement of HMe2SiCl by HCl (c 1.0 mol/L in dioxane) failed to replicate the anticipated reactivity, indicating that HMe2SiCl is not just a simple HCl carrier (Scheme 6A). The preformed TMS-protected propargyl alcohol (1x) was a competent substrate, suggesting its involvement in this deoxygenative hydrochlorination reaction. In addition, 1,3-diphenyl allene (1y) only proceeded via partial hydrogenation to give the corresponding alkene under standard reaction conditions, while chloroallene (1z) did afford the anticipated alkenyl chloride (2b) in 85% yield. Thus, both TMS-protected propargyl alcohol (1x) and chloroallene (1z) were conceived as possible reaction intermediates. As illustrated in the isotopic labeling experiment in Scheme 6B, using Et3SiD/Me3SiCl, exclusive deuterium incorporation was found to be uniformly located at the original propargyl positions for the monoalkenyl halide (d-2a), 1,3-dihalide (d-4a), and 1,5-dihalide (d-6a). Regarding the cobalt catalyst, we found the brownish solution of Co(salen) in dichloroethane rapidly changed to deep green upon adding TMSCl, and then into pale green with the extra Et3SiH. Consequently, cyclic voltammetry studies revealed that the reversible redox couples of Co(salen) turned into irreversible oxidation peaks at 1.1 V and 1.5 V, respectively (Fig. 1A). These redox behaviors were distinct from Co(salen)‒Cl though it is readily formed in the presence of chloride in the air. Attempts to crystallize these newly formed species were unsuccessful yet.
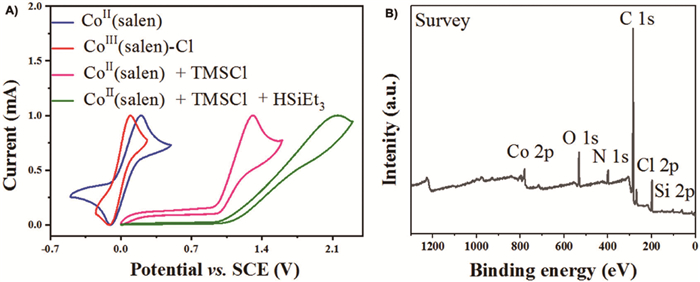
Nevertheless, the X-ray photoelectron spectroscopy (XPS) survey spectrum confirmed the existence of all elements from Co(salen) (Co, C, O, and N) and TMSCl (Si and Cl) (Fig. 1B).
Based on these experimental observations, a mechanistic rationale was proposed (Scheme 7). The propargyl alcohol (1a) is first activated by HMe2SiCl into its TMS-protected derivative (Ⅱ), which was confirmed by nuclear magnetic resonance spectroscopy. It is then transformed into chloroallene (Ⅳ) [43–45] probably via a cobalt-promoted formal SN2′ type nucleophilic chlorination (Ⅲ) [46,47] wherein the byproduct HSiMe2OH was detected by GC–MS. The innate charge dispersion of chloroallene (Ⅳ) [48] resulted in the highly regioselective protonation to form the allyl cation intermediates (Ⅴ). According to our DFT calculations (see Supporting information), the resultant cis-allyl cation (cis-Ⅴ) was 1.9 kcal/mol more stable than its trans-isomer (trans-Ⅴ), which accounts for the excellent stereoselectivity of this reaction. Finally, the hydride transfer reaction from penta-coordinated silane species to the cis-Ⅴ led to the final vinyl chloride product. This mechanistic rationale is reminiscent of the stepwise hydrogenation [49] of chloroallene (Ⅲ) with proton and hydride [50] and consistent with our deuterium experiments. The other possible reaction pathways were also considered but excluded. For instance, the reaction mechanism initiated from the hydride attack to chloroallene (Ⅳ) is disfavored because DFT calculations suggest this should result in an unstable vinyl anion that rapidly eliminates the chloride to an alkyne (see Supporting information).
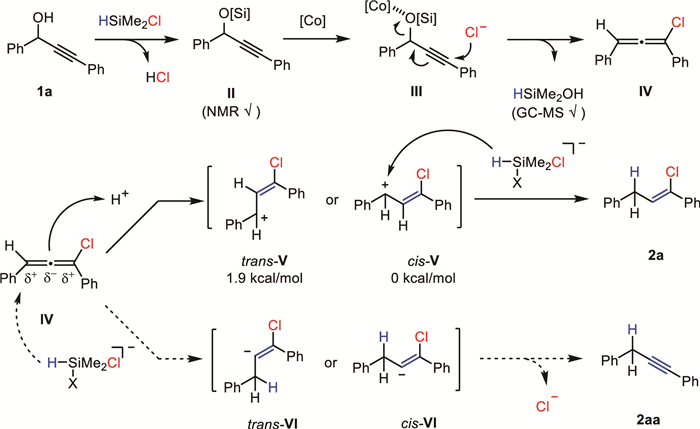
An alternative reaction pathway for the hydrogenation of chloroallene (Ⅴ) involving CoⅢ-H mediated hydrogen-atom transfer [51], however, favors the initial addition of hydrogen at the C2 position of allene [52,53], which is unlikely as substantiated by the deuterium experiment. We have also examined the possibilities of the mechanistically analog deoxygenative cyanation with TMSCN and azidation with TMSN3, which are unfortunately not successful (see Supporting information).
In summary, we have developed a unified deoxygenative hydrohalogenation of propargyl alcohols toward the facile preparation of various Z-alkenyl halides and unsaturated remote 1, n-dihalides with good regio- and stereoselectivity. This protocol utilizes halodimethylsilane, or the surrogate combination Et3SiH and Me3SiX, as the hydrogen and halogen sources. Access toward synthetically challenging 1,3-, 1,4- and 1,5-dibromides and diiodides is also readily achieved by using appropriate propargyl alcohol derivatives. The obtained alkenyl halides have been demonstrated to be useful and versatile synthons in diverse organic transformations. We anticipate this study should inspire and promote the development of modular and versatile syntheses of remotely functionalized and value-added halogenated molecules of complexity.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Zhihua Wang: Methodology, Data curation, Conceptualization. Xiang-Zhao Zhu: Data curation. Xinglei He: Data curation. Chen-Xu Gong: Data curation. Wang-Fu Liang: Data curation. Wenfeng Wang: Data curation. Yuqi Lin: Data curation. Ke-Yin Ye: Writing – review & editing, Writing – original draft.
Financial support from the National Natural Science Foundation of China (No. 22171046), the Hundred-Talent Project of Fujian (No. 50021113), and the Open Research Fund of School of Chemistry and Chemical Engineering, Henan Normal University (No. 2024Z08) is gratefully acknowledged.
Supplementary material associated with this article can be found, in the online version, at doi:
P.S. Charifson, W.P. Walters, J. Med. Chem. 57 (2014) 9701–9717. doi: 10.1021/jm501000a
W.Y. Fang, L. Ravindar, K.P. Rakesh, et al., Eur. J. Med. Chem. 173 (2019) 117–153. doi: 10.1016/j.ejmech.2019.03.063
G.W. Gribble, Environ. Chem. 123 (2015) 96–405.
W.J. Chung, C.D. Vanderwal, Angew. Chem. Int. Ed. 55 (2016) 4396–4434. doi: 10.1002/anie.201506388
N. Miyaura, A. Suzuki, Chem. Rev. 95 (1995) 2457–2483. doi: 10.1021/cr00039a007
C.C. Johansson Seechurn, M.O. Kitching, T.J. Colacot, V. Snieckus, Angew. Chem. Int. Ed. 51 (2012) 5062–5085. doi: 10.1002/anie.201107017
P. Ruiz-Castillo, S.L. Buchwald, Chem. Rev. 116 (2016) 12564–12649. doi: 10.1021/acs.chemrev.6b00512
C.M.R. Abreu, A.C. Fonseca, N.M.P. Rocha, et al., Prog. Poly. Sci. 87 (2018) 34–69. doi: 10.1016/j.progpolymsci.2018.06.007
A.F. Littke, C. Dai, G.C. Fu, J. Am. Chem. Soc. 122 (2000) 4020–4028. doi: 10.1021/ja0002058
W. Su, S. Urgaonkar, P.A. McLaughlin, J.G. Verkade, J. Am. Chem. Soc. 126 (2004) 16433–16439. doi: 10.1021/ja0450096
D. Ma, A.Q. Cai, Acc. Chem. Res. 41 (2008) 1450–1460. doi: 10.1021/ar8000298
G.W. Gribble, J. Nat. Prod. 55 (1992) 1353–1395. doi: 10.1021/np50088a001
G.W. Gribble, Acc. Chem. Res. 31 (1998) 141–152. doi: 10.1021/ar9701777
K. Takai, K. Nitta, K. Utimoto, J. Am. Chem. Soc. 108 (1986) 7408–7410. doi: 10.1021/ja00283a046
X. Zeng, S. Liu, G.B. Hammond, B. Xu, ACS Catal. 8 (2018) 904–909. doi: 10.1021/acscatal.7b03563
J. Derosa, A.L. Cantu, M.N. Boulous, et al., J. Am. Chem. Soc. 139 (2017) 5183–5193. doi: 10.1021/jacs.7b00892
P. Yu, A. Bismuto, B. Morandi, Angew. Chem. Int. Ed. 59 (2020) 2904–2910. doi: 10.1002/anie.201912803
M.J. Koh, T.T. Nguyen, H. Zhang, R.R. Schrock, A.H. Hoveyda, Nature 531 (2016) 459–465. doi: 10.1038/nature17396
L. Huo, X. Li, Y. Zhao, L. Li, L. Chu, J. Am. Chem. Soc. 145 (2023) 9876–9885. doi: 10.1021/jacs.3c02748
X. Ma, L. Li, M. Tan, et al., Chem 9 (2023) 1164–1181. doi: 10.1016/j.chempr.2023.01.005
Y.F. Zeng, W.W. Ji, W.X. Lv, et al., Angew. Chem. Int. Ed. 56 (2017) 14707–14711. doi: 10.1002/anie.201709070
J.L. Hofstra, K.E. Poremba, A.M. Shimozono, S.E. Reisman, Angew. Chem. Int. Ed. 58 (2019) 14901–14905. doi: 10.1002/anie.201906815
H. Qian, D. Huang, Y. Bi, G. Yan, Adv. Synth. Catal. 361 (2019) 3240–3280. doi: 10.1002/adsc.201801719
M. Dai, L. Song, L.A. Chen, Sci. China Chem. 67 (2024) 1384–1396. doi: 10.1007/s11426-023-1925-4
R.R. Fraser, F. Kong, Synth. Commun. 18 (1998) 1071–1077.
W.M. Schubert, W.A. Lanka, T.H. Liddicoet, Science 116 (1952) 124. doi: 10.1126/science.116.3005.124.a
G.W. Kabalka, M.L. Yao, S. Borella, et al., J. Org. Chem. 73 (2008) 2668–2673. doi: 10.1021/jo702493j
F. Toda, K. Tanaka, I. Sano, T. Isozaki, Angew. Chem. Int. Ed. 33 (1994) 1677–1786. doi: 10.1002/anie.199416771
K. Tamao, in Encyclopedia of Reagents for Organic Synthesis,
S.A. Snyder, Z.Y. Tang, R. Gupta, J. Am. Chem. Soc. 131 (2009) 5744–5745. doi: 10.1021/ja9014716
N. Fu, G.S. Sauer, S. Lin, J. Am. Chem. Soc. 139 (2017) 15548–15553. doi: 10.1021/jacs.7b09388
X. Dong, J.L. Roeckl, S.R. Waldvogel, B. Morandi, Science 371 (2021) 507–514. doi: 10.1126/science.abf2974
A. Saju, J.R. Griffiths, S.N. MacMillan, D.C. Lacy, J. Am. Chem. Soc. 144 (2022) 16761–16766. doi: 10.1021/jacs.2c08509
Y. Dai, F. Wang, S. Zhu, L. Chu, Chin. Chem. Lett. 33 (2022) 4074–4078. doi: 10.1016/j.cclet.2021.12.050
J.I. García, J.A. Mayoral, L. Salvatella, Acc. Chem. Res. 33 (2000) 658–664. doi: 10.1021/ar0000152
Q. Liu, Y. Mu, T. Koengeter, R.R. Schrock, A.H. Hoveyda, Nat. Chem. 14 (2022) 463–473. doi: 10.1038/s41557-022-00893-5
M. Wienhold, B. Kweon, C. McLaughlin, et al., Angew. Chem. Int. Ed. 62 (2023) e202304150. doi: 10.1002/anie.202304150
L.T. Cottrell, The Strengths of Chemical Bonds, Butterworths Scientific Publications, London, 1958.
G.F. Koser, Aldrichim. Acta 34 (2001) 89–102.
M.A. Raslan, S.M. Sayed, J. Heterocyclic. Chem. 57 (2020) 2862–2874. doi: 10.1002/jhet.3995
X. Xu, B. Wan, S.G. Franzblau, Q. You, Eur. J. Med. Chem. 46 (2011) 3551–3563. doi: 10.1016/j.ejmech.2011.05.018
A.C. Frisch, N. Shaikh, A. Zapf, M. Beller, Angew. Chem. Int. Ed. 41 (2002) 4056–4059. doi: 10.1002/1521-3773(20021104)41:21<4056::AID-ANIE4056>3.0.CO;2-8
M. Yasuda, S. Yamasaki, Y. Onishi, A. Baba, J. Am. Chem. Soc. 126 (2004) 7186–7187. doi: 10.1021/ja048688t
W.F. Jiang, X.S. Liu, X. Du, J.W. Wei, C.Y. Bao, Chin. Chem. Lett. 23 (2012) 999–1002. doi: 10.1016/j.cclet.2012.06.031
G.M. Shibuya, J.S. Kanady, C.D. Vanderwal, J. Am. Chem. Soc. 130 (2008) 12514–12518. doi: 10.1021/ja804167v
Y. Zhu, L. Sun, P. Lu, Y. Wang, ACS Catal. 4 (2014) 1911–1925. doi: 10.1021/cs400922y
J. Zhang, H. Shi, D. Zhang, et al., Green Synth. Catal. (2024), doi: 10.1016/j.gresc.2024.09.009.
C. Zhu, J.L. Schwarz, S. Embellín, S. Greßies, F. Glorius, Angew. Chem. Int. Ed. 57 (2018) 437–441. doi: 10.1002/anie.201710835
N. Archipowa, R.J. Kutta, D.J. Heyes, N.S. Scrutton, Angew. Chem. Int. Ed. 57 (2018) 2682–2686. doi: 10.1002/anie.201712729
B. Inés, D. Palomas, S. Holle, et al., Angew. Chem. Int. Ed. 51 (2012) 12367–12369. doi: 10.1002/anie.201205348
S.W.M. Crossley, C. Obradors, R.M. Martinez, R.A. Shenvi, Chem. Rev. 116 (2016) 8912–9000. doi: 10.1021/acs.chemrev.6b00334
H. Yan, Q. Liao, Y. Chen, et al., Angew. Chem. Int. Ed. 62 (2023) e202302483. doi: 10.1002/anie.202302483
H. Yan, J.R. Shan, L. Shi, ChemCatChem 16 (2016) e20230134.
Table 1. Optimization of reaction conditions.a
|
||
| Entry | Deviation from standard conditions | Yield (%)b |
| 1 | — | 95 (90)c |
| 2 | Without Co(salen) | 50 |
| 3 | Co(OAc)2 or CoCl2 | 68, 57 |
| 4 | Co(salen) B-D | 52–92 |
| 5 | Ni(salen) E | 21 |
| 6 | Cu(salen) F | 81 |
| 7 | Co(salen)Cl G | 89 |
| 8 | Mn(salen)Cl H | 93 |
| 9 | HSiPh2Cl | 21 |
| 10 | HSiCl3 | n.d. |
| 11 | Et3SiH + Me3SiCl | 90 |
| 12 | MeCN | 87 |
| 13 | DCM | 85 |
| 14 | DMSO, DMF, THF, Dioxane, Toluene | < 5 |
| a Reaction conditions: 1a (0.2 mmol, 1.0 equiv.), HSiMe 2Cl (0.6 mmol, 3.0 equiv.), Co(salen) A (0.01 mmol, 5 mol%), DCE (0.1 mol/L), 50 ℃, 3 h. b Yield determined by 1H NMR analysis using CH 2Br 2 as the internal standard. c Isolated yield. n.d. = not detected. |
||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们







