Scheme 1.
The synthesis of aminothioalkenes.
The sulfenylation of enamine esters with heterocyclic thiols or disulfides in water and application to DNA-compatible chemistry
Haoyu Tian , Xiaolin Cui , Guiwei Yao , Wenyan Wei , Junchao Lu , Senyao Zheng , Xingjian Wang , Xun Chen , Guangkuan Zhao , Dulin Kong

Enamines are vinylogous amines, characterized by an amino group attached at the α-position relative to an olefinic C=C double bond, resulting in conjugation between the two. These compounds are readily synthesized from aldehydes or ketones through reactions with either acyclic or cyclic secondary amines. Due to the conjugation between the amino group's lone pair of electrons and the C=C double bond, the electron density at the β-carbon atom is increased. This makes the β-carbon a nucleophilic center, susceptible to electrophilic attack. Enamines can be described as a resonance hybrid of two canonical structures, highlighting their potential as intermediates that can participate in various basic synthetic applications, such as alkylation, acylation, amidation, reduction, oxidation, conjugate addition, and cycloaddition reactions, which can be converted to many other useful compounds [1–13]. Leveraging their intriguing reactivity, the synthesis of novel derivatives can further enhance the utility of enamines. Particular attention has been paid to intergrate the sulfur atom into enamines, as a large number of drugs contain sulfide moieties on their skeletons [14–16].
Given the significant application potentiality of α-aminothioalkenes, various synthetic methods for oxidative C–S bond formation using thiols or disulfides have been developed in recent years. Loh et al. [17] introduced a direct, regioselective thiolation approach to synthesize β-amino sulfides via palladium-catalyzed C–S bond formation from stable enamines and thiols (Scheme 1a). However, this method has limitations, such as the use of toxic metal catalysts [18]. Concurrently, Wan et al. [19] reported KIO3-catalyzed aerobic cross-coupling reactions of enaminones and thiophenols using bio-based green solvent ethyl lactate as the reaction medium (Scheme 1b). Subsequently, Prabhu et al. [20] demonstrated an iodine-catalyzed sulfenylation of enaminones using DMSO as an oxidant, enabling the sulfenylation of enaminones with a broad range of heterocyclic thiols and thiones (Scheme 1c). Maddani et al. [21] later developed a more attractive cross-dehydrogenation coupling process using an HBr/DMSO oxidation system (Scheme 1d). In an effort to avoid oxidants, Lei [22] and Xie et al. [23] independently achieved electrochemical oxidative cross-coupling of aminoalkenes and thiophenols (Scheme 1e).
Compared to thiol derivatives, disulfides are considered a less toxic and more manageable sulfur source [24–26]. Utilizing disulfides in reactions can broaden the scope and make the process more adaptable to a wider range of applications. Consequently, various methods have been developed for synthesizing α-aminothioalkenes using disulfides. Firstly, Deng et al. [27] developed a silver-mediated oxidative vinylic C–H bond sulfenylation of enamides with disulfides (Scheme 1f). However, this reaction required 1.2 equiv. of the disulfide, as 1 equiv. of the arylthiol radical generated during the reaction is captured by the metal silver. Yu et al. [28] developed a direct oxidative C(sp2)-H α-sulfenylation of enaminones using 0.6 equiv. of disulfides. However, this reaction requires the use of a metal catalyst, specifically Cu(OAc)2 (Scheme 1g). As an enhancement, Du et al. [29] innovatively synthesized thioenamine derivatives through a tetrabutylammonium iodide/tert–butyl hydroperoxide-mediated oxidative coupling of enaminones with 0.6 equiv. of disulfides (Scheme 1h). Additionally, Du et al. [30] also described an oxidative sulfenylation reaction where disulfides with PhICl2 in DMF at room temperature led to the in situ formation of reactive sulfenyl chloride (RSCl), which reacted with enaminones to yield a series of α-thioenaminones (Scheme 1i). It should be noted that Deng et al. [31] developed an efficient multi-component synthesis of thioenamines starting from disulfides, amines, and alkynes mediated by iodine (Scheme 1j). Beyond the use of thiols and disulfides, Dong et al. [32] reported an iodine-catalyzed one-pot tandem synthesis of thioenamines using readily available phenyl isothiocyanate, alcohols, and enamines, further expanding research in organosulfur chemistry.
It is not surprising that most oxidative C–S bond formation have been carried out in organic solvents, with only a few exceptions. Therefore, exploring the study of aminothioalkenes in "green" solvents, particularly water, would be highly valuable [33–35]. Water is not only environmentally benign and abundant but also biocompatible, making it an ideal solvent for reactions involving biological molecules or processes. This makes water especially important in fields such as biochemistry and pharmaceuticals, where a natural and compatible solvent is crucial [36].
Heterocyclic sulfides, a significant class of sulfur compounds, play a crucial role in medicinal chemistry and are commonly found in a wide range of substances, from natural products to pharmaceutical agents [15,16]. As a result, considerable efforts have been devoted to the synthesis of heterocyclic sulfides within the field of heterocyclic chemistry [37–52]. Recently, we reported metal-free sulfenylation of phenol and arylamine derivatives [53], imidazoheterocycles [54], and sulfoxonium ylides [55] employing an oxidative C-S bond formation strategy with heterocyclic thiols. Building on our ongoing efforts to rapidly synthesize novel bioactive heterocycles, we investigated the use of enamine esters as key intermediates for creating heterocyclic sulfides through an oxidative C-S bond formation strategy with various heterocyclic thiols. In this study, we present a convenient and efficient method for the C–H bond sulfenylation of enamine esters under mild, environmentally friendly conditions (Scheme 1k). This approach is particularly noteworthy for its straightforward reaction conditions, broad substrate scope, and tolerance of a wide range of functional groups. Based on the proposed mechanism, we successfully developed a sulfenylation reaction of enamine esters with disulfides that proceeds without the need for an oxidant (Scheme 1l).
To determine the optimal reaction conditions, we selected enamine ester 1a and 1-phenyltetrazole-5-thiol 2a as model substrates (Table 1). Building on our previous work involving the sulfenylation of sulfoxonium ylides [55] using KIO3 as an oxidant, we initially tested the reaction of 1a and 2a in water with KIO3, yielding the desired sulfenylation product at a low 17% yield (entry 1). Encouraged by this result, we screened other common oxidants, including K2S2O8, KBrO3, TBHP, PIDA, I2, and H2O2. Among them, K2S2O8 emerged as the most effective, promoting the sulfenylation reaction with an impressive 89% yield (entries 2–9). Notably, in the absence of any oxidant, the reaction failed, and no product 3a was detected (entry 10). Next, we explored the effect of different solvents on the reaction. While the reaction proceeded in organic solvents such as DMSO, MeCN, and acetone, the yields of the target product were much lower (entries 12, 15, 16). Other solvents, including ethanol, DMF, 10% PEG-400 aqueous solution, and 1,4-dioxane, were entirely ineffective, with no desired product detected (entries 11, 13, 14, 17). Varying the amount of thiol 2a also impacted the yields; decreasing or increasing the thiol 2a amount led to reduced yields (entries 3 vs. 18–20). Further investigation into the effect of temperature revealed that reaction yield positively correlated with temperature. Lowering the temperature from 80 ℃ to 60, 50, 25 ℃ resulted in decreased yield, respectively (entries 3 vs. 21–23). However, increasing the temperature from 80 ℃ to 110 ℃ did not significantly improve the yield (entry 3 vs. 24). Consequently, we conducted subsequent experiments at 80 ℃. After thoroughly screening various parameters, we determined that the optimal conditions for the oxidative coupling reaction involved reacting enamine ester 1a (1 equiv.) and 1-phenyltetrazole-5-thiol 2a (1.2 equiv.) in the presence of K2S2O8 (1 equiv.) in 2 mL of H2O at 80 ℃ for 12 h (entry 3).
 DownLoad:
CSV
DownLoad:
CSV
 |
|||||
| Entry | Oxidant (equiv.) | Solvent | Temp. (℃) | 2a (equiv.) | Yield (%)b |
| 1 | KIO3 (1.0) | H2O | 80 | 1.2 | 17 |
| 2 | K2S2O8 (0.8) | H2O | 80 | 1.2 | 45 |
| 3 | K2S2O8 (1.0) | H2O | 80 | 1.2 | 89 |
| 4 | K2S2O8 (2.0) | H2O | 80 | 1.2 | 89 |
| 5 | TBHP (1.0) | H2O | 80 | 1.2 | nd |
| 6 | I2 (1.0) | H2O | 80 | 1.2 | nd |
| 7 | PIDA (1.0) | H2O | 80 | 1.2 | nd |
| 8 | KBrO3 (1.0) | H2O | 80 | 1.2 | 23 |
| 9 | H2O2 (1.0) | H2O | 80 | 1.2 | nr |
| 10 | – | H2O | 80 | 1.2 | nr |
| 11 | K2S2O8 (1.0) | Ethanol | 80 | 1.2 | nr |
| 12 | K2S2O8 (1.0) | DMSO | 80 | 1.2 | 45 |
| 13 | K2S2O8 (1.0) | DMF | 80 | 1.2 | nr |
| 14 | K2S2O8 (1.0) | 10% PEG-400 aqueous solution | 80 | 1.2 | nd |
| 15 | K2S2O8 (1.0) | MeCN | 80 | 1.2 | 23 |
| 16 | K2S2O8 (1.0) | Acetone | 80 | 1.2 | 17 |
| 17 | K2S2O8 (10) | 1,4-Dioxane | 80 | 1.2 | nr |
| 18 | K2S2O8 (1.0) | H2O | 80 | 0.8 | 71 |
| 19 | K2S2O8 (1.0) | H2O | 80 | 1.0 | 87 |
| 20 | K2S2O8 (1.0) | H2O | 80 | 1.5 | 85 |
| 21 | K2S2O8 (1.0) | H2O | 25 | 1.2 | 20 |
| 22 | K2S2O8 (1.0) | H2O | 50 | 1.2 | 49 |
| 23 | K2S2O8 (1.0) | H2O | 60 | 1.2 | 67 |
| 24 | K2S2O8 (1.0) | H2O | 110 | 1.2 | 91 |
| a Reaction conditions: 1a (0.5 mmol), 2a (x mmol), oxidant (y equiv.) in solvent (2 mL) for 12 h. b Isolated yields. nr: no reaction occurred. nd: no desired product was detected. |
|||||
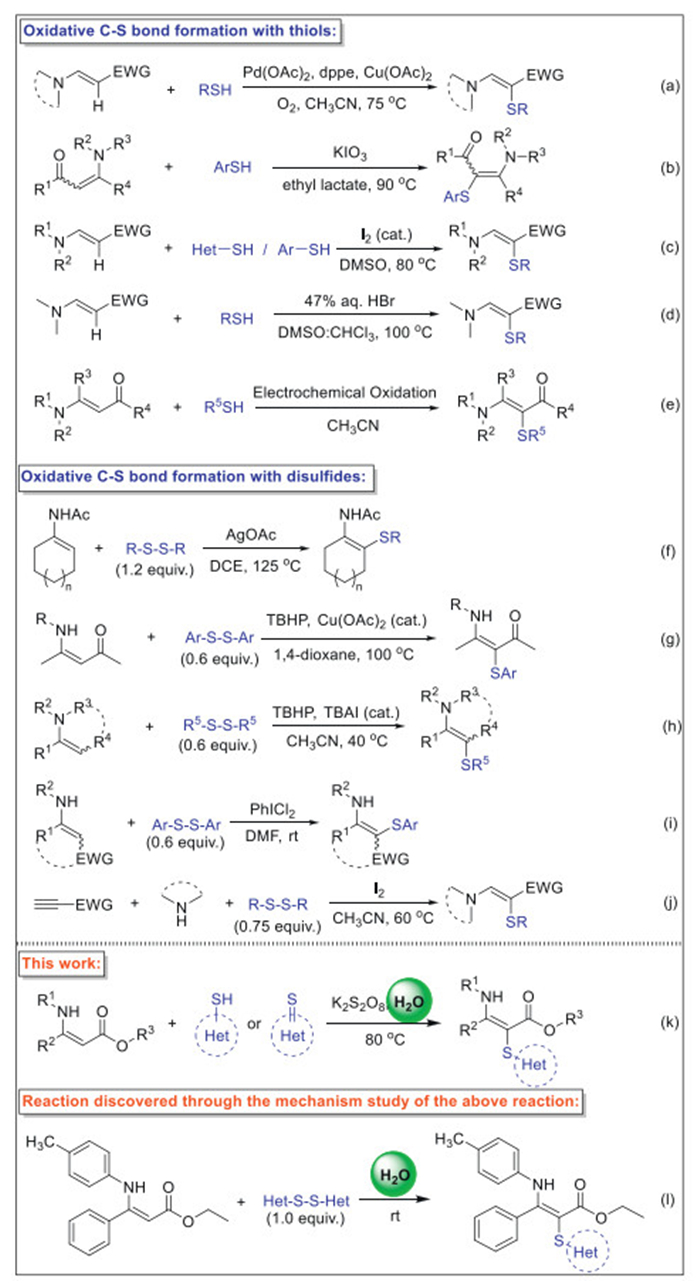
With the optimized reaction conditions in hand, we then proceeded to investigate the substrate generality of our reaction. The reactions of phenyl-, 4-methoxyphenyl-, 4-ethoxyphenyl-, 3-acetamidophenyl-, 4-hydroxylphenyl-, 2-hydroxyethyl-, and methyl-substituted tetrazole thiols with 1a resulted in the formation of the corresponding thioenamines with satisfactory yields, respectively (Scheme 2, 3a-3g). Encouragingly, other heterocyclic thiols, including 5-methyl-1,3,4-thiadiazole-2-thiol, 4-methylthiazole-2-thiol, 5-phenyl-1,3,4-oxadiazole-2(3H)-thiol, benzo[d]oxazole-2-thiol, benzo[d]thiazole-2-thiol, 1H-benzo[d]imidazole-2-thiol, pyridine-2-thiol, and pyrimidine-2-thiol also reacted smoothly with 1a to afford the desired α-thioenamine derivatives in good yields (3h-3o). Further investigation revealed that phenyl and aliphatic thiols exhibited lower reactivity compared to heterocyclic thiols. Reactions of 1a with 4-methoxybenzenethiol and 1-hexanethiol were attempted under various conditions, including extended reaction times, increased quantities of K2S2O8, and elevated temperatures. However, these thiols failed to yield the desired product. Subsequent mechanistic studies suggest that this outcome may result from the formation of intermediates such as 1,2-bis(4-methoxyphenyl)disulfane or 1-hexyl-2-pentyldisulfane, which appear to be less reactive with 1a than heterocyclic disulfides. Moreover, reactions of 1a with 1,2-bis(4-methoxyphenyl)disulfane and 1-hexyl-2-pentyldisulfane also proved unsuccessful, further supporting the conclusion that phenyl and aliphatic disulfides are less reactive than their heterocyclic counterparts.

Motivated by these results, we next explored the scope of the coupling reaction of 2a with a variety of enamine esters and we are gratifyingly pleased with the generality of this method (Scheme 3). In general, variety of substituents on the N-aryl group, including electron-rich and electron-poor, showed no significant influence on the results, and various enamine esters reacted smoothly to afford the desired α-thioenamine derivatives in good to excellent yields. Significantly, halogenated aromatic moieties remained intact under the reaction conditions, facilitating further structural elaboration of the products. In addition, by changing the N-substituent to an alkyl group, the desired thiolation product 3ag and 3ah was obtained respectively, in good yields (67% and 69%, respectively). In addition to enamine esters, enaminone was also found to suitable for this reaction to afford product 3as in an excellent yield of 92%. These active functional groups allow the construction of complex molecules containing a sulfur moiety after subsequent derivatization.
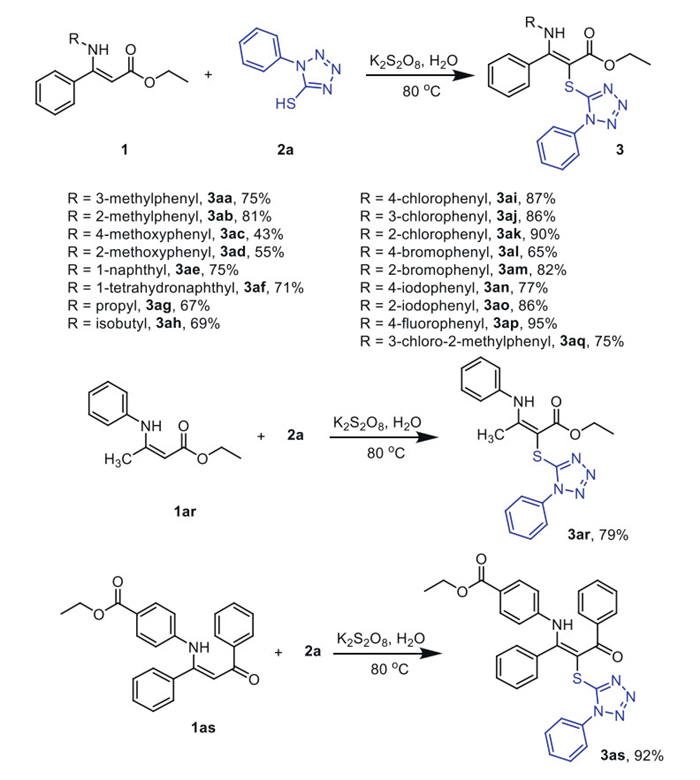
To understand the reaction mechanism, we conducted a series of control experiments (Scheme 4). First, in the existence of radical scavenger TEMPO (2,2,4,4-tetramethyl-1-piperidinyloxy) or BHT (butylated hydroxytoluene), the reaction of enamine ester (1a) and 1-phenyltetrazole-5-thiol (2a) under the standard conditions proceeded well to form 3a in 46% and 85% yield, respectively, indicating that the transformation might not go through a radical pathway (Scheme 4a). There was no reaction when 1a was used alone under the standard conditions, indicating that 1a cannot produce a reaction intermediate with K2S2O8 (Scheme 4b). With treatment, 2a only under standard conditions afforded disulfide compound 4a in a 94% yield (Scheme 4c). The yield decreased slightly under argon protection (Scheme 4d). In addition, in the absence of K2S2O8, the reaction between 1a and disulfide compound 4a yielded 3a at 91% (Scheme 4e), and under standard conditions, the same reaction yielded 3a at 83% (Scheme 4f), suggesting that the transformation may proceed through the disulfide intermediate 4a. Even if the sulfenylation of enamine ester into α-thioenamine derivatives is well-known, to our knowledge, this result shows that the formation of a α-thioenamine derivative can be carried out in the absence of any activating agent since simple stirring in H2O is sufficient to promote the sulfenylation reactions.
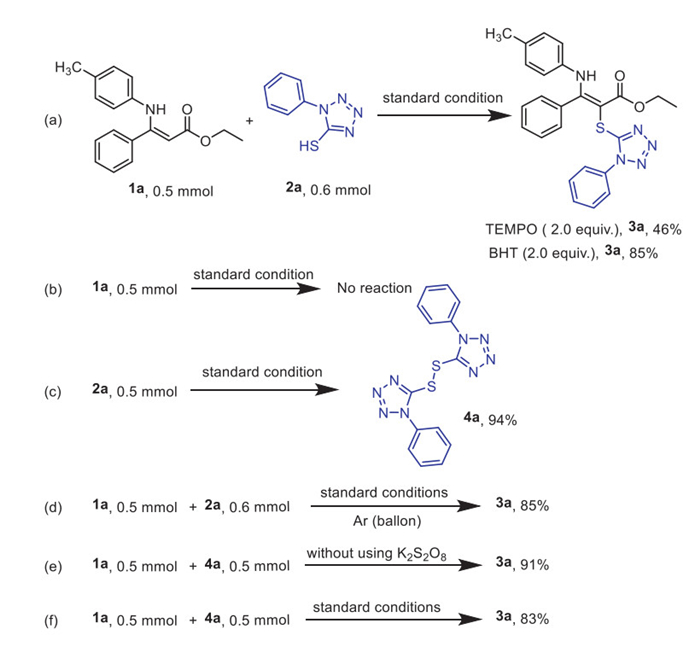
Based on the experimental results and previous studies [55], we propose a plausible mechanism outlined in Scheme 5. The process primarily involves two steps: first, the disulfide compound 4a is formed from 1-phenyltetrazole-5-thiol 2a through oxidation by K2S2O8. This active disulfide species then reacts with the nucleophilic α-carbon of the enamine ester 1a' to produce the desired product 3a, along with the regeneration of 1-phenyltetrazole-5-thiol 2a.
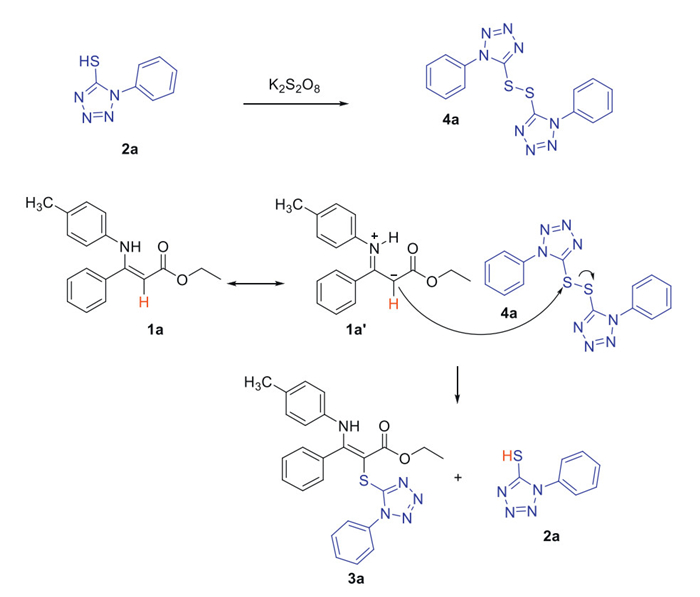
As a logical next step, we attempted the sulfenylation reaction between enamine esters and various disulfide compounds. We were pleased to find that various disulfides reacted smoothly with 1a, yielding the desired α-thioenamine compounds in excellent yields (Table 2). A comparison of the yields obtained using disulfides versus thiols clearly demonstrates that this oxidant-free process is highly favorable, as the yields of the sulfenylation derivatives are significantly higher. Although the reaction requires the use of 1 equiv. of disulfides, which lowers atom efficiency, the higher conversion efficiency and the absence of an oxidant make this reaction more valuable for practical applications.
DownLoad:
CSV
 |
||
| Het-S-S-Het | Product | Yield (%)b |
 |
3a | 93 |
 |
3g | 93 |
 |
3h | 94 |
 |
3i | 95 c |
 |
3j | 91 c |
 |
3k | 96 |
| a Reaction conditions: 1a (0.5 mmol), 4 (0.5 mmol) in H 2O (2 mL) at room temperature for 12 h. b Isolated yields. c In H 2O/MeCN (1:1, 2 mL) at 50 ℃ for 12 h. |
||
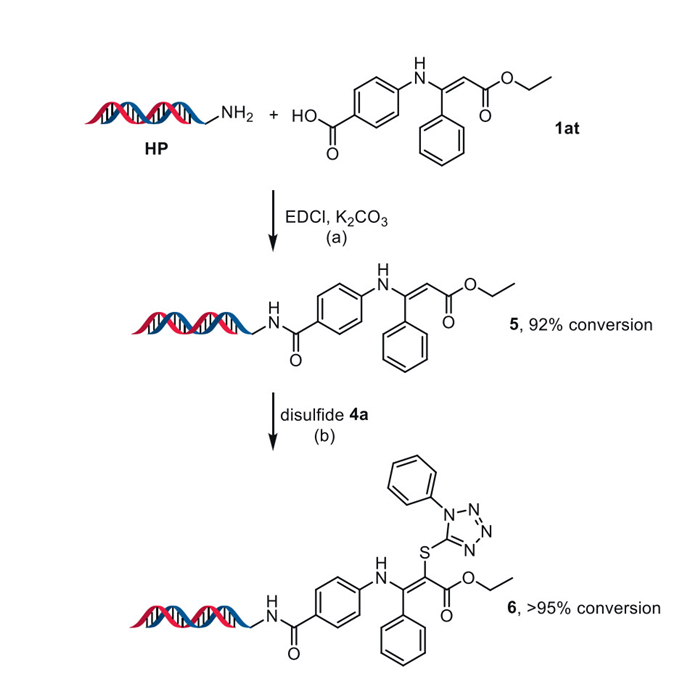
In recent years, DNA-encoded chemical library (DECL) technology, which enables the generation and selection of synthetic molecules with vast chemical diversity, has gained significant attention as a promising tool in drug discovery and chemical biology [56–58]. Incorporating heterocyclic sulfides into DNA-encoded compounds would expand the diversity of DECLs and pave the way for discovering sulfur-containing drugs [59]. Given that the reaction between enamine esters and disulfide compounds can occur in water without the need for additional additives, we were motivated to explore the potential of this methodology for the on-DNA synthesis of α-thioenamine compounds. Initially, the headpiece (HP) was attached to enamine ester 1at using EDCl as a promoter to produce DNA-tagged compound 5 in 92% conversion (Scheme 6a). This compound then reacted with disulfide 4a under the conditions we developed, in an H2O/MeCN (1:1) solvent system, where MeCN was used to dissolve compound 4a to achieve the appropriate concentration, yielding DNA-tagged thioenamine 6 in > 95% conversion (Scheme 6b).
In summary, we have developed a straightforward, environmentally friendly, and green method for synthesizing α-thioenamine derivatives through a K2S2O8-promoted cross-dehydrogenative coupling reaction between enamine esters and commercially available heterocyclic thiols. This reaction demonstrates excellent tolerance for enamine esters with various functional groups, yielding α-thioenamine derivatives in moderate to excellent yields. Mechanistic studies revealed that heterocyclic thiols react with K2S2O8 in water to form reactive disulfides in situ, which subsequently react with enamine esters to produce a range of α-thioenamines. Building on the proposed mechanism, we developed an oxidant-free sulfenylation reaction of enamine esters with disulfides. The effectiveness of this method was further demonstrated by the successful synthesis of DNA-tagged α-thioenamine, highlighting its considerable potential for various synthetic applications. These features make this approach highly promising for use in synthetic and medicinal chemistry.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Haoyu Tian: Methodology. Xiaolin Cui: Methodology. Guiwei Yao: Methodology. Wenyan Wei: Methodology. Junchao Lu: Investigation. Senyao Zheng: Investigation. Xingjian Wang: Investigation. Xun Chen: Resources, Conceptualization. Guangkuan Zhao: Project administration. Dulin Kong: Project administration.
We are very grateful for the financial support from the Key R & D Projects of Hainan Province (No. ZDYF2024SHFZ065), the Cultivation Research Foundation of Hainan Medical University (No. RZ2300002021 to Dulin Kong, No. RZ2400001591 to Guangkuan Zhao), and the Postgraduate Innovation Project of Hainan Medical University (No. HYYB2023A07 to Wenyan Wei).
Supplementary material associated with this article can be found, in the online version, at doi:
G. Stork, A. Brizzolara, H. Landesman, J. Szmuszkovicz, R. Terrell, J. Am. Chem. Soc. 85 (1963) 207–222. doi: 10.1021/ja00885a021
S. Mukherjee, J.W. Yang, S. Hoffmann, B. List, Chem. Rev. 107 (2007) 5471–5569. doi: 10.1021/cr0684016
J.H. Hansen, M. Zeeshan, Mini-Rev. Org. Chem. 11 (2014) 432–444. doi: 10.2174/1570193X1104140926170017
I.V. Efimov, D.I. Zhilyaev, L.N. Kulikova, L.G. Voskressensky, Eur. J. Org. Chem. 26 (2023) e202201450. doi: 10.1002/ejoc.202201450
G. Bernadat, G. Masson, Synlett 25 (2014) 2842–2867. doi: 10.1055/s-0034-1379166
X.L. Lian, Z.H. Ren, Y.Y. Wang, Z.H. Guan, Org. Lett. 16 (2014) 3360–3363. doi: 10.1021/ol501394k
J.L. Zhan, L. Zhu, J.N. Bai, et al., Org. Biomol. Chem. 21 (2023) 8984–8988. doi: 10.1039/d3ob01662e
X.H. Cai, M.Z. Yang, H. Guo, Curr. Org. Synth. 16 (2019) 70–97. doi: 10.2174/1570179415666181107122814
L.H. Zhu, D.H. Wang, Z.B. Jia, et al., ACS Catal. 8 (2018) 5466–5484. doi: 10.1021/acscatal.8b01263
N.K. Fu, L. Zhang, S.Z. Luo, Org. Biomol. Chem. 16 (2018) 510–520. doi: 10.1039/c7ob02615c
T. Courant, G. Dagousset, G. Masson, Synthesis 47 (2015) 1799–1826. doi: 10.1055/s-0034-1378706
N.U.D. Reshi, V.B. Saptal, M. Beller, J.K. Bera, ACS Catal. 11 (2021) 13809–13837. doi: 10.1021/acscatal.1c04208
B.Y. Han, Z.F. Wang, Y. Huang, Chem. Rec. 23 (2023) e202300099. doi: 10.1002/tcr.202300099
K.S. Carroll, Curr. Opin. Chem. Biol. 79 (2024) 102422. doi: 10.1016/j.cbpa.2023.102422
K.A. Scott, J.T. Njardarson, Top. Curr. Chem. 376 (2018) 5. doi: 10.1007/s41061-018-0184-5
M. Feng, B. Tang, S.H. Liang, X. Jiang, Curr. Top. Med. Chem. 16 (2016) 1200–1216. doi: 10.2174/1568026615666150915111741
Y.J. Jiang, G.H. Liang, C. Zhang, T.P. Loh, Eur. J. Org. Chem. (2016) 3326–3330. doi: 10.1002/ejoc.201600588
M. Saroha, J. Sindhu, S. Kumar, et al., ChemistrySelect 6 (2021) 13077–13208. doi: 10.1002/slct.202102042
J.P. Wan, S.S. Zhong, L.L. Xie, et al., Org. Lett. 18 (2016) 584–587. doi: 10.1021/acs.orglett.5b03608
Y. Siddaraju, K.R. Prabhu, J. Org. Chem. 82 (2017) 3084–3093. doi: 10.1021/acs.joc.7b00073
G.S. Sorabad, M.R. Maddani, Asian J. Org. Chem. 8 (2019) 1336–1343. doi: 10.1002/ajoc.201900402
F.L. Lu, K. Zhang, X.Y. Wang, et al., Chem. Asian J. 15 (2020) 4005–4008. doi: 10.1002/asia.202001116
D.D. Li, J.P. Jia, X.W. Zhao, et al., ChemistrySelect 6 (2021) 6460–6463. doi: 10.1002/slct.202101541
H. Fang, J.C. Zhao, P. Qian, J.L. Han, Y. Pan, Asian J. Org. Chem. 3 (2014) 1266–1269. doi: 10.1002/ajoc.201402169
J.B. Azeredo, M. Godoi, G.M. Martins, C.C. Silveira, A.L. Braga, J. Org. Chem. 79 (2014) 4125–4130. doi: 10.1021/jo5000779
M. Majek, A. Jacobi von Wangelin, Chem. Commun. 49 (2013) 5507–5509. doi: 10.1039/c3cc41867g
L. Yang, Q. Wen, F.H. Xiao, G.J. Deng, Org. Biomol. Chem. 12 (2014) 9519–9523. doi: 10.1039/C4OB01970A
B. Hu, P. Zhou, K.R. Rao, et al., Tetrahedron Lett. 59 (2018) 1438–1442. doi: 10.1016/j.tetlet.2018.02.079
J.Y. Sun, D. Zhang-Negrerie, Y.F. Du, Adv. Synth. Catal. 358 (2016) 2035–2040. doi: 10.1002/adsc.201501099
Z.H. Shang, Q.Y. Chen, L.L. Xing, et al., Adv. Synth. Catal. 361 (2019) 4926–4932. doi: 10.1002/adsc.201900940
F. Xiao, D. Wang, S. Yuan, H. Huang, G.J. Deng, RSC Adv. 8 (2018) 23319–23322. doi: 10.1039/c8ra04374d
Y.X. Wu, S.Y. Wu, Z.B. Dong, J. Org. Chem. 87 (2022) 15350–15357. doi: 10.1021/acs.joc.2c01924
Y.M. Lin, G.P. Lu, C. Cai, W.B. Yi, Org. Lett. 17 (2015) 3310–3313. doi: 10.1021/acs.orglett.5b01488
Y.M. Lin, G.P. Lu, G.X. Wang, W.B. Yi, Adv. Synth. Catal. 358 (2016) 4100–4105. doi: 10.1002/adsc.201600846
Z.B. Xu, G.P. Lu, C. Cai, Org. Biomol. Chem. 15 (2017) 2804–2808. doi: 10.1039/C6OB02823C
P.R. Fitzgerald, B.M. Paegel, Chem. Rev. 121 (2021) 7155–7177. doi: 10.1021/acs.chemrev.0c00789
J.Y. Mao, T.Z. Jia, G. Frensch, P.J. Walsh, Org. Lett. 16 (2014) 5304–5307. doi: 10.1021/ol502470e
M.A. Fernandez-Rodriguez, Q. Shen, J.F. Hartwig, J. Am. Chem. Soc. 128 (2006) 2180–2181. doi: 10.1021/ja0580340
R. Das, D. Chakraborty, Tetrahedron Lett. 53 (2012) 7023–7027. doi: 10.1016/j.tetlet.2012.09.127
J. Shanmugapriya, K. Rajaguru, S. Muthusubramanian, N. Bhuvanesh, Eur. J. Org. Chem. (2016) 1963–1967. doi: 10.1002/ejoc.201600160
C.F. Lee, Phos. Sulfur Silicon Relat. Elem. 194 (2019) 678–681. doi: 10.1080/10426507.2019.1603228
C. Uyeda, Y. Tan, G.C. Fu, J.C. Peters, J. Am. Chem. Soc. 135 (2013) 9548–9552. doi: 10.1021/ja404050f
X. Ku, H. Huang, H. Jiang, H. Liu, J. Comb. Chem. 11 (2009) 338–340. doi: 10.1021/cc800182q
J.R. Wu, C.H. Lin, C.F. Lee, Chem. Commun. (2009) 4450–4452. doi: 10.1039/b907362k
A. Correa, M. Carril, C. Bolm, Angew. Chem. Int. Ed. 47 (2008) 2880–2883. doi: 10.1002/anie.200705668
F.J. Guo, J. Sun, Z.Q. Xu, et al., Catal. Commun. 96 (2017) 11–14. doi: 10.1016/j.catcom.2017.02.007
H. Xu, C.Y. He, B.J. Huo, et al., Org. Chem. Front. 10 (2023) 5171–5179. doi: 10.1039/d3qo01236k
Y.C. Wong, T.T. Jayanth, C.H. Cheng, Org. Lett. 8 (2006) 5613–5616. doi: 10.1021/ol062344l
S.D. Timpa, C.J. Pell, O.V. Ozerov, J. Am. Chem. Soc. 136 (2014) 14772–14779. doi: 10.1021/ja505576g
M. Arisawa, T. Suzuki, T. Ishikawa, M. Yamaguchi, J. Am. Chem. Soc. 130 (2008) 12214–12215. doi: 10.1021/ja8049996
Y. Siddaraju, K.R. Prabhu, Org. Lett. 18 (2016) 6090–6093. doi: 10.1021/acs.orglett.6b03084
Y. Siddaraju, K.R. Prabhu, J. Org. Chem. 83 (2018) 2986–2992. doi: 10.1021/acs.joc.7b03290
D. Kong, T. Huang, M. Liang, M. Wu, Q. Lin, Org. Biomol. Chem. 17 (2019) 830–834. doi: 10.1039/c8ob02800a
X.L. Cui, H.Y. Tian, S.J. Wang, X. Chen, D.L. Kong, J. Org. Chem. 88 (2023) 8576–8582. doi: 10.1021/acs.joc.3c00499
H. Tian, Q. Wang, W. Wei, et al., J. Org. Chem. 89 (2024) 15523–15528. doi: 10.1021/acs.joc.4c01592
S. Brenner, R.A. Lerner, Proc. Natl. Acad. Sci. U. S. A. 89 (1992) 5381–5383. doi: 10.1073/pnas.89.12.5381
A.A. Peterson, D.R. Liu, Nat. Rev. Drug Discov. 22 (2023) 699–722. doi: 10.1038/s41573-023-00713-6
G. Zhang, J. Zhang, Y.T. Gao, Y.F. Li, Y.Z. Li, Expert Opin. Drug Discovery 17 (2022) 55–69. doi: 10.1080/17460441.2021.1969359
S.L. Yang, G.X. Zhao, Y.T. Gao, et al., Chem. Sci. 13 (2022) 2604–2613. doi: 10.1039/d1sc06268a

Scheme 2 Substrate scope of heterocyclic thiols. Reaction conditions: 1a (0.5 mmol), 2 (0.6 mmol), K2S2O8 (0.5 mmol) in H2O (2 mL) at 80 ℃ for 12 h. Isolated yields.

Scheme 3 Substrate scope of enamine esters. Reaction conditions: 1 (0.5 mmol), 2a (0.6 mmol), K2S2O8 (0.5 mmol) in H2O (2 mL) at 80 ℃ for 12 h. Isolated yields.

Scheme 6 Synthesis of DNA-tagged thioenamine 6. Reaction conditions: (a) HP (50 nmol, 100 µL, 0.5 mmol/L in sodium borate buffer (10 mmol/L, pH 9.18), 1.0 equiv.), 1at (2.5 µmol, 50 µL, 50 mmol/L in DMA, 50 equiv.), EDCl (5 µmol, 50 µL, 100 mmol/L in DMA, 100 equiv.), K2CO3 (100 nmol, 10 µL, 10 mmol/L in H2O, 2.0 equiv.) at room temperature for 12 h; (b) 5 (10 nmol, 20 µL, 0.5 mmol/L in H2O, 1.0 equiv.), 4a (1 µmol, 20 µL, 50 mmol/L in MeCN, 100 equiv.) at room temperature for 0.5 h. Conversions were determined by UPLC-MS.
Table 1. Optimization of the reaction conditions.a
|
|||||
| Entry | Oxidant (equiv.) | Solvent | Temp. (℃) | 2a (equiv.) | Yield (%)b |
| 1 | KIO3 (1.0) | H2O | 80 | 1.2 | 17 |
| 2 | K2S2O8 (0.8) | H2O | 80 | 1.2 | 45 |
| 3 | K2S2O8 (1.0) | H2O | 80 | 1.2 | 89 |
| 4 | K2S2O8 (2.0) | H2O | 80 | 1.2 | 89 |
| 5 | TBHP (1.0) | H2O | 80 | 1.2 | nd |
| 6 | I2 (1.0) | H2O | 80 | 1.2 | nd |
| 7 | PIDA (1.0) | H2O | 80 | 1.2 | nd |
| 8 | KBrO3 (1.0) | H2O | 80 | 1.2 | 23 |
| 9 | H2O2 (1.0) | H2O | 80 | 1.2 | nr |
| 10 | – | H2O | 80 | 1.2 | nr |
| 11 | K2S2O8 (1.0) | Ethanol | 80 | 1.2 | nr |
| 12 | K2S2O8 (1.0) | DMSO | 80 | 1.2 | 45 |
| 13 | K2S2O8 (1.0) | DMF | 80 | 1.2 | nr |
| 14 | K2S2O8 (1.0) | 10% PEG-400 aqueous solution | 80 | 1.2 | nd |
| 15 | K2S2O8 (1.0) | MeCN | 80 | 1.2 | 23 |
| 16 | K2S2O8 (1.0) | Acetone | 80 | 1.2 | 17 |
| 17 | K2S2O8 (10) | 1,4-Dioxane | 80 | 1.2 | nr |
| 18 | K2S2O8 (1.0) | H2O | 80 | 0.8 | 71 |
| 19 | K2S2O8 (1.0) | H2O | 80 | 1.0 | 87 |
| 20 | K2S2O8 (1.0) | H2O | 80 | 1.5 | 85 |
| 21 | K2S2O8 (1.0) | H2O | 25 | 1.2 | 20 |
| 22 | K2S2O8 (1.0) | H2O | 50 | 1.2 | 49 |
| 23 | K2S2O8 (1.0) | H2O | 60 | 1.2 | 67 |
| 24 | K2S2O8 (1.0) | H2O | 110 | 1.2 | 91 |
| a Reaction conditions: 1a (0.5 mmol), 2a (x mmol), oxidant (y equiv.) in solvent (2 mL) for 12 h. b Isolated yields. nr: no reaction occurred. nd: no desired product was detected. |
|||||
 下载: 导出CSV
下载: 导出CSV
Table 2. Sulfenylation reaction between enamine ester and disulfide compound.a
|
||
| Het-S-S-Het | Product | Yield (%)b |
|
3a | 93 |
|
3g | 93 |
|
3h | 94 |
|
3i | 95 c |
|
3j | 91 c |
|
3k | 96 |
| a Reaction conditions: 1a (0.5 mmol), 4 (0.5 mmol) in H 2O (2 mL) at room temperature for 12 h. b Isolated yields. c In H 2O/MeCN (1:1, 2 mL) at 50 ℃ for 12 h. |
||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


