Received Date:
10 December 2024 Accepted Date:
20 February 2025 Revised Date:
18 February 2025 Available Online:
15 February 2026
Abstract:
The von Hippel-Lindau tumor suppressor (VHL) has been extensively used to develop degraders targeting numerous proteins of interest. However, studies on the rational design of VHL-proteolysis-targeting chimeras (PROTACs) remain scarce. This study aimed to develop strategies to investigate VHL-recruiting PROTACs connecting with varying attachment sites on VHL ligands, which could be utilized for KRASG12C degraders development and expanded to additional targets. We developed a molecular dynamics (MD)-based strategy to explore the stability of ternary complexes induced by KRASG12C PROTACs with four distinct attachment sites of VH032. We found a potent degrader namely YN14-H, linked to hydroxyl group on VH032 benzene ring, exhibited the most superior ability of inducing ternary complexes, reflected by the lowest dissociation constant (Kd) for ternary complex induction and the highest AlphaScreen (AS)-based interaction. YN14-H inhibited cell growth with low nanomolar half maximal inhibitory concentration (IC50) and half maximal degradation concentration (DC50) values as well as >98% of maximum degradation (Dmax) in NCI-H358 and MIA PaCa-2 cells harboring KRASG12C-mutation. Mechanistically, YN14-H significantly induced apoptosis and inhibited the migratory capacity. Notably, YN14-H demonstrated favorable pharmacokinetic properties and excellent antitumor activity in vivo. Furthermore, bromodomain-containing protein 7 (BRD7) and Bruton tyrosine kinase (BTK) degraders attached to distinct sites on VH032 further verified the rationality and universality of our MD-based strategies. Our findings demonstrated that YN14-H could serve as a promising candidate for the treatment of tumors with KRASG12C-mutation and present a strategy for the rational design of VHL-recruiting PROTACs that target additional proteins at distinct attachment sites.
Over the past two decades, proteolysis-targeting chimeras (PROTACs) have emerged as a promising strategy for inducing the degradation of specific proteins through recruitment to the ubiquitin-proteasome system [1,2]. In particular, PROTACs have made significant progress in the treatment of various human diseases, such as tumors [3-5], viruses [6,7], and immune-related diseases [8]. PROTACs are heterobifunctional compounds comprising three key components: a target protein-binding moiety, a recruiting unit for E3 ubiquitin ligase, and a linker that connects these two units [9]. The von Hippel-Lindau tumor suppressor (VHL) protein was the first E3 ubiquitin ligase to be harnessed for target protein degradation and has been demonstrated to be effective in degrading nuclear, cytoplasmic, and membrane-associated proteins [10], such as Kirsten rat sarcoma (KRAS) [11,12], Bruton tyrosine kinase (BTK) [13,14], and bromodomain containing protein 7 (BRD7) [15].
KRAS plays a pivotal role in the regulation of cell growth signaling. When KRAS is mutated, it abnormally activates downstream signaling pathways, resulting in the occurrence and progression of tumors [16]. The mutated protein KRASG12C (glycine 12 to cysteine) has been detected in various tumor types, such as lung, colorectal, and undifferentiated endometrial cancer [17,18]. Previous studies have shown that VHL-based PROTACs can degrade endogenous KRASG12C in cancer cells [11,12]. In our laboratory, we developed a highly potent and selective VHL-recruiting KRASG12C degrader, YN14, that exhibits great promise as a therapeutic strategy for tumors harboring KRASG12C both in vitro and in vivo [19].
Most VHL-recruiting PROTACs focus on linker optimization to promote their degradation efficacy. Moreover, previous studies have demonstrated that the attachment sites for the E3 ligand have significant implications for the physicochemical properties and spatial orientation of the protein of interest (POI)-PROTAC-E3 ternary complex, influencing the overall bioactivity of the degraders [20,21]. VHL-PROTACs which utilize the same warhead but with distinct attachment sites, can selectively degrade closely related proteins and exhibit varying degradation activities against the same target protein [22,23]. Despite these findings, studies regarding the rational design of VHL-PROTACs based on different attachment sites remain scarce. Furthermore, to the best of our knowledge, all VHL-based KRASG12C PROTACs utilize a linker attached to the acetamide group of VH032, and no alternative attachment sites have been explored.
The formation of the ternary complexes is the key to the success of PROTAC-induced protein degradation [3]. Nevertheless, the lack of effective ternary complex modeling techniques has limited the application of computer-aided drug discovery tools to this novel therapeutic tool. In this study, we developed a strategy that integrates protein-protein docking, molecular dynamics (MD) simulations, and precise binding energy calculations via umbrella sampling to evaluate the stability of ternary complexes induced by PROTACs with different attachment sites on VHL ligands. This strategy was used to investigate the degradation potency of novel PROTACs targeting KRASG12C at various attachment sites with VHL. Previously reported BRD7 and BTK degraders attached to distinct sites on VH032 further verified the rationality and universality of our MD-based strategies. We believe that this strategy can effectively guide the discovery of KRASG12C PROTACs with higher activity and facilitate the rational design of VHL-based PROTACs that target other proteins at different attachment sites.
MD simulations have been extensively utilized to assess the stability of ternary complexes induced by PROTACS. As shown in Fig. S1 (Supporting information), the structures of the ternary complexes obtained from protein-protein docking underwent 500 ns MD simulations. Various methods, such as the molecular mechanics/Poisson-Boltzmann surface area (MM/PBSA) and generalized Born and surface area (MM/GBSA) methods, have been employed to calculate the binding energy (ΔG) [24]. However, calculating ΔG using the MM/PBSA method is trajectory-dependent. Different trajectories can yield varying results, which poses a challenge in obtaining reliable and consistent measurements. A commonly employed solution to address this issue is umbrella sampling, which allows the computation of the free energy along the reaction coordination [25,26]. As such, umbrella sampling was used to calculate the free energy of the ternary complex along the dissociation pathway. The well-equilibrated structures obtained from the unbiased MD simulations served as the starting points for umbrella sampling simulations, and the center of mass distance between the two proteins was selected to define the relaxed conformation (RC). The potential of mean force (PMF) curves for VHL unbinding from protein-protein interactions (PPIs) exhibited a consistent pattern, beginning at zero and progressively increasing until a stable plateau was reached. The average ΔG in each window was plotted as a function of the reaction coordinates to provide valuable insights into the thermodynamics of VHL dissociation. Furthermore, this analysis aided in identifying the key residues and interactions that contribute to the binding and dissociation of VHL.
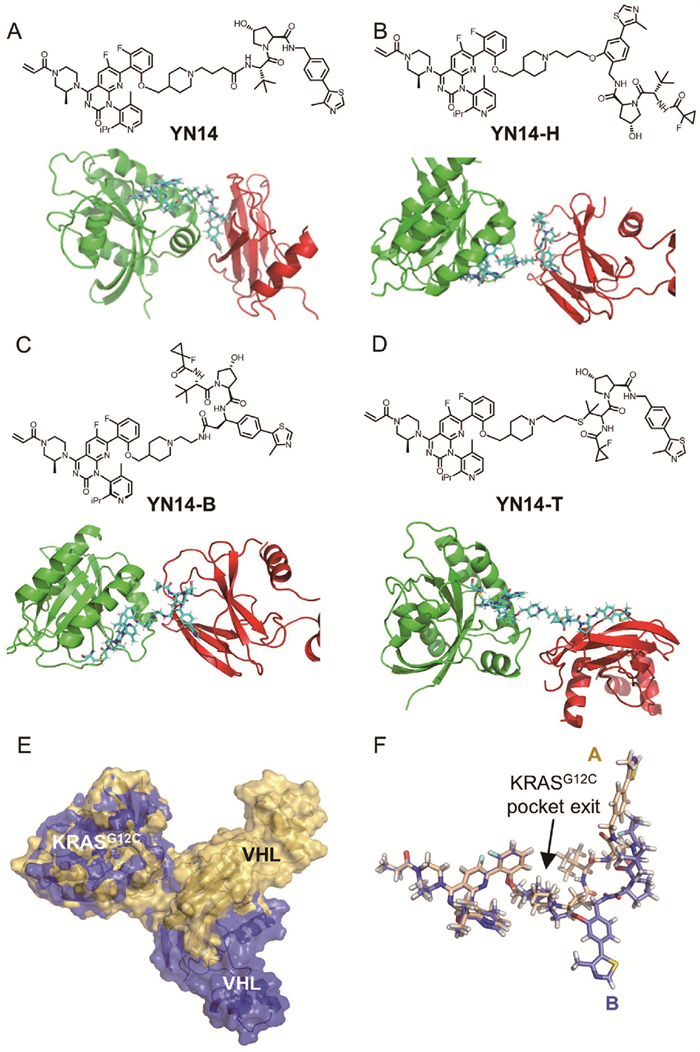
Based on our previously reported potent KRASG12C degrader YN14, we designed three novel degraders with different attachments. YN14-H, YN14-B, and YN14-T are linked by hydroxyl group on benzene ring, alkyl group on benzyl, and sulfhydryl group on tertiary butyl, respectively (Figs. 1A–D). To investigate the interaction between KRASG12C, VHL, and its degraders, we conducted protein-protein docking and unbiased MD simulations. Notably, docking and short (500 ns) MD simulations revealed distinct conformations of the ternary complexes induced by different PROTACs with different linker attachments. In particular, YN14 and YN14-H recruited VHL with entirely different conformations and residual interactions (Fig. 1E). However, closer examination of the conformation of each PROTAC in these models revealed the YN14-H directly interacted with VHL outside the KRASG12C pocket, similar to YN14, which is linked by an amide group, to recruit both VHL and KRASG12C (Fig. 1F).
Figure 1
Figure 1.
Ternary complexes constructed by protein-protein docking and unbiased MD simulations. (A–D) The chemical structures of PROTACs with different linker attachments and resulting ternary complexes. KRASG12C, VHL, and PROTACs were subjected to protein-protein docking and 500 ns MD simulations to relax the ternary structure. (E) VHL recruitment by the degraders YN14-H and YN14; the KRASG12C-YN14-VHL ternary complex is shown in yellow, while the KRASG12C-YN14-H-VHL ternary complex is shown in blue. By aligning the KRASG12C structures from both ternary complex MD simulations, the divergent VHL structures can be visualized. (F) At the exit of the KRASG12C binding pocket, YN14 and YN14-H display distinct interactions with the KRASG12C-VHL PPI interface.
The root mean square deviation (RMSD) of the protein was calculated throughout the 500 ns trajectory to assess convergence of the simulation system [27]. Convergence was observed in all the systems after 300 ns of simulation (Fig. 2A). Additionally, the ΔG of the YN14-H-KRASG12C-VHL complex was determined to be −158.12 kJ/mol. The YN14, YN14-B, and YN14-T-KRASG12C-VHL complexes exhibited higher ΔG values at −123.68, 170.66 and −46.10 kJ/mol, respectively (Fig. 2B). These results suggest that the presence of the degrader YN14-H promoted a more stable interaction between KRASG12C and VHL proteins.
Figure 2
Figure 2.
Analysis of results from unbiased MD simulations. (A) The RMSD of ternary complexes. (B) The binding free energy (ΔG, kJ/mol) of PROTAC-recruited ternary complexes. (C) The H bonds of KRASG12C and VHL in the whole trajectory induced by different degraders. (D) Distinct interaction modes of KRASG12C and VHL at PPI interfaces induced by degraders YN14, YN14-H, YN14-B, and YN14-T.
To gain a comprehensive understanding of the recruitment process, identify the key hydrogen bonds (H-bonds) formed within the system is essential. H-bonds play a critical role in maintaining the stability of protein-protein complexes. The analysis of H-bonds shows that YN14-H can induce the formation of 10 stable H-bond interactions between KRASG12C and VHL, whereas the fluctuating number of H-bonds induced by YN14, YN14-B, and YN14-T indicates the instability of the ternary complexes during the MD simulations (Fig. 2C). The PPI interfaces are depicted in Fig. 2D. The degrader YN14-H induced a wider range of interactions between KRASG12C and VHL, such as H-bonds between the residue pairs Tyr64-Arg247, Ala66-Gln212, Arg102-His249 and salt bridge of Asp69-Arg208. These interactions contributed to the formation of the ternary complex by the degrader YN14-H.
As shown in Fig. S2 (Supporting information), the formation of the ternary complex KRASG12C-PROTACs-VHL resulted in ΔG values of −23.6, −24.8, −18.8, and −21.7 kcal/mol for YN14, YN14-H, YN14-B, and YN14-T, respectively (Fig. S2A), indicating that the stability order of ternary complexes is YN14-H > YN14 > YN14-T > YN14-B. In the steered simulations, the initial step involved the disengagement of the H bond between the partial group of VH032 in the degrader YN14-H and Ser250, Tyr251, and His254 of VHL (Fig. S2C). This disengagement led to the first peak in the rupture force at approximately 138 ps (Fig. S2B). The second rupture force peak arose from the electrostatic arm of Asp69-Arg208, which holds the two proteins together. Breaking the electrostatic zipper structure formed between Asp69-Arg208 led to a rapid return of forces to the basal level (Fig. S2D), accompanied by a jump in extension. The terminal state was observed when the degrader YN14-H was mostly unfolded and lacked protein-protein contacts (Fig. S2E).
To validate our computational findings, we synthesized four degraders. The degraders YN14 and YN14-B were synthesized according to the procedure outlined in Scheme S1 (Supporting information). Intermediate 2a was subjected to nucleophilic substitution with different linkers to obtain intermediates 10a and 11a, followed by acid hydrolysis to obtain intermediates 12a and 13a. The target compound YN14 was then produced by amide coupling of 13a and VH032. Similarly, amidation of 12a with 3b yielded intermediate 4b. YN14-B was then synthesized by the amidation of intermediate 5b, which was prepared from intermediate 4b. The synthesis of the key intermediates (2a and VHL-SH) is shown in Scheme S2 (Supporting information). Briefly, nucleophilic substitution of compound 1 with AMG510 was performed to obtain intermediate 1a, followed by acidic deprotection to form intermediate 2a. Amidation of 3a with 4a generated intermediate 5a, and subsequent deprotection of the Boc group yielded compound 6a. Amide coupling of 6a with compound 2 followed by hydrolysis with sodium hydroxide produced the key intermediate 8a. The amide condensation of 8a with 3 gave intermediate 9a, which, after trifluoroacetic acid deprotection, afforded the target compound VHL-SH.
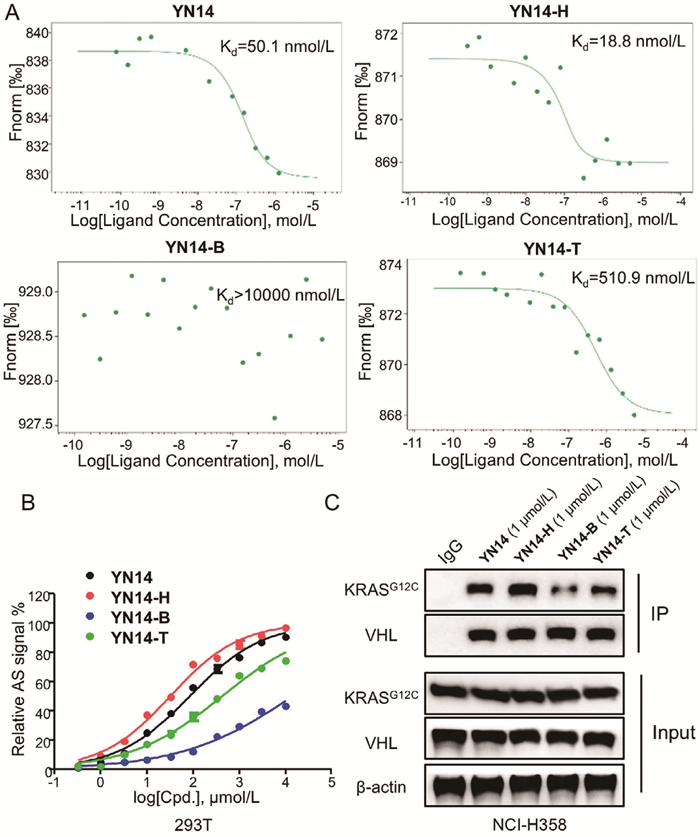
As depicted in Scheme S3 (Supporting information), VH101 and VHL-SH reacted with 1‑bromo-3-chloropropane to produce intermediates 2c and 3cvia nucleophilic substitution. Subsequently, further nucleophilic substitution of intermediates 2c and 3c with intermediate 2a yielded the desired target compounds YN14-H and YN14-T, respectively. We then characterized the ability to induce ternary complex formation using biochemical and cell-based experiments. Firstly, microscale thermophoresis (MST) experiments were performed to assess the ability of the four degraders for inducing ternary complex formation [28]. As shown in Fig. 3A and Table S1 (Supporting information), the affinity of ternary complex induced by YN14 and YN14-H was 50.1 and 18.8 nmol/L, respectively, indicating a stronger ability to induce ternary complex formation compared to YN14-B (over 10,000 nmol/L) and YN14-T (510.9 nmol/L). Then, the AlphaScreen (AS) biochemical interaction assay revealed that YN14-H significantly induced interaction between KRASG12C and VHL, which was comparable to YN14 and stronger than YN14-T, while YN14-B exhibited weak effects (Fig. 3B). We next evaluated the capacity of KRASG12C to interact with VHL following degraders treatment. We observed increased levels of endogenous KRASG12C that co-immunoprecipitated with VHL in the presence of four degraders, and YN14-H exhibited the most significant effect (Fig. 3C). Moreover, the capacity of degraders in inducing the degradation of the KRASG12C protein in MIA PaCa-2 cells in a dose-dependent manner was demonstrated, as depicted in Table S2 and Fig. S6 (Supporting information). The half-maximal degradation concentrations (DC50) of YN14-H were determined to be 28.9 and 18.1 nmol/L in NCI-H358 and MIA PaCa-2 cells, respectively, with a maximal degradation rate (Dmax) exceeding 95%. This result demonstrated that YN14-H is the most potent KRASG12C degrader compared to YN14 (67.4 and 26.2 nmol/L, in NCI-H358 and MIA PaCa-2 cells, respectively), YN14-B (10,000 nmol/L in NCI-H358 and MIA PaCa-2 cells), and YN14-T (108.9 and 201.1 nmol/L, in NCI-H358 and MIA PaCa-2 cells, respectively).
Figure 3
Figure 3.
Ternary complex formation and degradation effects of the four KRASG12C degraders. (A) Ternary complex formation induced by degraders via MST. The Kd determinations were performed in binding assay. (B) Dose-dependent interaction assay using AS technology. The VHL-KRASG12C complex formation was analyzed. The relative AS signals treated with YN14, YN14-H, YN14-B and YN14-T, respectively, are expressed as the luminescence signal relative to the luminescence signal of DMSO. (C) Immunoprecipitation of KRASG12C in NCI-H358 cells treated with DMSO, YN14, YN14-H, YN14-B and YN14-T at 1 µmol/L for 8 h, respectively.
The assessment of the antiproliferative effects of the degraders across various cell lines revealed significant outcomes. Notably, both YN14 and YN14-H exhibited strong anti-proliferative effects in NCI-H358 and MIA PaCa-2 cells (Table S3 and Fig. S7A in Supporting information). Notably, the half-maximal inhibitory concentration (IC50) of YN14-H was 0.042 µmol/L in NCI-H358 cells and 0.021 µmol/L in MIA PaCa-2 cells, surpassing that of YN14 (0.091 and 0.045 µmol/L, in NCI-H358 and MIA PaCa-2 cells, respectively), YN14-B (0.345 and 0.317 µmol/L, in NCI-H358 and MIA PaCa-2 cells, respectively), and YN14-T (0.150 and 0.105 µmol/L, in NCI-H358 and MIA PaCa-2 cells, respectively). The degrader YN14-H displayed superior anticancer activity compared to our previously reported highly potent KRASG12C degrader YN14. Furthermore, the inhibitory effects on pancreatic adenocarcinoma ASPC-1 cells with the KRASG12D mutation, hepatocellular carcinoma HepG2 cells harboring wild-type KRAS (KRASWT), and normal 293T cells were investigated. Notably, all KRASG12C degraders exhibited no cytotoxicity up to 10 µmol/L in these cells, underscoring their high selectivity and low cytotoxicity profiles.
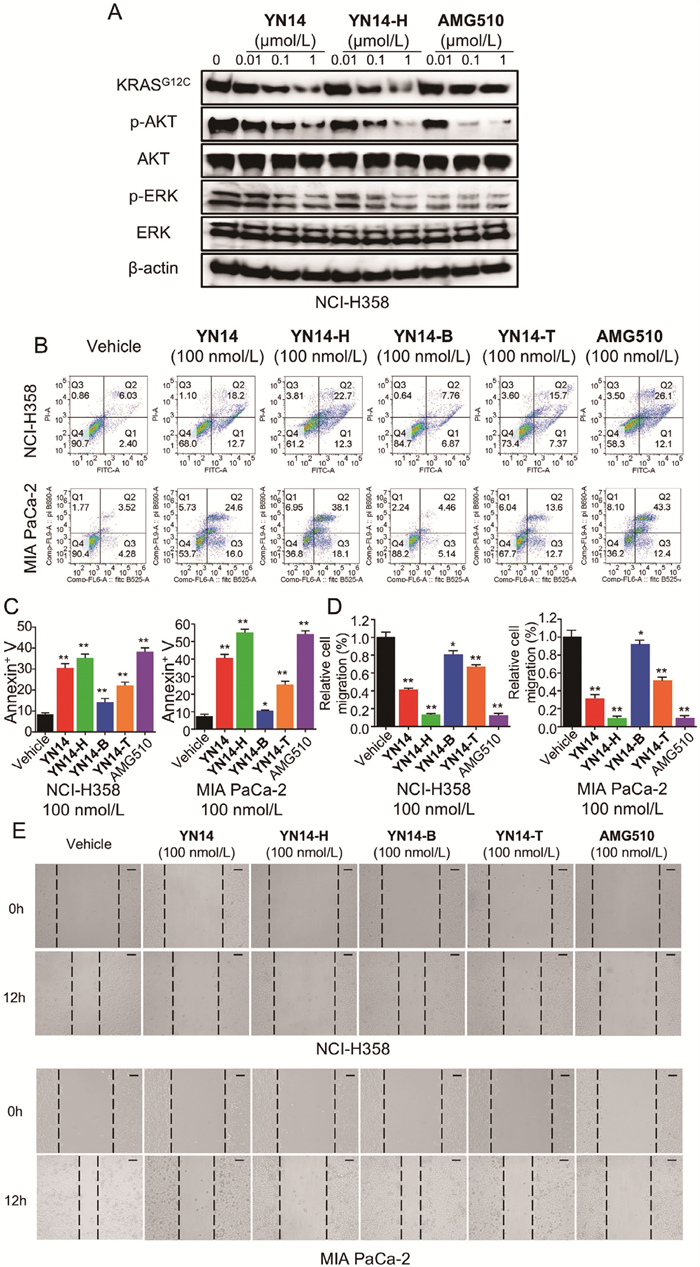
Based on the above data, YN14-H was selected for further study. KRAS is known to trigger a series of downstream RAF-mitogen-activated protein kinase (MEK)-extracellular regulated protein kinases (ERK) and phosphatidylinositol 3-kinase (PI3K)-AKT-mammalian target of rapamycin (mTOR) signaling pathways to regulate cell proliferation and survival [29]. As revealed by Fig. 4A and Fig. S7B (Supporting information), compared with YN14, YN14-H showed more potent inhibitory effects on the levels of phosphorylated AKT (p-AKT) and phosphorylated ERK (p-ERK) in NCI-H358 and MIA PaCa-2 cells in a concentration-dependent manner, which was similar to AMG510. The exploration of the degraders’ abilities to induce apoptosis and inhibit cell migration was also explored. As shown in Figs. 4B and C, following treatment with YN14, YN14-H, YN14-B and YN14-T, the apoptosis rates of NCI-H358 cells induced by treatment with 100 nmol/L of the degraders for 48 h were 30.52%, 35.19%, 14.14%, and 22.14%, respectively, and those of MIA PaCa-2 cells were 40.52%, 55.19%, 10.47%, and 25.47%, respectively. Additionally, the effects of the four degraders on cell migration were examined. The wound healing assay highlighted the significant inhibitory effects of YN14 and YN14-H on the migratory capacity of NCI-H358 and MIA PaCa-2 cells at 100 nmol/L concentration (Figs. 4D and E). The above biological experiment results demonstrated that compound YN14-H displayed the highest efficacy in inducing stable ternary complexes and the most efficient degradation activity among the four PROTACs, with different attachment sites on VH032. Notably, this finding aligns with those of our previous computational studies.
Figure 4
Figure 4.
Antiproliferative effect of the KRASG12C degraders. (A) The effects of YN14, YN14-H and AMG510 on KRASG12C, AKT and ERK phosphorylation in NCI–H358 cells. Cells were treated compounds for 48 h at the indicated concentration. (B, C) Representative images of flow cytometry analysis of apoptosis in NCI-H358 and MIA PaCa-2 cells treated with the vehicle control (DMSO) and indicated degraders at the indicated doses for 48 h. Histograms showing the relative cell percentage of apoptosis in NCI-H358 and MIA PaCa-2 cells are on the right. (D, E) Wound healing assay of NCI-H358 and MIA PaCa-2 cells treated with DMSO and the four degraders at the indicated doses for 12 h. Histograms showing the relative cell migration are on the right. Scale bar: 100 µm. Data are represented as the mean ± SD (n = 3). *P < 0.05, **P < 0.01 (t-test).
As YN14 and YN14-H exhibit superior antiproliferative activity in vitro, we next evaluated their biological efficacy in vivo. Animal research has been approved by the Animal Care Committee of the Beijing Institute of Biotechnology. All operations were following the Animal Care and Use Committee Guidelines of China. The pharmacokinetics (PK) properties of PROTAC at different attachment sites would be varied greatly, we sought to explore the PK properties of YN14 and YN14-H. In this study, thirty male BALB/c mice were randomly divided into 6 groups with 5 mice per group, and then administered with YN14, YN14-H or AMG510 via intraperitoneal (i.p.) injection, respectively. Detailed PK parameters were provided in Table S4 (Supporting information), for AMG510, it showed a short elimination half-life (<0.5 h) in mice after i.p. administration for both low and high doses. Also, both Cmax and area under the curve (AUC) were increased linearly according to the dose. The pharmacokinetic properties described in this study were consistent with previous literature reports. In comparison, the AUC of YN14 was significantly higher than AMG510, and showed linearity with the dose, and YN14 showed a longer half-life exceeding 5 h (t1/2 = 5.90 h). Moreover, YN14-H showed an even longer half-life exceeding 8 h (t1/2 = 8.81 h) at the dose of 10 mg/kg compared with YN14, which may be resulted from the higher metabolic stability of ether bond than the amide bond. However, the Cmax for YN14-H was relatively lower than both AMG510 and YN14, and it did not increase with dose escalation. The AUC was still significantly higher compared to YN14, and essentially exhibited linearity with the dose. It could be speculated that YN14-H may experience elimination saturation. Both YN14 and YN14-H demonstrated a significant increase in plasma elimination half-life and total exposure compared to AMG510. Between YN14 and YN14-H, with equimolar dosing, the Cmax achieved by YN14-H was significantly lower than that of YN14, particularly with high-dose conditions, where the Cmax of YN14-H is less than half of YN14 (1090.66 ± 171.68 vs. 2373.70 ± 461.34 ng/mL). The reduction in Cmax can significantly decrease the probability of post-administration side effects. Concurrently, the elimination rate of YN14-H is markedly lower than that of YN14, regardless of whether it is at low or high doses, resulting in a significantly prolonged half-life in the body, and an increased exposure time as mean residence time (MRT) is 2–3 times greater than that of YN14 with both doses (5.92 ± 0.48 vs. 2.36 ± 0.39 h, and 14.20 ± 1.43 vs. 4.69 ± 1.00 h). Due to the significantly increased half-life, despite the lower Cmax, the AUC for YN14-H is >1.5 times that of YN14.
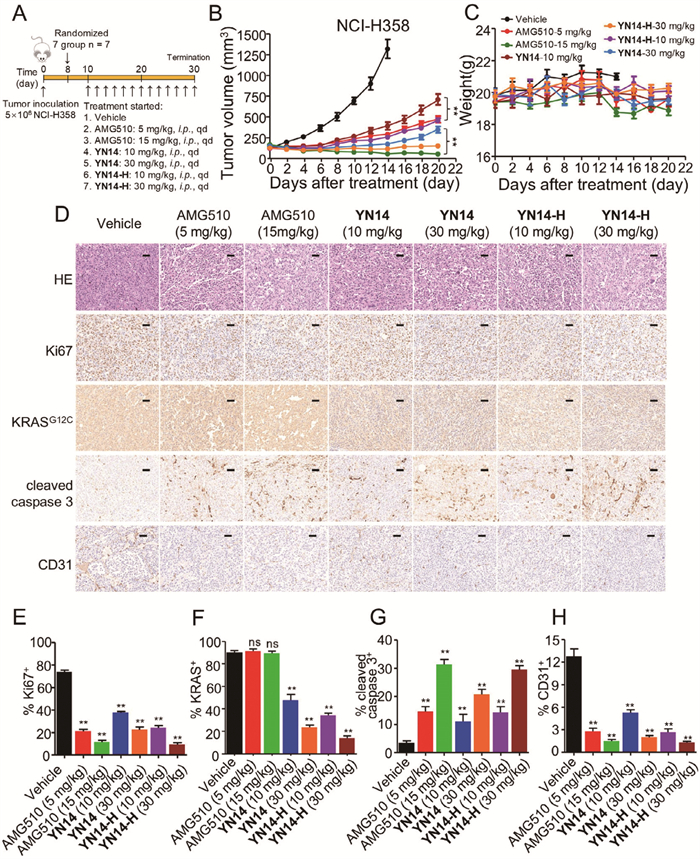
Given the superior PK properties of YN14-H in vivo, we further assessed its anticancer efficacy in BALB/c nude mice bearing NCI-H358 xenograft tumors (Fig. 5A). As shown in Fig. 5B and Fig. S7C (Supporting information), daily i.p. administration of YN14-H induced more potent suppression of NCI-H358 tumor growth with tumor growth inhibition (TGI%) rates of 88.68% at dose of 10 mg/kg compared with 77.59% of YN14 after 14 days of treatment. Moreover, YN14-H at dose of 30 mg/kg resulted in marked tumor regression, as reflected by the smaller tumor volume compared with that on the initial day. which also surpassed that of YN14 (95.4%, 30 mg/kg). As the administration period was extended from 14 days to 20 days, the difference in antitumor efficacy between the same doses of YN14 and YN14-H became more pronounced. By day 20, the volume of the YN14 (10 mg/kg) group reached approximately 750 mm3, whereas the YN14-H (10 mg/kg) achieved stronger inhibitory efficacy with 500 mm3, which was comparative to AMG510 (5 mg/kg). Moreover, YN14-H (30 mg/kg) demonstrated consistent inhibitory effects with complete tumor regression. No obvious body weight loss and no other obvious toxic signs were observed in nude mice during the treatment period (Fig. 5C), indicating no toxic events triggered by YN14 and YN14-H in the nude mice.
Figure 5
Figure 5.
Pharmacokinetic (PK) properties and anticancer efficacy of YN14, YN14-H and AMG510 in vivo. (A) Treatment schedule for the NCI-H358 cells xenograft tumors model treated with vehicle, AMG510, YN14 and YN14-H, respectively. (B) The change of tumor volume was measured every 2 days (n = 7). Data are mean ± SD, **P < 0.01 (one-way ANOVA). (C) The change of body weight of all mice was measured every 2 days (n = 7). (D) Representative images of H&E, Ki67, KRASG12C, cleaved caspase 3 and CD31-staining in harvested tumors from each group are shown. Scale bar: 50 µm. Histograms show the quantification of Ki67+ (E), KRASG12C+ (F), cleaved caspase 3+ (G) and CD31+ (H) cells. Error bars denote standard deviations (independent experiments, n = 3). Data are represented as the mean ± SD. Student’s t-test. ns, no significance. **P < 0.01.
To elucidate the molecular changes upon in vivo administration of AMG510, YN14 and YN14-H in tumors, we next assessed hematoxylin and eosin (H&E), Ki67, KRASG12C, cleaved caspase 3 and CD31-stained immunohistochemical sections of the tumors (Figs. 5D–H). In consistent with the in vitro studies, the number of tumor foci in the AMG510, YN14 and YN14-H groups was much lower than that in the vehicle group. Additionally, single doses of AMG510, YN14 and YN14-H groups significantly reduced the percentage of Ki67- and CD31-positive tumor cells and increased the percentage of cleaved caspase 3-positive tumor cells. Notably, single doses of 10 and 30 mg/kg of YN14 dramatically reduced the levels of KRASG12C, and same dose of YN14-H showed higher potency compared with YN14, demonstrating that YN14-H exhibited more efficient degradation effect than YN14 in vivo. In summary, YN14-H demonstrates significant anticancer efficacy and PK properties advantages over YN14, indicating a better potential for future development.
Previous studies have reported different degradation abilities of BRD7 PROTACs 5 and 26, which are attached to distinct sites on VH032; degrader 26 has been shown to be superior to degrader 5 [15]. These degraders were used to verify the universality of the aforementioned MD-based strategies. First, we constructed the BRD7 and VHL complex using protein-protein docking, and the resulting conformational differences are shown in Figs. S8A and B (Supporting information). Next, unbiased MD simulations were employed to construct ternary complexes with relaxed conformations, and the ΔG was calculated using the MM/PBSA method. As shown in Figs. S8C and D (Supporting information), the formation of the ternary complex BRD7-PROTACs-VHL resulted in ΔG values of −23.3 and −30.6 kcal/mol for degraders 5 and 26, respectively, indicating that the higher stability of the resulting ternary complexes was induced by degrader 26. We also applied our computational strategy to the reported BTK degraders SJ638 and SJ690, which are attached to distinct sites on VH032 [13], and the calculated results were consistent with their biological activity (Figs. S7E–H in Supporting information). The observed stability order aligned with the corresponding biological activities of these compounds, thus validating the rationality and universality of our method.
In this study, we employed an MD-based strategy to explore the stability of ternary complexes of four KRASG12C PROTACs with distinct attachment sites of VHL032. We found the degrader YN14-H exhibited superior ability of inducing ternary complexes compared with YN14, YN14-B, and YN14-T reflected by the lowest ΔG of the four degraders during MD simulation. Subsequent biochemical interaction and cell-based functional assays revealed that YN14-H induced the strongest interaction between KRASG12C and VHL and exhibited most effective degradation as well as antiproliferative efficacy with nanomolar potency. YN14-H also demonstrated superior PK properties and antitumor activity in vivo. Furthermore, we also validated the universality of our calculate strategy with previously reported BRD7 and BTK PROTACS. Overall, our results demonstrate YN14-H as a promising candidate for the treatment of tumors with KRASG12C-mutation, and this MD-based strategy can be utilized to optimize KRASG12C PROTACs and facilitate the rational design of VHL-based PROTACs targeting other proteins at distinct attachment sites.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Shiyang Sun: Writing – review & editing, Writing – original draft, Methodology, Formal analysis, Data curation. Ning Yang: Writing – original draft, Validation, Methodology, Formal analysis, Data curation. Yaqiu Mao: Validation, Data curation. Ting Wei: Validation, Methodology, Data curation. Pengli Wei: Validation, Methodology, Data curation. Tingting Yang: Software, Data curation. Yixin Zhang: Validation, Methodology, Data curation. Jian Yan: Methodology, Data curation. Changkai Jia: Validation, Data curation. Yi Li: Formal analysis. Xu Cai: Formal analysis, Data curation. Zhiyuan Zhao: Validation, Formal analysis. Xuesong Feng: Methodology, Formal analysis. Xiaomei Zhuang: Methodology, Formal analysis, Data curation. Wenpeng Zhang: Writing – review & editing, Supervision, Investigation, Data curation. Junhai Xiao: Supervision, Project administration, Data curation. Pengyun Li: Writing – review & editing, Project administration, Methodology, Investigation, Funding acquisition, Conceptualization. Zhibing Zheng: Writing – review & editing, Supervision, Project administration, Conceptualization. Song Li: Supervision, Investigation.
Acknowledgments
This study was supported by National Natural Science Foundation of China (No. 82404417), State Key Laboratory of National Security Specially Needed Medicines Program (No. LTMC2022ZZ006). We would like to thank Editage (www.editage.cn) for English language editing.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.110992.
N.A. Zografou-Barredo, A.J. Hallatt, J. Goujon-Ricci, C. Cano, Bioorg. Med. Chem. 88-89 (2023) 117334. doi: 10.1016/j.bmc.2023.117334
[24]
W. Li, J. Zhang, L. Guo, Q. Wang, J. Chem. Inf. Model. 62 (2022) 523–532. doi: 10.1021/acs.jcim.1c01150
[25]
J. Zhang, Y. Cong, L. Duan, J.Z.H. Zhang, J. Chem. Inf. Model. 63 (2023) 5297–5308. doi: 10.1021/acs.jcim.3c00813
[26]
K.K. Bhanja, M. Sharma, N. Patra, J. Phys. Chem. B 127 (2023) 10749–10765. doi: 10.1021/acs.jpcb.3c04337
[27]
J. Yan, S. Sun, W. Zhang, et al., J. Cell. Biochem. 122 (2021) 1207–1215. doi: 10.1002/jcb.29941
[28]
T. Bartoschik, A. Zoephel, K. Rumpel, et al., Methods. Mol. Biol. 2365 (2021) 115–133. doi: 10.1007/978-1-0716-1665-9_6
[29]
K. Parikh, G. Banna, S.V. Liu, et al., J. Hematol. Oncol. 15 (2022) 152.
Figure 1
Ternary complexes constructed by protein-protein docking and unbiased MD simulations. (A–D) The chemical structures of PROTACs with different linker attachments and resulting ternary complexes. KRASG12C, VHL, and PROTACs were subjected to protein-protein docking and 500 ns MD simulations to relax the ternary structure. (E) VHL recruitment by the degraders YN14-H and YN14; the KRASG12C-YN14-VHL ternary complex is shown in yellow, while the KRASG12C-YN14-H-VHL ternary complex is shown in blue. By aligning the KRASG12C structures from both ternary complex MD simulations, the divergent VHL structures can be visualized. (F) At the exit of the KRASG12C binding pocket, YN14 and YN14-H display distinct interactions with the KRASG12C-VHL PPI interface.
Figure 2
Analysis of results from unbiased MD simulations. (A) The RMSD of ternary complexes. (B) The binding free energy (ΔG, kJ/mol) of PROTAC-recruited ternary complexes. (C) The H bonds of KRASG12C and VHL in the whole trajectory induced by different degraders. (D) Distinct interaction modes of KRASG12C and VHL at PPI interfaces induced by degraders YN14, YN14-H, YN14-B, and YN14-T.
Figure 3
Ternary complex formation and degradation effects of the four KRASG12C degraders. (A) Ternary complex formation induced by degraders via MST. The Kd determinations were performed in binding assay. (B) Dose-dependent interaction assay using AS technology. The VHL-KRASG12C complex formation was analyzed. The relative AS signals treated with YN14, YN14-H, YN14-B and YN14-T, respectively, are expressed as the luminescence signal relative to the luminescence signal of DMSO. (C) Immunoprecipitation of KRASG12C in NCI-H358 cells treated with DMSO, YN14, YN14-H, YN14-B and YN14-T at 1 µmol/L for 8 h, respectively.
Figure 4
Antiproliferative effect of the KRASG12C degraders. (A) The effects of YN14, YN14-H and AMG510 on KRASG12C, AKT and ERK phosphorylation in NCI–H358 cells. Cells were treated compounds for 48 h at the indicated concentration. (B, C) Representative images of flow cytometry analysis of apoptosis in NCI-H358 and MIA PaCa-2 cells treated with the vehicle control (DMSO) and indicated degraders at the indicated doses for 48 h. Histograms showing the relative cell percentage of apoptosis in NCI-H358 and MIA PaCa-2 cells are on the right. (D, E) Wound healing assay of NCI-H358 and MIA PaCa-2 cells treated with DMSO and the four degraders at the indicated doses for 12 h. Histograms showing the relative cell migration are on the right. Scale bar: 100 µm. Data are represented as the mean ± SD (n = 3). *P < 0.05, **P < 0.01 (t-test).
Figure 5
Pharmacokinetic (PK) properties and anticancer efficacy of YN14, YN14-H and AMG510 in vivo. (A) Treatment schedule for the NCI-H358 cells xenograft tumors model treated with vehicle, AMG510, YN14 and YN14-H, respectively. (B) The change of tumor volume was measured every 2 days (n = 7). Data are mean ± SD, **P < 0.01 (one-way ANOVA). (C) The change of body weight of all mice was measured every 2 days (n = 7). (D) Representative images of H&E, Ki67, KRASG12C, cleaved caspase 3 and CD31-staining in harvested tumors from each group are shown. Scale bar: 50 µm. Histograms show the quantification of Ki67+ (E), KRASG12C+ (F), cleaved caspase 3+ (G) and CD31+ (H) cells. Error bars denote standard deviations (independent experiments, n = 3). Data are represented as the mean ± SD. Student’s t-test. ns, no significance. **P < 0.01.

 DownLoad:
DownLoad:


 下载:
下载:






