
Figure 1.
From inspiration to reaction design. (A) State of the art in radical cross-coupling reactions. (B) Reactivity of organoborons in cross-coupling reactions. (C) This work: Deboronative cross-coupling.
Deboronative cross-coupling enabled by nickel metallaphotoredox catalysis
Fuyang Yue , Mingxing Li , Fei Yuan , Hongjian Song , Yuxiu Liu , Qingmin Wang

In the past 10 years, photoredox catalysis has become a widely accepted, powerful tool for organic synthesis [1–9]. With the development of transition-metal/photoredox dual catalysis, many unprecedented, previously infeasible cross-coupling reactions have become possible [10–16]. Metallaphotocatalysis, which takes advantage of the synergistic effects of combining photocatalysis and transition-metal catalysis, has strongly influenced the development of organic chemistry (Fig. 1A, left) [13–18]. It is worth noting that photocatalysts can generate free radicals and regulate the oxidation state of metal catalysts, abilities that have greatly expanded the range of electrophilic and nucleophilic reagents that can be used in cross-coupling reactions [10–22]. Early in the development of photocatalysts, they were used for single electron transfer processes involving substrates containing redox agents, ensuring critical mesolytic fragmentation to provide shell-opening intermediates. Then, the free radical can be intercepted by a transition-metal catalyst and coupled with a suitable electrophilic reagent to form a C(sp3)–C(sp2) bond [10–16]. In pioneering work, the groups of MacMillan [23] and others [24] utilized the hydrogen atom transfer ability of photocatalysts to expand the range of radical precursors that can be used in metallaphotoredox reactions to include simple alkanes. Similarly, metallaphotoredox catalysts have been applied to halogen atom transfer reactions of halogenated compounds [25–28]. In the past few years, chemists have devoted attention to olefin bifunctionalization through metal/photoredox dual catalysis [29–32] and have developed many methods for intermolecular carboarylation of olefins (Fig. 1A, right). Although excellent methods for transition-metal/photoredox synergistic catalysis have been developed, the need for additional free-radical precursors and methods for their activation remains a driving force for research in this field.
The development of transition-metal-catalyzed cross-coupling reactions is beneficial for the synthesis of drugs and agricultural chemicals [33,34]. Despite significant progress in this field, introducing C(sp3) hybridized fragments by means of transition-metal-catalyzed cross-coupling reactions remains a major challenge in organic synthesis. The B-center Suzuki–Miyaura cross-coupling (SMC) reaction is a powerful method for generating C(sp2)–C(sp2) bonds through cross-coupling of aryl boron compounds with aryl halides (Fig. 1B) [35]. This reaction has many advantages, such as its good functional group tolerance and the low toxicity of the boron by-products [36–38]. However, although this method has proven to be highly effective for the formation of C(sp2)–C(sp2) bonds, extending it to C(sp3) centers remains challenging because of the low rates of oxidative addition and transition metallization, the tendency of alkyl metal intermediates to undergo β-hydride elimination, and the limitations of the method itself, including the need to use air- and moisture-sensitive organometallic compounds, high temperatures, and long reaction times; these features hinder the practical applications of the method. Therefore, the development of mild, selective B-alkyl SMC reactions has become an attractive option [36–38].
The groups of Molander [39] and Akita [40] pioneered the use of photoredox catalysis to convert trifluoroborates into alkyl radicals under mild conditions, opening up a new application of alkyl boron compounds. This breakthrough has accelerated the development of more types of boron reagents, such as borate esters and their metal salts [41–44]. In recent years, various elegant photomediated B-alkyl SMC reactions have been reported [45,46]. However, in these reactions, the precursors of the boron centers are limited to potassium trifluoroborates and borate esters. Because of the high oxidation potentials and weak acidity of stable, commercially available simple alkyl boronic acids, they are not often used as alkyl group precursors, which has hindered their utility for free-radical cross-coupling reactions [47]. Chemists have developed many excellent methods for reducing the oxidation potentials of alkyl boronic acids [47,48]. However, most methods require solvents or additives that make the methods suitable only for simple reactions such as Michael additions. In contrast, the necessary solvents and additives make transition-metal-catalyzed reactions infeasible, thereby greatly limiting the utility of alkyl boronic acids for the modification of drug molecules.
In order for alkyl boronic acids to participate in the photocatalytic cycles of visible-light-promoted SMC reactions, the oxidation potentials of these compounds must be reduced. However, most existing methods for this purpose use an additive such as water or a Lewis base (4-dimethylaminopyridine), but most photocatalytic/nickel-catalyzed reactions are very sensitive to water, and Lewis bases may affect coordination between the ligand and nickel. We envisioned that it might be possible to reduce the oxidation potentials of alkyl boronic acids with reagents that are typically used for metal/photocatalysis, such as inorganic bases [48]; in this way, free-radical SMC reactions involving metal/photoredox dual catalysis could be achieved (Fig. 1C).
Indeed, we herein report a method for deboronative radical cross-coupling reactions between free alkyl boronic acids and aryl bromides via photoredox/nickel dual catalysis. We used K3PO4 to reduce the oxidation potential of the alkyl boronic acids, allowing them to participate in the photocatalytic cycle and avoiding the need for any additional reaction components. Our method demonstrated good functional group tolerance, and the mild conditions permitted the functionalization of drug molecules. In addition, the method could be used for three-component carboacylation/carboarylation reactions of alkenes. Specifically, we achieved enantioselective three-component carboarylation of alkenes by using a chiral biimidazoline ligand. Additionally, we directly synthesized ibuprofen by modifying the two reactive sites of 1-(4-bromophenyl) ethan-1-ol by means of a one-pot two-step method.
For our initial experiments, we chose 1-(4-bromophenyl)ethan-1-one (1a) and (1-(tert–butoxycarbonyl)piperidin-4-yl)boronic acid (2a) as model substrates (Table 1). We conducted extensive screening to identify the optimal combination of activation reagent, photocatalyst, metal catalyst, ligand, and solvent for the desired transformation (for details, see Supporting information). To our delight, when a solution of the substrates in dry 1:3 ethyl acetate (EA)/dimethoxyethane (DME) ([1a] = 0.1 mol/L) containing Ir[dF(CF3)ppy]2(dtbbpy)PF6 (2 mol%; ppy = phenylpyridine, dtbbpy = 4,4′-di–tert–butyl–2,2′-dipyridyl) as a photocatalyst, NiBr2·glyme (10 mol%) as a metal catalyst, dtbbpy (L1, 10 mol%) as a ligand, and K3PO4 as an activation reagent was irradiated with a 425 nm LED at room temperature (~30 ℃) under argon for 24 h, desired cross-coupling product 3a was obtained in 87% yield (entry 1). Various other photocatalysts were tested, including 4CzIPN (1,2,3,5-tetrakis(carbazol-9-yl)−4,6-dicyanobenzene, entry 2; see Supporting information for additional details), but Ir[dF(CF3)ppy]2(dtbbpy)PF6 was found to be optimal. When we replaced K3PO4 with different activation reagents (entries 3–5), the reaction failed to proceed, suggesting that other activation methods may be incompatible with this dual catalytic system. Alternative nickel catalysts were assessed: Ni(dppf)Cl2 also delivered target product 3a, albeit in only 37% yield (entry 6); whereas NiCl2·6H2O failed to catalyze the reaction (entry 7; see Supporting information for additional details). Solvent screening revealed that reactions in CH3CN or DMSO gave little to none of the target product (entries 8 and 9; for other tested solvents, see Supporting information). Omitting the photocatalyst, light, or the nickel catalytic system resulted in no product formation (entries 10 and 11), as did omission of K3PO4 (entry 12). The reaction also failed to proceed under air or in the presence of water (entry 13). Additional details and optimization experiments can be found in Supporting information.
 DownLoad:
CSV
DownLoad:
CSV
 |
||
| Entry | Deviation from standard conditions | Yield (%)b |
| 1 | None | 87 (83c) |
| 2 | 4CzIPN as photocatalyst | 56 |
| 3 | 2,6-Lutidine instead of K3PO4 | NR |
| 4 | 4-Dimethylaminopyridine instead of K3PO4 | Trace |
| 5 | H2O/DMSO instead of K3PO4 | NR |
| 6 | Ni(dppf)Cl2 instead of NiBr2·glyme | 37 |
| 7 | NiCl2·6H2O instead of NiBr2·glyme | NR |
| 8 | CH3CN as solvent | Trace |
| 9 | DMSO as solvent | NR |
| 10 | No photocatalyst or light | NR |
| 11 | No NiBr2·glyme or dtbbpy | NR |
| 12 | No K3PO4 | NR |
| 13 | Under air or in the presence of H2O | NR |
| a Standard conditions: 1a (0.1 mmol), 2a (0.2 mmol), photocatalyst (2 mol%), K 3PO 4 (0.2 mmol), NiBr 2·glyme (10 mol%), dtbbpy (10 mol%), 1:3 EA/DME (2 mL, [ 1a] = 0.05 mol/L), Ar, 425 nm LED, ~30 ℃, 24 h. b Yields were determined by 1H NMR spectroscopy with dibromomethane as an internal standard. NR = no reaction. c Isolated yield. |
||
Having optimized the reaction conditions (Table 1, entry 1), we next assessed the scope. As outlined in Fig. 2, reactions of 1a with boronic acids bearing linear alkyl chains of various lengths efficiently delivered the corresponding products (3b–3f). A branched alkyl boronic acid and a benzyl boronic acid were also compatible with this catalytic system, reacting with 1a and to give 3g and 3h, respectively. We next examined a series of secondary alkyl boronic acids as coupling partners for reactions with aryl halides. Acyclic secondary boronic acids were successfully coupled, giving 3i and 3j. Furthermore, cyclic boronic acids with a five-, six- or seven-membered ring gave corresponding coupling products 3k–3m in 80%–85% yields. Compounds with a tetrahydropyran (3n), or an indane (3p) ring were also viable substrates. We then explored reactions of various aryl bromide coupling partners 1. In general, aryl bromides with an electron-withdrawing functional group fared best, giving the corresponding products in moderate to good yields. para-Substituted bromobenzenes bearing an ester (3p), a ketone (3q), a sulfone (3r), an aldehyde (3s), a cyclic ketone (3t, 3u) a sulfonamide (3v, 3w), or a silicyne (3x) were well tolerated. Interestingly, our method is complementary to Weix's method for aryl bromide/aldehyde reductive coupling, which does not tolerate unhindered aldehydes.

In addition, a meta-substituted bromobenzene delivered 3y in 75% yield, and bromobenzene itself was also compatible with this reaction, giving moderate yields of 3z and 3aa. Gratifyingly, aryl bromides with an electron-donating group, such as phenyl, methoxy, or trifluoromethoxy, were also viable substrates, furnishing desired products 3ab–3ae in 54%–81% yields. Demonstrating the excellent functional group tolerance and selectivity of this reaction, we found that substrates with a free OH group gave the desired products (3af and 3ag), and no C–O coupling by-products were detected. Considering the importance of nitrogen-containing heterocycles in the preparation of bioactive molecules, we were pleased to find that heteroaryl bromides were also suitable for the cross-coupling reaction, affording expected products 3ah–3aj in 65%–81% yields.
Because the construction of quaternary centers is a common challenge in the synthesis of structurally complex compounds, including natural products [49], we went on to determine whether our activation method could be used to generate molecules with a quaternary carbon center. Using the optimal conditions described above, we were unable to obtain a good yield of target product 3ak, but when we re-optimized the reaction conditions for this substrate (see Supporting information for more details), we obtained 3ak in 80% yield. Under the re-optimized conditions, reactions of tert–butyl boronic acid with para-substituted bromobenzenes bearing an ester (3al), an amide (3am, 3an), an aldehyde (3ao) or an ether (3ap) were well tolerated.
To further explore the functional group tolerance of this SMC reaction, we carried out reactions of an array of bromobenzenes 4 derived from drug molecules, pesticide molecules, and natural products (Fig. 3). These reactions furnished the corresponding C(sp3)–B arylation products (5a–5aa) in 40%–81% yields. Our findings confirm that our SMC reaction had excellent functional group tolerance and was compatible with functionality such as halogen atoms, amides, aniline and other heteroaromatic rings, esters, ethers, olefins, and ketones.

Chiral α-aryl carbonyl compounds are found in a wide variety of multifunctional intermediates in the synthesis of bioactive molecules, including drugs [50]. Therefore, the development of catalytic asymmetric methods for directly constructing enantioenriched α-aryl carbonyl compounds has attracted much attention, but because such stereocenters readily racemize under basic conditions, their synthesis remains a significant challenge. Recently, photoredox/nickel dual catalysis has emerged as a powerful strategy for directly constructing enantioenriched α-aryl carbonyls, as demonstrated by the groups of Chu [51], Mao [52], Nevado [53], and Shu [54]. Given the importance of these carbonyl compounds and the many limitations of the existing methods for their synthesis, we attempted to use our photoredox/nickel dual catalysis method to directly convert alkyl boronic acids into α-aryl carbonyl compounds.
We found that combining a photocatalyst with a nickel catalyst bearing a chiral biimidazoline ligand enabled asymmetric dicarbofunctionalization reactions of alkenes 6 with free boronic acids 2 and aryl bromides 1 to afford enantioenriched α-aryl carbonyl compounds 7 (Fig. 4A). We began our investigation by identifying suitable conditions for the reaction of tert–butyl boronic acid, tert–butyl acrylate, and methyl 4-bromobenzoate as model substrates. After extensive evaluation of the reaction parameters, we found that irradiation of the substrates in an acetone/DME = 1/3 solution containing NiBr2·DME (10 mol%), Ir[dF(CF3)ppy]2(dtbbpy)PF6 (2 mol%), K3PO4 (2 equiv.), and chiral biimidazoline L2 (11 mol%) with blue LEDs at 10 ℃ for 24 h under argon provided methyl (R)−4-(1-(tert–butoxy)−4,4-dimethyl-1-oxopentan-2-yl)benzoate (7a) in 83% isolated yield with 91% enantiomeric excess (ee). Using these optimized conditions, we investigated the scope of this transformation. Aryl bromides bearing a ketone (7b), a sulfone (7c), a nitrile (7d), a trifluoromethyl group (7e), or an aldehyde (7f) were all successfully converted to the corresponding dicarbofunctionalization products in good yields with high ee values. A pyridyl bromide also underwent the desired reaction, delivering target product 7g in a good yield with high ee. We used dtbbpy (L1) as a ligand for reactions to generate racemic products 7h–7o.
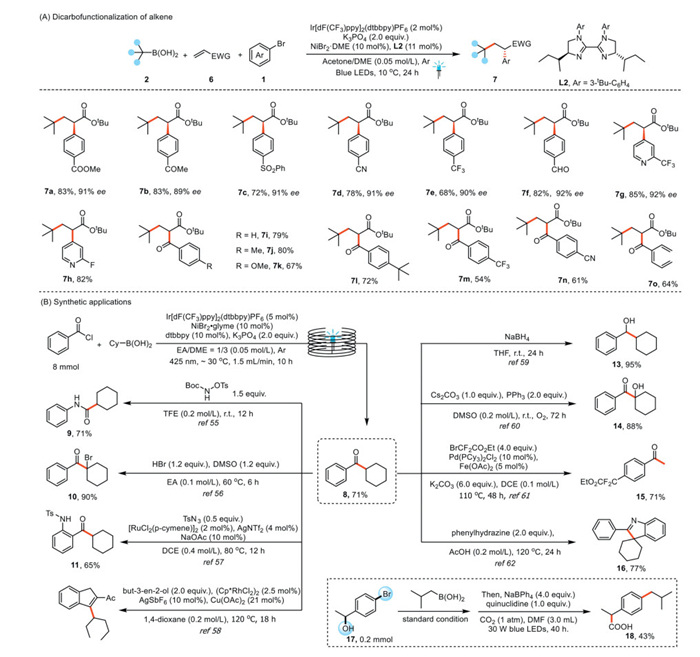
To further illustrate the synthetic potential of our method, we replaced the aryl bromide with benzoyl chloride and conducted a gram-scale continuous-flow B-alkyl SMC reaction to synthesize cyclohexyl(phenyl)methanone (8, Fig. 4B). Using a flow rate of 1.5 mL/min and a residence time of 10 h, we were able to scale up the reaction to 8 mmol. Subsequently, we explored the synthetic potential of ketone 8 by conducting a series of transformations [55–62] to generate products 9–16. Because continuous conversion [63] of two reactive sites has recently received considerable attention from chemists, we also carried out a reaction of 1-(4-bromophenyl)ethan-1-ol (17) and achieved continuous conversion [64] of its two reactive sites to afford ibuprofen (18).
To gain insight into the mechanism of this SMC reaction, we conducted a series of control experiments. First, because we believed that K3PO4 and the alkyl boronic acid formed an alkyl boronic acid–K3PO4 complex, which has a low oxidation–reduction potential (Eox ~+1.1 V) [48], we carried out titration experiments that were monitored by 11B NMR and 31P NMR; the obtained spectra showed clear changes in the B and P chemical shifts upon complex formation (Fig. 5A). We also performed Stern–Volmer fluorescence quenching experiments (Fig. 5B), which showed that the excited state of the photocatalyst was quenched by the alkyl boronic acid–K3PO4 complex rather than by the alkyl boronic acid or K3PO4 alone. To confirm that the reaction involved free radicals, we conducted electron paramagnetic resonance experiments (Fig. 5C) and successfully detected the formation of cyclohexyl radicals. In addition, a reaction of 1b and 2b was completely inhibited by TEMPO (2,2,6,6-tetramethylpiperidine 1-oxyl). Moreover, both TEMPO-alkylated compound 20 (the product of a two-component reaction; Fig. 5D, left) and TEMPO-alkylated compound 21 (the product of a three-component reaction; Fig. 5D, right) were detected by high-resolution mass spectrometry, suggesting a radical mechanism. This suggestion was supported by intramolecular radical clock experiments (Fig. 5E). Specifically, when hex–5-en-1-yl boronic acid (2d) was allowed to react with 1b, direct arylation product 22 was not detected; instead, cyclized cross-coupling product 23 was obtained in 75% yield. We also investigated the catalytic activity of a presynthesized 4-COCH3C6H4Ni(Ⅱ)L1Br complex. Reaction of 2a alone with a stoichiometric amount of the presynthesized complex (0.2 mmol), instead of NiBr2·DME/L1, afforded only a trace of 3a. One possible reason for this outcome is that in the absence of aryl bromide 1a, the presynthesized complex was unstable and strongly absorbed visible light. In contrast, reaction of 1a and 2a with catalysis by the presynthesized complex (10 mol%) afforded 3a in 47% yield. Taken together, the results of our control experiments indicate that an aryl Ni(Ⅱ) intermediate may not have been involved in the main catalytic cycle.
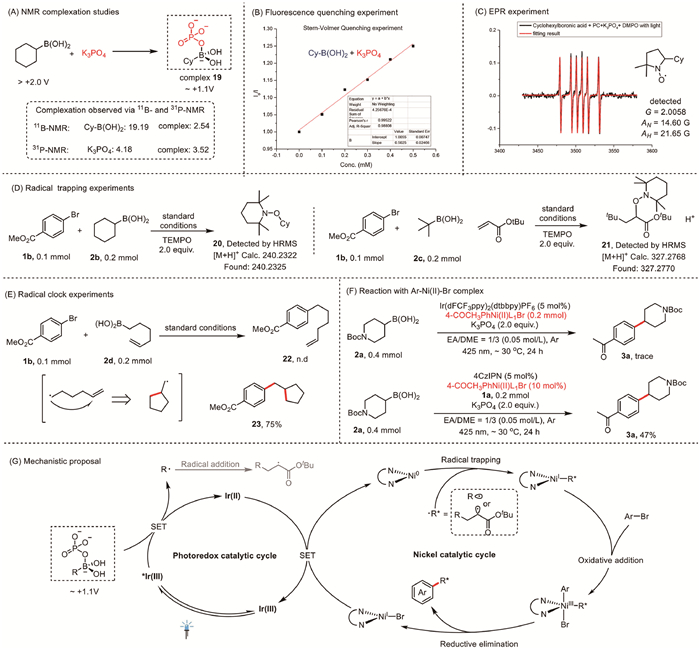
On the basis of our experimental results, we propose that these C(sp2)–C(sp3) coupling reactions proceed via the mechanism shown in Fig. 5G. First, the alkyl boronic acid and K3PO4 form an alkyl boronic acid–K3PO4 complex. Then, because of the high oxidation potential of the excited triplet state of [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 [*Ir(Ⅲ)], the first step of the photoredox catalytic cycle is oxidation of the alkyl boronic acid–K3PO4 complex, which furnishes an alkyl radical and an Ir(Ⅱ) complex. Trapping of the alkyl radical by a Ni(0) species generates an alkyl–Ni(Ⅰ) intermediate. If an olefin is present in the reaction mixture, the alkyl radical first adds to the olefin to form a new radical that is then captured by the Ni(0) species. Oxidative addition of the aryl bromide to the Ni(Ⅰ) species generates an alkyl–Ni(Ⅲ)–aryl intermediate. Subsequent reductive elimination produces the desired dicarbofunctionalization product and a Ni(Ⅰ) species.
In this study, we developed a method for deboronative cross-coupling reactions of alkyl boronic acids by means of photoredox/nickel dual catalysis. We reduced the oxidation potential of the alkyl boronic acids by generating alkyl boronic acid–K3PO4 complexes; this obviated the need for additional reaction components, high reaction temperatures, and expensive catalysts, which limit the utility of previously reported approaches. Our method allowed us to synthesized compounds with quaternary carbon centers, and when we introduced a chiral ligand, we were able to synthesize chiral α-aryl carbonyl compounds. Our method demonstrated strong functional group tolerance, and the fact that the synthesis of the alkyl boronic acid substrates is straightforward makes our method ideal for functionalizing biologically relevant alkyl substrates for medicinal chemistry and total synthesis applications.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Fuyang Yue: Writing – review & editing, Writing – original draft, Methodology, Investigation. Mingxing Li: Methodology, Investigation. Fei Yuan: Methodology, Investigation. Hongjian Song: Writing – review & editing. Yuxiu Liu: Writing – review & editing. Qingmin Wang: Writing – review & editing, Supervision, Project administration, Funding acquisition, Conceptualization.
We are grateful to the National Natural Science Foundation of China (No. 22271166) and the Frontiers Science Center for New Organic Matter, Nankai University (No. 63181206), for generous financial support.
Supplementary material associated with this article can be found, in the online version, at doi:
Z. Zhang, V. Gevorgyan, Chem. Rev. 124 (2024) 7214–7261. doi: 10.1021/acs.chemrev.3c00869
S. Ni, R. Halder, D. Ahmadli, et al., Nat. Catal. 7 (2024) 733–741. doi: 10.1038/s41929-024-01160-1
F. Yue, H. Ma, P. Ding, et al., ACS Cent. Sci. 9 (2023) 2268–2276. doi: 10.1021/acscentsci.3c00993
F. Yue, M. Li, K. Yang, et al., Chem. Sci. 15 (2024) 14241–14247. doi: 10.1039/d4sc02889a
D. Rana, P.M. Pflüger, N.P. Hölter, G. Tan, F. Glorius, ACS Cent. Sci. 10 (2024) 899–906.
Y. Liang, T. Bian, K. Yadav, et al., ACS Cent. Sci. 10 (2024) 1191–1200. doi: 10.1021/acscentsci.4c00094
M. Lu, J. Goh, M. Maraswami, et al., Chem. Rev. 122 (2022) 17479–17646. doi: 10.1021/acs.chemrev.2c00032
W. Liu, P. Liu, L. Lv, C.J. Li, Angew. Chem. Int. Ed. 57 (2018) 13499–13503. doi: 10.1002/anie.201807181
D. Zheng, A. Studer, Angew. Chem. Int. Ed. 131 (2019) 15950–15954. doi: 10.1002/ange.201908987
Y. Chen, X. Wang, X. He, Q. An, Z. Zuo, J. Am. Chem. Soc. 143 (2021) 4896–4902. doi: 10.1021/jacs.1c00618
C. Zhu, H. Yue, L. Chu, M. Rueping, Chem. Sci. 11 (2020) 4051–4064. doi: 10.1039/d0sc00712a
F. Song, F. Wang, L. Guo, et al., Angew. Chem. Int. Ed. 59 (2020) 177–181. doi: 10.1002/anie.201909543
J. Khamrai, I. Ghosh, A. Savateev, M. Antonietti, B. König, ACS Catal. 10 (2020) 3526–3532. doi: 10.1021/acscatal.9b05598
L. Huang, T. Ji, M. Rueping, J. Am. Chem. Soc. 142 (2020) 3532–3539. doi: 10.1021/jacs.9b12490
M.S. Oderinde, M. Frenette, D.W. Robbins, B. Aquila, J.W. Johannes, J. Am. Chem. Soc. 138 (2016) 1760–1763. doi: 10.1021/jacs.5b11244
X. Lang, J. Zhao, X. Chen, Chem. Soc. Rev. 45 (2016) 3026–3038. doi: 10.1039/C5CS00659G
K.L. Skubi, T.R. Blum, T.P. Yoon, Chem. Rev. 116 (2016) 10035–10074. doi: 10.1021/acs.chemrev.6b00018
C.K. Prier, D.A. Rankic, D.W.C. MacMillan, Chem. Rev. 113 (2013) 5322–5363. doi: 10.1021/cr300503r
J.K. Matsui, S.B. Lang, D.R. Heitz, G.A. Molander, ACS Catal. 7 (2017) 2563–2575. doi: 10.1021/acscatal.7b00094
K.J. Romero, M.S. Galliher, D.A. Pratt, C.R.J. Stephenson, Chem. Soc. Rev. 47 (2018) 7851–7866. doi: 10.1039/c8cs00379c
L. Buzzetti, A. Prieto, S.R. Roy, P. Melchiorre, Angew. Chem. Int. Ed. 56 (2017) 15039–15043. doi: 10.1002/anie.201709571
R.A. Garza-Sanchez, A. Tlahuext-Aca, G. Tavakoli, F. Glorius, ACS Catal. 7 (2017) 4057–4061. doi: 10.1021/acscatal.7b01133
I.B. Perry, T.F. Brewer, P.J. Sarver, et al., Nature 560 (2018) 70–75. doi: 10.1038/s41586-018-0366-x
D. Ravelli, S. Protti, M. Fagnoni, Acc. Chem. Res. 49 (2016) 2232–2242. doi: 10.1021/acs.accounts.6b00339
A. Luridiana, D. Mazzarella, L. Capaldo, et al., ACS Catal. 12 (2022) 11216–11225. doi: 10.1021/acscatal.2c03805
F. Juliá, T. Constantin, D. Leonori, Chem. Rev. 122 (2022) 2292–2352. doi: 10.1021/acs.chemrev.1c00558
V. Bacauanu, S. Cardinal, M. Yamauchi, et al., Angew. Chem. Int. Ed. 57 (2018) 12543–12548. doi: 10.1002/anie.201807629
Q.Q. Zhou, S.J.S. Dü sel, L.Q. Lu, B. König, W.J. Xiao, Chem. Commun. 55 (2019) 107–110. doi: 10.1039/c8cc08362b
M.W. Campbell, J.S. Compton, C.B. Kelly, G.A. Molander, J. Am. Chem. Soc. 141 (2019) 20069–20078. doi: 10.1021/jacs.9b08282
A. García-Domínguez, R. Mondal, C. Nevado, Angew. Chem. Int. Ed. 58 (2019) 12286–12290. doi: 10.1002/anie.201906692
S. Xu, H. Chen, Z. Zhou, W. Kong, Angew. Chem. Int. Ed. 60 (2021) 7405–7411. doi: 10.1002/anie.202014632
M.W. Campbell, M. Yuan, V.C. Polites, O. Gutierrez, G.A. Molander, J. Am. Chem. Soc. 143 (2021) 3901–3910. doi: 10.1021/jacs.0c13077
R. Zhang, G. Li, M. Wismer, et al., ACS Med. Chem. Lett. 9 (2018) 773–777. doi: 10.1021/acsmedchemlett.8b00183
P. Li, J.A. Terrett, J.R. Zbieg, ACS Med. Chem. Lett. 11 (2020) 2120–2130. doi: 10.1021/acsmedchemlett.0c00436
S.R. Chemler, D. Trauner, S.J. Danishefsky, Angew. Chem. Int. Ed. 40 (2001) 4544–4568. doi: 10.1002/1521-3773(20011217)40:24<4544::AID-ANIE4544>3.0.CO;2-N
R. Jana, T.P. Pathak, M.S. Sigman, Chem. Rev. 111 (2011) 1417–1492. doi: 10.1021/cr100327p
G.A. Molander, T. Ito, Org. Lett. 3 (2001) 393–396. doi: 10.1021/ol006896u
B. Wang, H.X. Sun, Z.H. Sun, G.Q. Lin, Adv. Synth. Catal. 351 (2009) 415–422. doi: 10.1002/adsc.200800630
J.C. Tellis, D.N. Primer, G.A. Molander, Science 345 (2014) 433–436. doi: 10.1126/science.1253647
Y. Yasu, T. Koike, M. Akita, Adv. Synth. Catal. 354 (2012) 3414–3420. doi: 10.1002/adsc.201200588
F. Lima, M.A. Kabeshov, D.N. Tran, et al., Angew. Chem. Int. Ed. 55 (2016) 14085–14089. doi: 10.1002/anie.201605548
C. Shu, A. Noble, V.K. Aggarwal, Angew. Chem. Int. Ed. 58 (2019) 3870–3874. doi: 10.1002/anie.201813917
D. Kaiser, A. Noble, V. Fasano, V.K. Aggarwal, J. Am. Chem. Soc. 141 (2019) 14104–14109. doi: 10.1021/jacs.9b07564
Y. Sato, K. Nakamura, Y. Sumida, et al., J. Am. Chem. Soc. 142 (2020) 9938–9943. doi: 10.1021/jacs.0c04456
E. Speckmeier, T.C. Maier, J. Am. Chem. Soc. 144 (2022) 9997–10005. doi: 10.1021/jacs.2c03220
T. Wan, L. Capaldo, J. Djossou, et al., Nat Commun. 15 (2024) 4028. doi: 10.1038/s41467-024-48212-5
S. Pillitteri, P. Ranjan, E.V. Van der Eycken, U.K. Sharma, Adv. Synth. Catal. 364 (2022) 1643–1665. doi: 10.1002/adsc.202200204
F. Yue, H. Ma, H. Song, et al., Chem. Sci. 13 (2022) 13466–13474. doi: 10.1039/d2sc05521j
Z. Dong, D.W.C. MacMillan, Nature 598 (2021) 451–456. doi: 10.1038/s41586-021-03920-6
P.J. Harrington, E. Lodewijk, Org. Process Res. Dev. 1 (1997) 72–76. doi: 10.1021/op960009e
L. Guo, M. Yuan, Y. Zhang, et al., J. Am. Chem. Soc. 142 (2020) 20390–20399. doi: 10.1021/jacs.0c08823
P. Qian, H. Guan, Y.E. Wang, et al., Nat. Commun. 12 (2021) 6613–6621. doi: 10.1038/s41467-021-26794-8
X. Du, I. Cheng-Sánchez, C. Nevado, J. Am. Chem. Soc. 145 (2023) 12532–12540. doi: 10.1021/jacs.3c00744
M.S. Liu, W. Shu, JACS Au 3 (2023) 1321–1327. doi: 10.1021/jacsau.3c00069
J.L. Jat, P. Kumar, S. Verma, et al., New J. Chem. 46 (2022) 14782–14785. doi: 10.1039/d2nj02755k
S. Song, X. Li, X. Sun, Y. Yuan, N. Jiao, Green Chem. 17 (2015) 3285–3289. doi: 10.1039/C5GC00528K
J. Kim, J. Kim, S. Chang, Chem. Eur. J. 19 (2013) 7328–7333. doi: 10.1002/chem.201301025
Z. Shi, M. Boultadakis-Arapinis, F. Glorius, Chem. Commun. 49 (2013) 6489–6491. doi: 10.1039/c3cc43903h
A.R. Schmitzer, S. Franceschi, E. Perez, et al., J. Am. Chem. Soc. 123 (2001) 5956–5961. doi: 10.1021/ja000378x
Y.F. Liang, N. Jiao, Angew. Chem. Int. Ed. 53 (2014) 548–552. doi: 10.1002/anie.201308698
Y.J. Mao, B.X. Wang, Q.Z. Wu, et al., Chem. Commun. 55 (2019) 2019–2022. doi: 10.1039/c8cc09129c
H. Lee, Y.K. Sim, J.W. Park, C.H. Jun, Chem. Eur. J. 20 (2014) 323–333. doi: 10.1002/chem.201302699
A. Long, C.J. Oswood, C.B. Kelly, M.C. Bryan, D.W.C. MacMillan, Nature 628 (2024) 326–332. doi: 10.1038/s41586-024-07181-x
W. Li, Y. Wu, S. Li, et al., J. Am. Chem. Soc. 144 (2022) 8551–8559. doi: 10.1021/jacs.1c12463

Figure 1 From inspiration to reaction design. (A) State of the art in radical cross-coupling reactions. (B) Reactivity of organoborons in cross-coupling reactions. (C) This work: Deboronative cross-coupling.

Figure 2 Illustration of the broad applicability, selectivity, and functional group tolerance of the C(sp2)–C(sp3) cross-coupling reaction. Reaction conditions, unless otherwise noted: 1 (0.1 mmol), 2 (0.2 mmol), Ir[dF(CF3)ppy]2(dtbbpy)PF6 (2 mol%), K3PO4 (0.2 mmol), NiBr2·glyme (10 mol%), dtbbpy (10 mol%), 1:3 EA/DME (2 mL, [1] = 0.05 mol/L), Ar, 425 nm LED, ~30 ℃, 24 h. aNi(TMHD)2 (10 mol%) instead of NiBr2·glyme, additive of ZnBr2 (10 mol%), 48 h.

Figure 3 Cross-coupling reactions of natural products and pharmaceutical derivatives. Reaction conditions: 4 (0.1 mmol), 2 (0.2 mmol), Ir[dF(CF3)ppy]2(dtbbpy)PF6 (2 mol%), K3PO4 (0.2 mmol), NiBr2·glyme (10 mol%), dtbbpy (10 mol%), 1:3 EA/DME (2 mL, [4] = 0.05 mol/L), Ar, 425 nm LED, ~30 ℃, 24 h. For details, see Supporting information.

Figure 5 Mechanism experiment: (A) NMR complexation studies, (B) fluorescence quenching experiment, (C) EPR experiment, (D) radical trapping experiments, (E) radical clock experiments, (F) reaction with Ar-Ni(Ⅱ)-Br complex and (G) mechanistic proposal.
Table 1. Optimization of conditions for cross-coupling of aryl bromide 1a and piperidinyl boronic acid 2a.a
|
||
| Entry | Deviation from standard conditions | Yield (%)b |
| 1 | None | 87 (83c) |
| 2 | 4CzIPN as photocatalyst | 56 |
| 3 | 2,6-Lutidine instead of K3PO4 | NR |
| 4 | 4-Dimethylaminopyridine instead of K3PO4 | Trace |
| 5 | H2O/DMSO instead of K3PO4 | NR |
| 6 | Ni(dppf)Cl2 instead of NiBr2·glyme | 37 |
| 7 | NiCl2·6H2O instead of NiBr2·glyme | NR |
| 8 | CH3CN as solvent | Trace |
| 9 | DMSO as solvent | NR |
| 10 | No photocatalyst or light | NR |
| 11 | No NiBr2·glyme or dtbbpy | NR |
| 12 | No K3PO4 | NR |
| 13 | Under air or in the presence of H2O | NR |
| a Standard conditions: 1a (0.1 mmol), 2a (0.2 mmol), photocatalyst (2 mol%), K 3PO 4 (0.2 mmol), NiBr 2·glyme (10 mol%), dtbbpy (10 mol%), 1:3 EA/DME (2 mL, [ 1a] = 0.05 mol/L), Ar, 425 nm LED, ~30 ℃, 24 h. b Yields were determined by 1H NMR spectroscopy with dibromomethane as an internal standard. NR = no reaction. c Isolated yield. |
||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
