
Scheme 1.
Copper-mediated ethoxycarbonyl difluoromethylation.

Structural motifs containing fluorine are crucial in biological active compounds, particularly within the realms of pharmaceuticals and agrochemicals. The incorporation of fluorine typically leads to a significant increase in lipophilicity, metabolic stability, and bioavailability to their non-fluorinated counterparts [1–6]. Among the array of fluorinated groups, the difluoromethylene group (CF2) holds a distinguished position and is considered as a bioisostere of carbonyl and oxygen by medicinal chemists [7,8]. In this context, the ethoxycarbonyl difluoromethyl group (-CF2CO2Et), a pre-functionalized difluoromethylene precursor, has garnered considerable attention, largely due to the fact that the carboxyl group could be readily transformed into a variety of different functional groups, providing a versatile synthetic handle for the preparation of a range of CF2-containing compounds.
During the last two decades, a variety of methods for integrating the ethoxycarbonyl difluoromethyl group into diverse chemical structures have been documented [9–20]. Prominent among these is the copper-mediated/-catalyzed ethoxycarbonyl difluoromethylation, which has been extensively studied with broad applicability (Scheme 1, top) [21]. Generally, this copper-mediated process can be divided into two main categories. In the first category, a nucleophile is coupled with ethyl halodifluoroethylacetate in the presence of a copper catalyst to produce the targeted ethoxylcarbonyl difluoromethylated compounds. For instance, Kobayashi and co-workers reported in 1986 on the copper powder-mediated coupling of aryl, alkynyl, allyl and alkyl halides with ethyl iododifluoroacetate [22]. The reaction was conducted by mixing copper powder and ethyl iododifluoroacetate in DMSO at room temperature, followed by the addition of the electrophile. Later, in 2012, Lu, Shen and coworkers described a ligandless, copper-mediated, aerobic fluoroalkylation of arylboronic acids with ethyl iododifluoroacetate under mild conditions, using a mixed solvent of DMSO/DMF [23]. Nevertheless, it was noted that under these conditions, the more cost-effective ethyl bromodifluoroacetate resulted in low yields. Following this, Kumadaki and Ashwood's teams found that raising the temperature to 50–55 ℃ allowed a smooth copper-mediated coupling reaction with ethyl bromodifluoroacetate in satisfactory yields [24,25]. In the second category, a nucleophilic ethoxycarbonyl difluoromethylating reagent, specifically α-silyldifluoroacetate, has been shown to react with a variety of aryl or vinyl electrophiles to produce the ethoxylcarbonyl difluoromethylated compounds. Amii and co-workers first reported this type of reaction in 2011, using 20 mol% CuI as the catalyst and CsF as the activator to cleave the C-Si bond in α-silyldifluoroacetate at 60 ℃ [26,27]. Building on this, in 2016, the group of Hartwig demonstrated that using α-silyldifluoroamides as nucleophilic reagents and 20 mol% CuOAc as a catalyst could effectively couple a variety of electron-rich/-poor or sterically hindered aryl, heteroaryl, and vinyl iodides when the reaction was carried out in toluene at 100 ℃ [28].
Mechanistically, it was proposed that copper-mediated/-catalyzed ethoxycarbonyl difluoromethylation initiates with the formation of an active CuⅠ species [CuⅠCF2CO2Et], which then engages with an electrophile to afford the ethoxycarbonyl difluoromethylated product. Early study by Kobayashi showed that copper power reacted with methyl iododifluoroacetate in DMSO or HMPA at room temperature, resulting in the formation of an active specie [CuⅠCF2CO2Me] with a chemical shift at −106 ppm in 19F NMR spectroscopy. Addition of alkenyl iodide to this intermediate led to the disappearance of the initial signal and the emergence of a new signal that is corresponding to the coupling product [22]. Nevertheless, the precise structure of this active ethoxycarbonyl difluoromethylated Cu(Ⅰ) species [CuⅠCF2CO2Et] has yet to be determined. Clearly, isolation, characterization and investigation of the reactivity of the active [CuⅠCF2CO2R] species in copper-mediated ethoxycarbonyl difluoromethylation is urgently needed.
We herein report the isolation and characterization of three ethoxycarbonyl difluoromethylated CuⅠ species [Q]+ [CuⅠ(CF2CO2Et)(Cl)]- 1a (Q = Ph4P) and [Q]+ [CuⅠ(CF2CO2Et)2]- (Q = Ph4P (1b) or nBu4 N (1c)). Stoichiometric reactions of complexes 1b and 1a with 4-NO2C6H4I showed that the reactivity of 1b is much higher than that of 1a. Further studies showed that the highly reactive Cu(Ⅰ) species 1b underwent oxidative addition with ICH2CN to generate the Cu(Ⅲ) complex [Q]+ [CuⅢ(Ⅰ)(CH2CN)(CF2CO2Et)2]-, which was characterized by 19F NMR spectroscopy but could not be isolated. Instead, the introduction of a bipyridine ligand or its derivative into the mixture of 1b and ICH2CN led to the formation of the isolable Cu(Ⅲ) intermediate [(bpy)CuⅢ(CF2CO2Et)2(CH2CN)], which subsequently underwent reductive elimination to give NCCH2CF2CO2Et in 65%−92% yields when methyl 2-iodobenzoate was used to trap the Cu(Ⅰ) species. Based on these mechanistic studies, we achieved the first copper-catalyzed ethoxycarbonyl difluoromethylation of benzyl and allyl chlorides and bromides with good yields. Furthermore, we found that complex 1b could serve as an efficient ethoxycarbonyl difluoromethylating reagent, reacting with a variety of electrophiles such as alkyl iodides, aryl iodides, activated (hetero)aryl bromides and chlorides, acid chlorides, disulfide, hypervalent iodide. In addition, we developed an oxidative ethoxycarbonyl difluoromethylation of complex 1c with various lithium aryl butyl boronates, resulting in the formation of ethoxycarbonyl difluoromethylated arenes in good yields (Scheme 1, bottom).
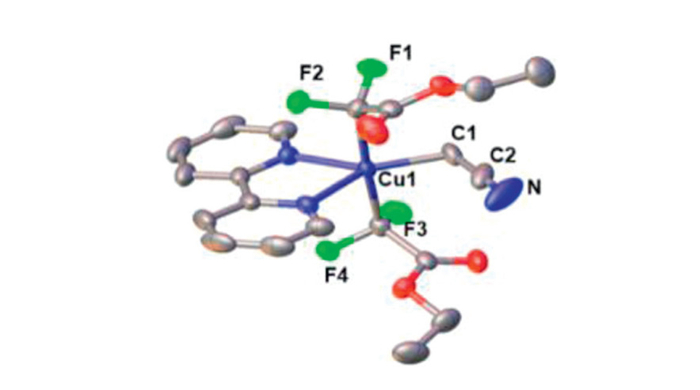
In our quest to isolate the ethoxycarbonyl difluoromethyl-ated Cu(Ⅰ) species, we sought to determine the optimal conditions for the formation of either Q+ [CuⅠ(CF2CO2Et)(Cl)]- 1a (Q = Ph4P) or Q+ [CuⅠ(CF2CO2Et)2]- 1b through the reaction of CuCl with TMSCF2CO2Et in the presence of an activator in various solvents at room temperature. We discovered that the reaction time and the amount of TMSCF2CO2Et/KF is crucial for the smooth generation of both complexes. Stirring a mixture of CuCl with 2.0 equiv. of TMSCF2CO2Et and 3.0 equiv. of KF in DME generated a new signal with a chemical shift of −106.9 ppm in 85% yield after 0.5 h at room temperature (Scheme 2a). With extended reaction times, the peak intensity gradually diminished, and a new peak at −110.7 ppm emerged. By increasing TMSCF2CO2Et to 3.0 equiv. and KF to 6.0 equiv., the species with chemical shift at −110.7 ppm was formed in 80% yield after 1.0 h at room temperature (Scheme 2b), while the other species with chemical shift at −106.9 ppm was obtained in only 5%. Addition of 1.0 equiv. of [Ph4P]+Cl- to the solution, followed by stirring for an additional 5.0–10.0 min and standard workup, yielded 1a in 82% yield and 1b in 78% yield (85% when the cation is nBu4 N). Both complexes were characterized by 1H, 19F and 31P NMR spectroscopies as well as elemental analysis. The structures were validated by single crystal X-ray diffraction (Fig. 1). The solid-state structure of complex 1b (Fig. 1, right) exhibits a linear configuration with the C1-Cu-C2 bond angle of 175.67 (13)°. The two Cu-C bond lengths in complex 1b are 1.937(4) Å and 1.959(4) Å, respectively. These Cu-C bond lengths was longer than known [Ph4P]+ [CuⅠ(CF3)2]- (1.930 Å) [29–31] and are close to those in [Ph4P]+ [CuⅠ(CF2H)2]- (1.934 Å, 1.971 Å) [32].


Complexes 1a and 1b are readily soluble in solvents including dichloromethane, chloroform, THF, DMF, dimethylacetamide (DMAc), acetonitrile but are insoluble in less polar solvents such as toluene, diethyl ether, hexane. When stored in the solid state at −30 ℃ under an argon atmosphere, both complexes 1a and 1b remain stable and do not decompose for over three months. However, it is noted that upon exposure to air, both complexes tend to gradually discolor to green, whether in the solid state or in solution. Additional investigations revealed that the 19F NMR spectrum of complex 1a in HMPA exhibits a chemical shift at −107.1 ppm in 19F spectroscopy using benzotrifluoride as an internal standard, a value that is similar to the species reported previously by Kabayashi and co-workers (Fig. S1 in Supporting information) [22,24–25,33].
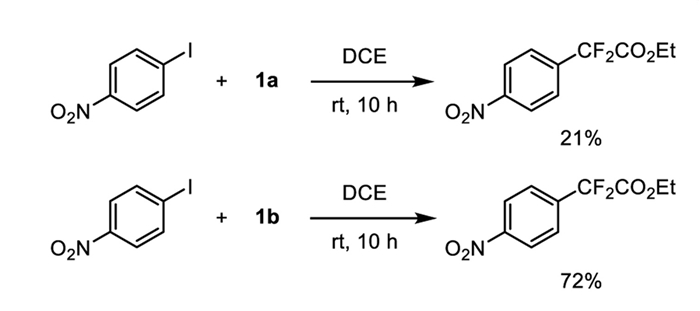
With both complexes 1a and 1b in hand, we proceeded to evaluate their reactivity in the ethoxycarbonyl difluoromethylation of aryl iodides. For this purpose, we selected the reaction with the electron-deficient substrate 1-iodo-4-nitrobenzene as a benchmark to assess their efficacy. When complex 1b (1.0 equiv.) was allowed to react with 1-iodo-4-nitrobenzene in DCE at ambient temperature for 10 h, it yielded the corresponding product ethyl 2,2-difluoro-2-(4-nitrophenyl)acetate with a yield of 72% yield. In contrast, the analogous reaction with complex 1a occurred in 21% yield (Scheme 3). These findings indicate that the reactivity of complex 1b is much higher than that of complex 1a for ethoxycarbonyl difluoromethylation of the aryl iodides.
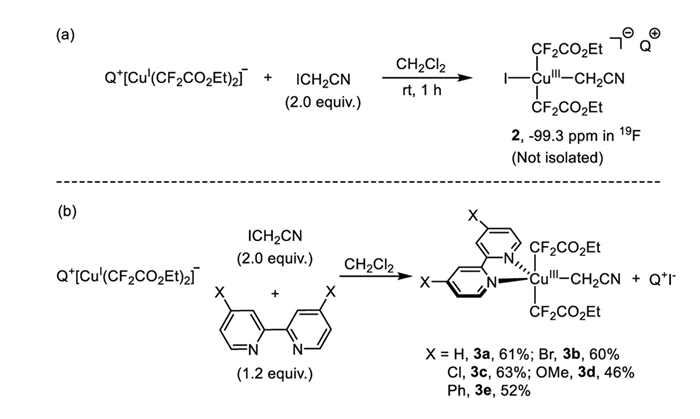
Recently, we reported that Q+ [CuⅠ(CF3)2]- undergoes oxidative addition with iodoacetonitrile, leading to the formation of Cu(Ⅲ) complex Q+ [CuⅢ(CF3)3(CH2CN)]- in high yields [34–38]. We thus would like to probe the potential of complex 1b to engage in a similar oxidative addition with iodoacetonitrile. Mixing complex 1b with 2.0 equiv. of iodoacetonitrile in CH2Cl2 occurred smoothly at room temperature after 1 h to full conversion and gave a new species with chemical shift at −99.3 ppm as identified by 19F NMR spectroscopy. We hypothesize that this new species is likely the oxidative addition product Cu(Ⅲ) complex Q+ [CuⅢ(X)(CH2CN)(CF2CO2Et)2]- 2 (Scheme 4a). Despite our efforts, attempts to isolate and characterize this species in its pure form were unsuccessful. The complex proved to be unstable, decomposing during the crystallization process, which prevented further structural characterization. This instability highlights the challenges in handling higher oxidation state copper species and underscores the need for further research into stabilizing such intermediates for detailed analysis. Subsequently, we focused on examining the oxidative addition reaction of complex 1b in the presence of a bipyridine ligand. Our investigation revealed that when complex 1b was combined with 2.0 equiv. iodoacetonitrile and 1.2 equiv. 2,2′-bipyridine in CH2Cl2, it generated a new species with a chemical shift at −96.8 ppm after 30 min at room temperature. Following standard workup procedures, this complex was successfully isolated, and its structure was identified as [(bpy)CuⅢ(CF2CO2Et)2(CH2CN)] 3a, based on 1H, 19F NMR spectroscopies as well as elemental analysis. The structure of complex 3a was unambiguously confirmed through X-ray diffraction of its single crystals (Fig. 2). The X-ray structure of 3a shows that it adopts a trigonal bipyramid geometry. The Cu-CH2CN bond length was determined to be 1.967(2) Å, while the Cu-CF2CO2Et bond length was found to be 1.997(2) Å. Under identical conditions, Cu(Ⅲ) complexes 3b-e with different bipyridine ligands were synthesized with moderate yields ranging of 40%−63% (Scheme 4b).
Having established the solid- and solution-phase structures of the Cu(Ⅲ) complexes 3a-e, we next sought to investigate their reactivity in C(sp3)-CF2CO2Et bond-forming reductive elimination (Scheme 5) [39–48]. Upon thermolysis of complex 2 in DMF at 60 ℃ for 2 h, the reductive elimination product NCCH2CF2CO2Et (−102.9 ppm, t, J = 15.3 Hz) was obtained in 20% yield (Scheme 5a).
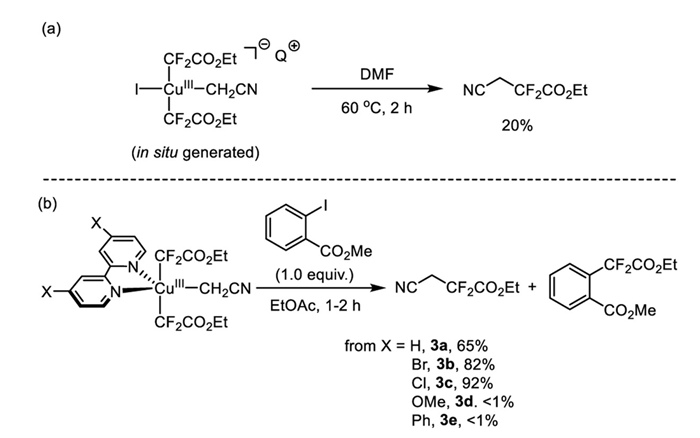
However, extended heating to 5 h led to the disappearance of the reductive elimination product, likely due to side reactions at elevated temperatures (Figs. S2-S4 in Supporting information). Likewise, heating a solution of 3a in ethyl acetate at 40 ℃ for 5 h resulted in complete consumption of the starting material and the formation of reductive elimination product NCCH2CF2CO2Et in 34% yield, along with a mixture of by-product Cu(Ⅰ) species. The yield was improved to 65% with the addition of methyl 2-iodobenzoate (1.0 equiv.) as a trap for the Cu(Ⅰ) species. Under the same conditions, the EtOAc solution of Cu(Ⅲ) complexes 3b, 3c with electron-poor bipyridine ligands were heated at 40 ℃ for 1 h, yielding the reductive elimination product in 82% and 92% yields, respectively (Scheme 5b). In contrast, trace amounts of reductive elimination products were observed for the reactions of Cu(Ⅲ) complexes 3e and 3f with electron-rich or neutral ligand, even after extensive optimization of reaction parameters, including solvents and reaction temperature.
Copper-mediated/-catalyzed cross-coupling of (hetero)aryl halides with XCF2CO2Et (X = Br, I) or TMSCF2CO2Et have been reported previously [26–28]. However, copper-catalyzed cross-coupling of alkyl halides with TMSCF2CO2Et has not been explored until now. With the fundamental steps of oxidative addition and reductive elimination in copper-catalyzed ethoxycarbonyl difluoromethylation established, we aimed to translate them into a copper-catalyzed ethoxycarbonyl difluoromethylation of alkyl halides using TMSCF2CO2Et as the ethoxycarbonyl difluoromethyl source.
Our initial experiments demonstrated that alkyl difluoroacetylation product could be obtained in 45% yield when benzyl bromide, an activated alkyl bromide, was treated with 1.5 equiv. of TMSCF2CO2Et in CH3CN after 24 h at room temperature, using 20 mol% of Cu(CH3CN)4PF6 as the catalyst. Although stoichiometric reactions indicated that the addition of ligand could promote the reductive elimination, our studies did not show acceleration with the addition of bipyridine or its derivatives. We then optimized the reaction parameters, including solvent, reaction time, and temperature, to improve the efficiency of the reaction (Table S3 in Supporting information for details). Ultimately, we found that using 30 mol% of Cu(CH3CN)4PF6 as the catalyst, 2.0 equiv. of KF as the activator, and CH3CN as the solvent, the reaction went to full conversion after 48 h to afford the difluoroacetylation product 4a in 70% yield, as determined by 19F NMR spectroscopy. A control experiment confirmed that without the copper catalyst, the reaction did not occur at all. Our previous studies demonstrated that the halogen ion had a detrimental effect on the copper catalyst, likely causing the low TON of the reaction [30]. Nevertheless, benzylic bromides with both electron-rich or electron-poor substituents could be ethoxylcarbonyl difluoromethylated in good yields (Scheme 6, 4a-d). In addition, benzo[c][1,2,5]oxadiazole derived benzyl bromide also reacted to afford the corresponding product in 54% yield (Scheme 6, 4e). Electron-deficient (hetero)aryl substituted with bromomethyl groups, such as 2-(bromomethyl)−6-methylpyridine 4f and 3-(bromomethyl)−1,2-benzoxazole 4g, could also be converted to the corresponding ethoxylcarbonyl difluoromethylated products smoothly in moderate yields. Likewise, benzylic chlorides reacted effectively under the identical conditions to afford the corresponding ethoxylcarbonyl difluoromethylated products in acceptable yields (Scheme 6, 4a-c). Further investigation showed that allylic bromide or chlorides were also suitable for ethoxylcarbonyl difluoromethylation under the same reaction conditions, affording compounds 4h-i in 50%−90% yields.
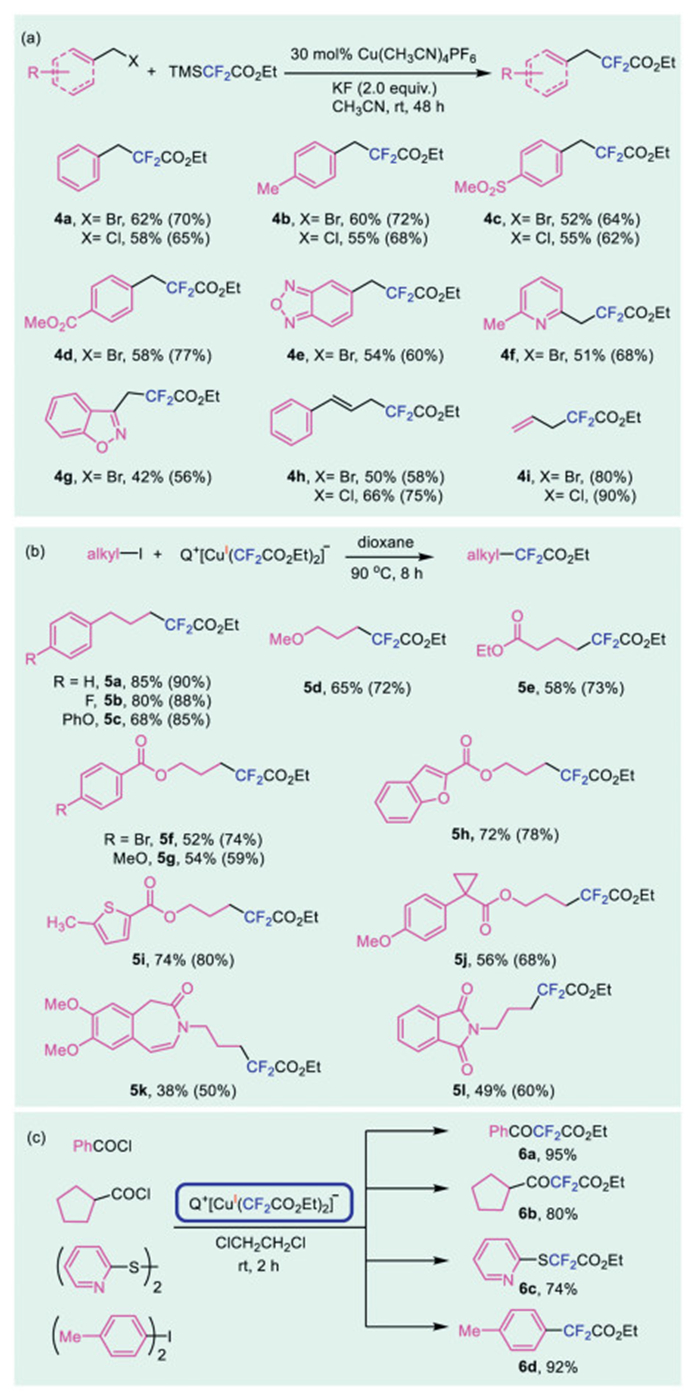
Having successfully developed a copper-catalyzed ethoxylcarbonyl difluoromethylation of activated alkyl halides including benzyl/allyl bromides/chlorides, we sought to broaden the scope of this methodology to encompass unactivated alkyl halides. However, our initial attempts to apply the catalytic reaction conditions to unactivated substrates, such as 3-phenyl propyl iodide with TMSCF2CO2Et were unsuccessful, with reaction conversion remaining below 10%, even when the reaction temperature was increased to 80–100 ℃. Thus, stoichiometric amount of complex 1b was employed to enhance the ethoxylcarbonyl difluoromethylation of activated alkyl halides. After a thorough screening of the various reaction conditions (see Table S4 in Supporting information for details), we found that treating 3-phenyl propyl iodide with 1.5 equiv. of 1b in 1,4-dioxane led to smooth conversion, affording the desired ethoxycarbonyl difluoromethylated compound 5a in 90% yield after 8 h at 90 ℃. As depicted in Scheme 6, the reaction protocol was found to be broadly applicable to a range of unactivated alkyl iodides. Moreover, the protocol was tolerant of common function groups, including ester (5e-j), amide (5k-l), fluoride (5b), bromide (5f). Furthermore, other electrophiles such as acyl chloride, disulfide, diphenyliodonium also reacted with complex 1b to give the target products 6a-d in 74%−95% yields (Scheme 6c).
Previous reports on copper-mediated/-catalyzed ethoxycarbonyl difluoromethylation were largely limited to electro-poor (hetero)aryl iodides [26–28]. In an effort to expand the scope of the reaction, we aimed to include electron-rich aryl iodides as well as activated (hetero)aryl bromides or chlorides. We found that the reaction of electro-poor aryl iodides with complex 1b occurred smoothly in DCE at 40 ℃ to give ethoxycarbonyl difluoromethylated products 7a-e in excellent yields (Scheme 7). By increasing the reaction temperature to 60 ℃, even electro-rich aryl iodides were able to react to produce the desired products in high yields (7f-j). Common functional groups as ester (7a, 7f), cyano (7b), bromide (7c), nitro (7d), Bn-protected phenoxide (7h) were compatible. Notably, aryl iodide with enolizable ketone, which are typically incompatible under the conditions of KF/TMSCF2CO2Et, could also be ethoxycarbonyl difluoromethylated in excellent yields (7e).
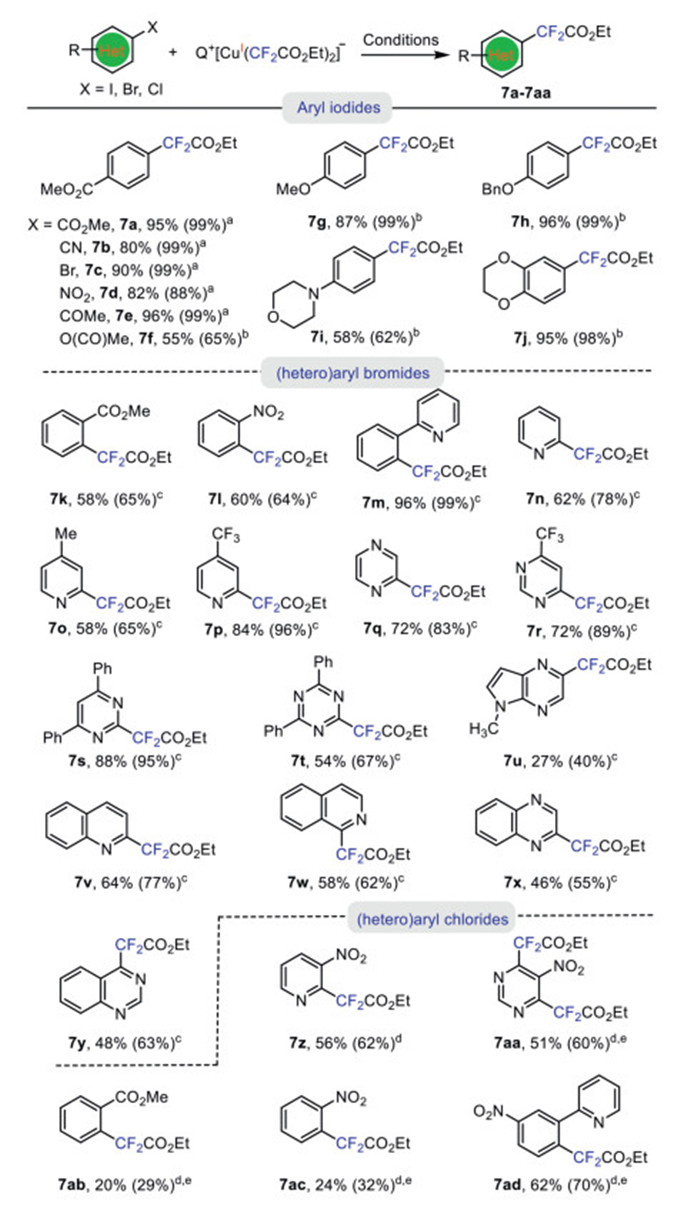
Aryl bromides and chlorides are typically much less reactive than aryl iodides. In general, palladium catalysis has been the mainstay for facilitating coupling reactions with these more challenging substrates [14,49]. Although the reactions of aryl bromides with complex 1b was initially slow, it was discovered that aryl bromides featuring a directing group, such as ester (7k), nitro (7l) or pyridine (7m) could be effectively converted into the desired ethoxycarbonyl difluoromethylated products 7k-m with yields ranging of 58%−96% (Scheme 7).
Conversely, the reactivity of activated heteroaryl bromides was notably higher, reacting with complex 1b at 90 ℃ to give the corresponding ethoxycarbonyl difluoromethylated products 7n-y in good yields. For instance, 2–bromo-substituted substrates were converted into compounds 7n-p in 58%−84% yields, while reactions of 2-bromopyrazine, pyrimidines with bromide at 2- or 4-position proceeded in 72%−88% yields (Scheme 7, 7q-s). A variety of other activated haloheteroarenes, including 1,3,5-triazine, 5λ2-pyrrolo[2,3-b]pyrazine, quinoline, isoquinoline, quinoxaline, quinazoline with the bromide at the ortho-position, were all successfully ethoxycarbonyl difluoromethylated in acceptable to good yields (Scheme 7, 7q-y). Furthermore, activated (hetero)aryl chlorides, with a chlorine atom at the ortho-position of pyridine and with a directing group, such as NO2, were able to be coupled with 1b to give the corresponding ethoxycarbonyl difluoromethylated compounds (7z and 7aa) in moderate yields. Likewise, aryl chlorides featuring a directing group, such as ester (7ab), nitro (7ac) or pyridine (7ad) could be effectively converted into the desired ethoxycarbonyl difluoromethylated products with yields ranging of 20%−62% (Scheme 7, 7ab-ad). Thus, the scope of copper-mediated ethoxycarbonyl difluoromethylation could be significantly broadened for the synthesis of ethoxycarbonyl difluoromethylated heteroarenes.
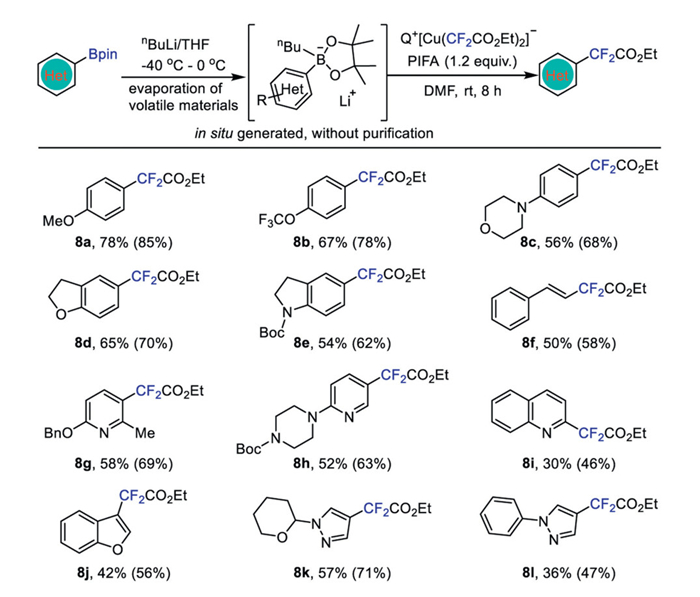
The oxidative fluoroalkylation of nucleophilic substrates, such as aryl boronic acid or its equivalent, in conjunction with a nucleophilic fluoroalkylating reagent is a widely applicable method for synthesizing fluoroalkylated compounds. This approach is advantageous due to the lack of air sensitivity [32,50]. Our initial attempts involving the direct use of arylboronic acids or arylboronic acid pinacol esters with complex 1b did not yield promising results. We hypothesized that a slow transmetalation step might be hindering the reaction. To address this, we employed lithium 4-methoxyphenyl nbutyl pinacol borate, which was in situ generated from 4-methoxyphenylboronic acid pinacol ester with equimolar amount of nBuLi, to enhance the transmetalation step [32,51]. We were pleased to find that with this modified approach, the desired product was observed in 18% yield when the reaction was conducted in DMF after 8.0 h at room temperature. The yield could be improved to 40% when complex 1c with nBu4N+ cation was used as the nucleophilic ethoxycarbonyl difluoromethyl source and Selectfluor was used as the oxidant. Further optimization with bis(trifluoroacetoxy)iodobenzene (PIFA) as the oxidant led to a significantly higher yield of 85% (Table S7 in Supporting information for details).
To assess the versatility of this protocol, a variety of lithium (hetero)aryl nbutyl pinacol esters were subjected to the optimized conditions (Scheme 8). Generally, electron-rich, electron-neutral and electron-deficient (hetero)aryl boronic acid pinacol esters were all successfully converted to the corresponding ethoxycarbonyl difluoromethylated products in moderate to high yields. Moreover, aryl boronic acid pinacol esters with a cyclicheteroatom moieties, such as morpholine (8c), 2,3-dihydrobenzofuran (8d), N-Boc indoline (8e), could be effectively transformed into the ethoxycarbonyl difluoromethylated (hetero)arenes in good yields. Additionally, vinyl boronic acid pinacol ester was found to be ethoxycarbonyl difluoromethylated in 65% yield (8f). Electron-deficient (hetero)aryl boronic acid pinacol esters such as pyridine (8g-h), quinolone (8i), and (hetero)aryl boronic acid pinacol esters such as benzofuran (8j), pyrazole (8k-l), were all effectively ethoxycarbonyl difluoromethylated in decent yields.
In summary, our curiosity about the active species in copper-mediated ethoxycarbonyl difluoromethylation of organohalides led us to isolate and characterize the first ethoxycarbonyl difluoromethylated Cu(Ⅰ) species Q+ [CuⅠ(CF2CO2Et)(Cl)]- 1a and Q+ [CuⅠ(CF2CO2Et)2]- 1b. Stoichiometric reactions of complexes 1a and 1b with electron poor aryl iodide 1-iodo-4-nitrobenzene showed that complex 1b is much more reactive than complex 1a. In addition, oxidative addition of complex 1b with iodoacetonitrile in the presence of a bidentate nitrogen ligand led to the formation of stable Cu(Ⅲ) complexes [(L)CuⅢ(CF2CO2Et)2(CH2CN)] 3a-e that underwent reductive elimination to give NCCH2CF2CO2Et in high yields.
These stoichiometric reactions laid the foundation for the development of the first copper-catalyzed ethoxycarbonyl difluoromethylation of benzylic or allylic halides with TMSCF2CO2Et. Extended research revealed that complex 1b, due to its high reactivity, serves as a powerful ethoxycarbonyl difluoromethylating reagent to allow a broad range of substrates, including electron-deficient and electron-rich aryl iodides, activated heteroaryl chlorides and aryl bromides with a directing group, heteroaryl bromides, alkyl iodides, as well as other electrophiles such as acid chloride, disulfide, diphenyliodonium, to undergo ethoxycarbonyl difluoromethylation in moderate to excellent yields. Moreover, complex 1c effectively participated in oxidative ethoxycarbonyl difluoromethylation with a variety of lithium nbutyl arylboronic acid pinacol esters, alkenyl and heteroarene in the presence of an oxidant. Ongoing studies in our laboratory are delving into the mechanistic aspect of the copper-mediated or -catalyzed fluoroalkylation reactions, with the findings to be reported in due course.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Yixiao Zhao: Writing – review & editing, Writing – original draft, Methodology, Investigation. Gavin Chit Tsui: Writing – review & editing, Validation, Project administration, Funding acquisition. Qilong Shen: Writing – review & editing, Writing – original draft, Supervision, Resources, Project administration, Funding acquisition, Conceptualization.
We gratefully acknowledge the financial support from the National Key Research and Development Program of China (No. 2021YFF0701700), the National Natural Science Foundation of China (No. 22061160465) and the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB0590000). We also thank the Research Grants Council of Hong Kong (NSFC/RGC Joint Research Scheme, No. N_CUHK403/20), the Chinese University of Hong Kong (Faculty of Science - Direct Grant for Research) for funding.
Supplementary material associated with this article can be found, in the online version, at doi:
Y. Zhou, J. Wang, Z. Gu, et al., Chem. Rev. 116 (2016) 422–518. doi: 10.1021/acs.chemrev.5b00392
Y. Yu, A. Liu, G. Dhawan, et al., Chin. Chem. Lett. 32 (2021) 3342–3354. doi: 10.1016/j.cclet.2021.05.042
J. He, Z. Li, G. Dhawan, et al., Chin. Chem. Lett. 34 (2023) 107578. doi: 10.1016/j.cclet.2022.06.001
A.S. Nair, A.K. Singh, A. Kumar, et al., Processes 10 (2022) 2054. doi: 10.3390/pr10102054
D. O'Hagan, R.J. Young, Med. Chem. Res. 32 (2023) 1231–1234. doi: 10.1007/s00044-023-03094-y
E. Henary, S. Casa, T.L. Dost, J.C. Sloop, M. Henary, Pharmaceuticals 17 (2024) 281. doi: 10.3390/ph17030281
G.M. Blackburn, D.A. England, F. Kolkmann, J. Chem. Soc., Chem. Commun. (1981) 930–932.
D. O'Hagan, Y. Wang, M. Skibinski, A.M.Z. Slawin, Pure Appl. Chem. 84 (2012) 1587–1595. doi: 10.1351/pac-con-11-09-26
K. Araki, M. Inoue, Tetrahedron 69 (2013) 3913–3918. doi: 10.1016/j.tet.2013.03.041
S.Z. Ge, W. Chaladaj, J.F. Hartwig, J. Am. Chem. Soc. 136 (2014) 4149–4152. doi: 10.1021/ja501117v
Z. Feng, Q.Q. Min, Y.L. Xiao, B. Zhang, X.G. Zhang, Angew. Chem. Int. Ed. 53 (2014) 1669–1673. doi: 10.1002/anie.201309535
Y.L. Xiao, W.H. Guo, G.Z. He, Q. Pan, X.G. Zhang, Angew. Chem. Int. Ed. 53 (2014) 9909–9913. doi: 10.1002/anie.201405653
A. Tarui, S. Shinohara, K. Sato, M. Omote, A. Ando, Org. Lett. 18 (2016) 1128–1131. doi: 10.1021/acs.orglett.6b00232
T. Xia, L. He, Y.A. Liu, J.F. Hartwig, X. Liao, Org. Lett. 19 (2017) 2610–2613. doi: 10.1021/acs.orglett.7b00938
F. Zhang, Y.L. Xiao, X.G. zhang, Acc. Chem. Res. 51 (2018) 2264–2278. doi: 10.1021/acs.accounts.8b00230
A.D. Dilman, V.V. Levin, Acc. Chem. Res. 51 (2018) 1272–1280. doi: 10.1021/acs.accounts.8b00079
Q.H. Zhou, A. Ruffoni, R. Gianatassio, et al., Angew. Chem. Int. Ed. 52 (2013) 3949–3952. doi: 10.1002/anie.201300763
W.G. Zhang, Y. Wang, Tetrahedron Lett. 59 (2018) 1301–1308. doi: 10.1016/j.tetlet.2018.02.056
A. Lemos, C. Lemaire, A. Luxen, Adv. Synth. Catal. 361 (2019) 1500–1537. doi: 10.1002/adsc.201801121
D.Q. Dong, H. Yang, J.L. Shi, et al., Org. Chem. Front. 7 (2020) 2538–2575. doi: 10.1039/d0qo00567c
D.Q. Dong, S.H. Yang, P. Wu, et al., Molecules 27 (2022) 8461. doi: 10.3390/molecules27238461
T. Taguchi, O. Kitagawa, T. Morikawa, et al., Tetrahedron Lett. 27 (1986) 6103–6106. doi: 10.1016/S0040-4039(00)85409-X
Q. Qi, Q. Shen, L. Lu, J. Am. Chem. Soc. 134 (2012) 6548–6551. doi: 10.1021/ja301705z
K. Sato, M. Omote, A. Ando, I. Kumadaki, Chem. Pharm. Bull. 47 (1999) 1013–1016. doi: 10.1248/cpb.47.1013
I. Cottrell, C. Cowden, D. Wallace, M. Ashwood, Tetrahedron Lett. 43 (2002) 9271–9273. doi: 10.1016/S0040-4039(02)02276-1
K. Fujikawa, Y. Fujioka, A. Kobayashi, H. Amii, Org. Lett. 13 (2011) 5560–5563. doi: 10.1021/ol202289z
K. Fujikawa, A. Kobayashi, H. Amii, Synthesis 44 (2012) 3015–3018. doi: 10.1055/s-0032-1316761
S.I. Arlow, J.F. Hartwig, Angew. Chem. Int. Ed. 55 (2016) 4567–4572. doi: 10.1002/anie.201600105
H. Liu, Q. Shen, Org. Chem. Front. 6 (2019) 2324–2328. doi: 10.1039/c9qo00527g
H. Liu, J. Wu, Y.X. Jin, X.B. Leng, Q. Shen, J. Am. Chem. Soc. 143 (2021) 14367–14378. doi: 10.1021/jacs.1c07408
T. Dong, Q. Shen, G.C. Tsui, Chem. Sci. 15 (2024) 11550–11556. doi: 10.1039/d4sc02075h
H.W. Zhao, X. Leng, W. Zhang, Q. Shen, Angew. Chem. Int. Ed. 61 (2022) e202210151. doi: 10.1002/anie.202210151
T. Taguchi, O. Kitagawa, Y. Kobayashi, Chem. Lett. 18 (1989) 389–392. doi: 10.1246/cl.1989.389
Y. Luo, Y. Li, J. Wu, X.S. Xue, J.F. Hartwig, Q. Shen, Science 381 (2023) 1072–1079. doi: 10.1126/science.adg9232
A. Casitas, X. Ribas, Chem. Sci. 4 (2013) 2301–2318. doi: 10.1039/c3sc21818j
L. Liu, Z.F. Xi, Chin. J. Chem. 36 (2018) 1213–1221. doi: 10.1002/cjoc.201800365
H. Liu, Q. Shen, Coord. Chem. Rev. 442 (2021) 213923. doi: 10.1016/j.ccr.2021.213923
Q. Zhang, S. Tong, M.X. Wang, Acc. Chem. Soc. 55 (2022) 2796–2810. doi: 10.1021/acs.accounts.2c00316
A.M. Romine, N. Nebra, A.I. Konovalov, et al., Angew. Chem. Int. Ed. 54 (2015) 2745–2749. doi: 10.1002/anie.201411348
M. Paeth, S.B. Tyndall, L.Y. Chen, et al., J. Am. Chem. Soc. 141 (2019) 3153–3159. doi: 10.1021/jacs.8b12632
Z.H. Lu, H. Liu, S.Y. Liu, et al., Angew. Chem. Int. Ed. 58 (2019) 8510–8514. doi: 10.1002/anie.201904041
S.S. Liu, H. Liu, S.H. Liu, et al., J. Am. Chem. Soc. 142 (2020) 9785–9791. doi: 10.1021/jacs.0c03304
G.Y. Wang, M. Li, X.B. Leng, X.S. Xue, Q. Shen, Chin. J. Chem. 40 (2022) 1924–1930. doi: 10.1002/cjoc.202200230
W. Yan, S. Carter, C.T. Hsieh, et al., J. Am. Chem. Soc. 145 (2023) 26152–26159. doi: 10.1021/jacs.3c08510
D. Joven-Sancho, A. Eche-verri, N. Saffon-Merceron, J. Contreras-García, N. Nebra, Angew. Chem. Int. Ed. 62 (2023) e202319412.
Y.C. Weng, Y.X. Jin, J. Wu, et al., J. Am. Chem. Soc. 146 (2024) 23555–23565. doi: 10.1021/jacs.4c07552
G.Y. Wang, H.Y. Li, X.B. Leng, L. Lu, Q. Shen, Chin. J. Chem. 42 (2024) 1107–1113. doi: 10.1002/cjoc.202400041
W. Yan, A.T. Poore, L. Yin, et al., J. Am. Chem. Soc. 146 (2024) 15176–15185. doi: 10.1021/jacs.4c01668
S. Ge, S. Arlow, M. Mormino, J.F. Hartwig, J. Am. Chem. Soc. 136 (2014) 14401–14404. doi: 10.1021/ja508590k
L.L. Chu, F.L. Qing, Acc. Chem. Res. 47 (2014) 1513–1522. doi: 10.1021/ar4003202
W.C. Huang, X.L. Wan, Q. Shen, Angew. Chem. Int. Ed. 56 (2017) 11986–11989. doi: 10.1002/anie.201706868

Figure 1 ORTEP drawings of Q+ [CuⅠ(CF2CO2Et)(Cl)]- 1a (left) and Q+ [CuⅠ(CF2CO2Et)2]- 1b (right). Thermal ellipsoids are drawn at 30% probability.

Scheme 4 Oxidative addition of iodoacetonitrile to complex 1b in the absence or presence of bipyridine ligand.

Figure 2 ORTEP drawings of [(bpy)CuⅢ(CF2CO2Et)2(CH2CN)] (3a). Thermal ellipsoids are drawn at 30% probability.

Scheme 6 Copper-catalyzed or -mediated ethoxycarbonyl difluoromethylation of alkyl halides and other electrophiles. Isolated yields (yields in parenthesis were determined by 19F NMR spectroscopy using benzotrifluoride as an internal standard). (a) Alkyl halides (0.40 mmol), TMSCF2CO2Et (0.60 mmol), Cu(CH3CN)4PF6 (0.12 mmol), KF (0.80 mmol) in CH3CN (4.0 mL) at room temperature for 48 h. (b) Alkyl iodides (0.40 mmol), complex 1b (0.60 mmol) in 1,4-dioxane (4.0 mL), 90 ℃ for 8 h. (c) Other electrophiles (0.40 mmol), complex 1b (0.28 mmol) in DCE (4.0 mL) at room temperature for 2 h.

Scheme 7 Reaction of complex [Ph4P]+ [Cu(CF2CO2Et)2]- 1b with (hetero)aryl halides. Isolated yields (yields in parenthesis were de-termined by 19F NMR spectroscopy using benzotrifluoride as an internal standard). a Complex 1b (0.24 mmol), ArI (0.40 mmol) in DCE (4.0 mL) 40 ℃ for 16 h. b Complex 1b (0.28 mmol), ArI (0.40 mmol) in DCE (4.0 mL) 60 ℃ for 15 h. c Complex 1b (0.40 mmol), heteroaryl bromide (0.40 mmol) in 1,4-dioxane (4.0 mL) 80 ℃ for 12 h. d Complex 1b (0.40 mmol), heteroaryl chloride (0.40 mmol) in 1,4-dioxane (4.0 mL) 90 ℃ for 12 h. e Complex 1b (0.60 mmol).

Scheme 8 Oxidation difluoroacetylation of in situ generated lithium nbutyl arylboronic acid pinacol esters with complex 1c. Isolated yields (yields in parenthesis were determined by 19F NMR spectroscopy using benzotrifluoride as an internal standard). Aryl-Bpin (0.40 mmol), complex 1c (0.60 mmol), PIFA (0.48 mmol) in DMF (4.0 mL) at room temperature for 8 h.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:


