Citation:
Xinyi Wang, Iek Man Lei, Bei Li, Yunlu Dai. Progress in extracellular vesicle@STING towards immune regulation[J]. Chinese Chemical Letters,
2026, 37(2): 110990.
doi:
10.1016/j.cclet.2025.110990
Progress in extracellular vesicle@STING towards immune regulation
English
Progress in extracellular vesicle@STING towards immune regulation
Received Date:
11 December 2024 Accepted Date:
20 February 2025 Revised Date:
17 February 2025 Available Online:
15 February 2026
Abstract:
The stimulator of interferon genes (STING), as a critical innate immune sensor, has been widely and continually explored in immune-related disease treatment. As lipid bilayer-closed particles derived from cells, extracellular vesicles (EVs) inherently function in target-guided intercellular communication. To incorporate the native merits of EVs into STING pathways, i.e., engineered EV@STING, poor bioavailability and off-target issues that STING activators possess could be significantly overcome. In this review, emerged STING activators such as nitrogen-containing heterocyclic structures and the universal STING activation strategy (uniSTING) are firstly summarized. Diverse EVs sources from mesenchymal stem cells (MSCs) and innate and adaptive immune cells may evoke distinct regulatory results. Concurrently, how the EVs contents including double-stranded DNA (dsDNA), microRNA (miRNA), cyclic GMP-AMP synthase (cGAS) and 2′3′-cyclic GMP-AMP (2′3′-cGAMP) proteins participate in the regulation of STING activation are widely studied. After mastering the two pivotal aspects of EV@STING, their immunomodulatory roles including in pathogen infection, inflammatory diseases, and cancer therapy are comprehensively summed up and discussed. Finally, in cancer study field, therapeutic challenges and clinical translational opportunities of EV@STING are thoroughly evaluated.
Stimulator of interferon genes (STING) plays a pivotal role in innate immune response. Typically, it drives the generation of type Ⅰ interferons (IFNs) and inflammatory cytokines to fight against cell infection by pathogens. However, recent studies have revealed the pros and cons of STING signaling in the systemic immunity. Although protecting human beings from pathogen infection, STING activation aggravates the progress of inflammatory diseases such as systemic lupus erythematosus. In most cases, STING signaling improves antitumor immune response, but its persistent and acute activation may evoke an opposite effect [1]. Therefore, it is urgent to update the discoveries on STING pathway and their influences on immune regulation.
To investigate the clinical translational potential of diverse STING activators, the main barriers they encounter are poor bioavailability issues and off-target toxicity [2], which, however, have been proposed to be addressed by incorporating with extracellular vesicles (EVs). EVs released by cells participate in several homeostatic processes and the delivery of intercellular signals [3]. As natural membrane-derived vesicles, they exhibit good biocompatibility, high permeability, low immunogenicity and low toxicity. Moreover, EVs have great potential for clinical monitoring, diagnosis, and therapy [4-7]. To work for STING activator loading, the generated EV@STING could successfully overcome the off-target issues and reportedly show efficient antitumor therapeutic outcomes. The biology of the STING pathway and EVs in immunity have been introduced elsewhere individually [3,8]. The combination of both, despite therapeutically useful, has not been extensively summarized. We are thus inspired to concentrate on recent insights into the EV@STING and update their immunoregulatory progress in pathogen infection, inflammatory diseases, and cancer (Fig. 1). Furthermore, challenges and perspectives of EV@STING in the clinical translation are also discussed.
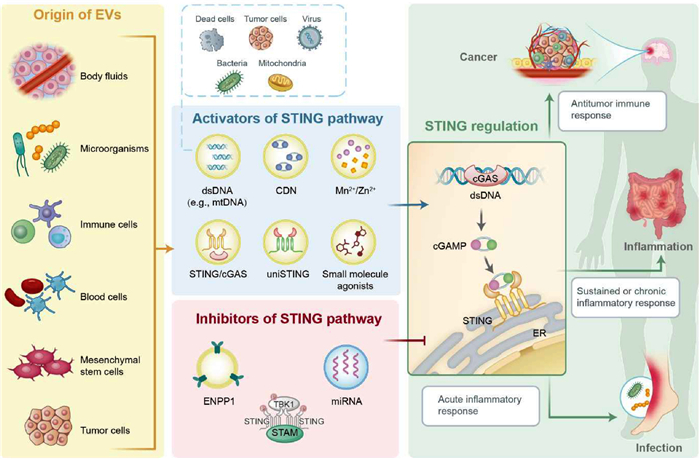
Figure 1
Figure 1.
Schematic illustration of EV@STING for immune regulation. EVs are lipid bilayer-closed particles that can be obtained from a variety of sources, including body fluids, microorganisms, and diverse cell sources. Many bioactive molecules have the capacity to regulate the STING pathway, which can be classified into activators and inhibitors. The activators of the STING pathway are comprised of dsDNA, STING agonists, metal ions, and STING mimic (i.e., uniSTING). Conversely, ENPP1, STAM, and certain specific miRNAs have an inhibitory effect on the STING pathway. The EVs, when loaded with different STING regulators, can exert immunomodulatory effects in cancer, inflammation, and infection.
As displayed in Fig. 2, in mammalian cells, the cyclic GMP-AMP synthase (cGAS) is primarily responsible for recognizing the invasion of pathogen DNA into the cytosol [9]. Following the binding of double-stranded DNA (dsDNA), cGAS undergoes conformational changes at the active site, which enable the conversion of ATP and GTP into 2′3′-cyclic GMP-AMP (2′3′-cGAMP), which is a second messenger molecule that functions as a potent agonist of STING [10,11]. cGAMP binds to the endoplasmic reticulum (ER)-resident adaptor protein STING, which induces a conformational change in STING that allows for TANK-binding kinase 1 (TBK1) binding and subsequent activation [12]. TBK1 phosphorylates interferon regulatory factor 3 (IRF3), which induces type Ⅰ IFNs [12,13]. Besides, STING also activates inhibitor of kappa B kinase (IKK) and subsequently nuclear factor kappa B (NFκB) for proinflammatory cytokine induction [14,15]. Accordingly, the cGAS-STING pathway is a crucial initiator of immune responses and plays a pivotal role in the pathogenesis of infections and diseases involving cytosolic DNA.
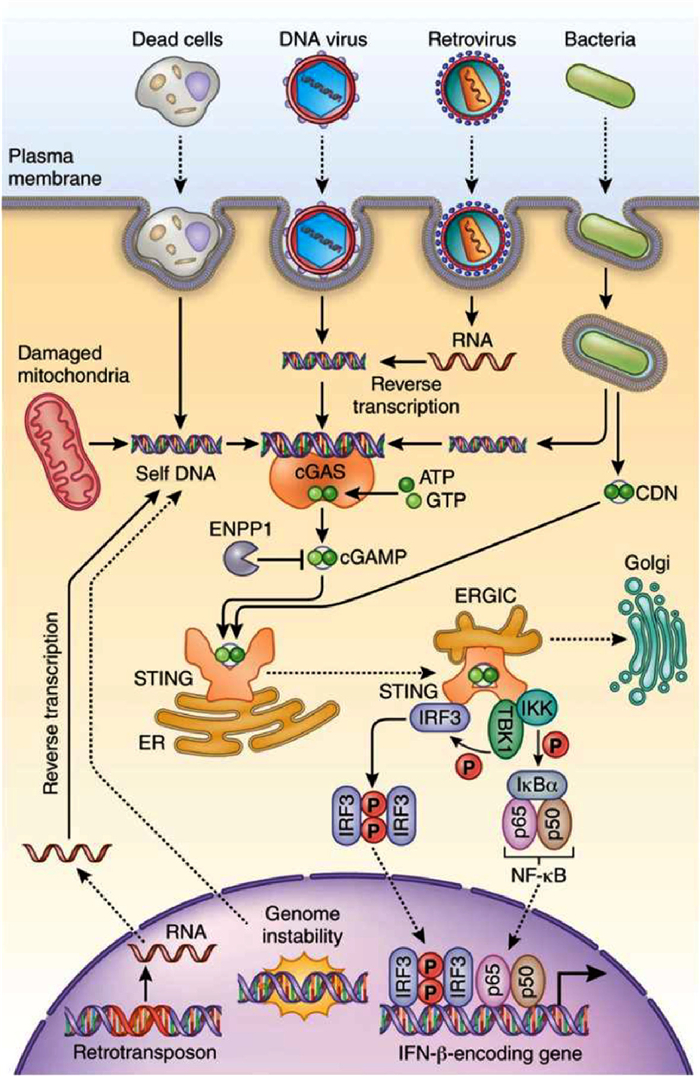
Figure 2
Figure 2.
The STING pathway. Following the binding of dsDNA, the activated cGAS convert ATP and GTP into cGAMP. cGAMP binds to the STING, which induces a conformational change in STING that allows for TBK1 binding and subsequent IRF3 and NF-κB phosphorylation. Phosphorylated IRF3 functions in conjunction with NF-κB to induce the expression of type Ⅰ IFNs and other immune-regulating molecules. Copied with permission [9]. Copyright 2016, Springer Nature.
Apart from the cGAS, STING pathway can be activated through other mechanisms. Natural cyclic dinucleotides (CDNs) can directly bind to STING protein and activate STING signaling. Some special non-cGAS DNA sensors are also active in the STING pathway. Interferon-inducible protein 16 (IFI16) senses cytoplasmic DNA in human cells, and may cooperate with cGAS for STING activation [16,17]. At the same time, IFI16 may work in a cGAS-independent manner. Dunphy et al. reported that the DNA repair proteins ataxia telangiectasia mutated (ATM) and poly-ADP-ribose polymerase 1 (PARP1), together with the DNA sensor IFI16 could activate STING in a non-canonical manner in cells treated with etoposide [18]. This alternative STING activation resulted in the predominant activation of NF-κB, which induced a distinct set of cytokines and chemokines compared to those typically observed in conventional DNA sensing did. Another important cytosolic DNA sensor, DEAD-box helicase 41 (DDX41), can directly bind both DNA and STING, thereby enhancing the production of type Ⅰ IFNs [19]. These alternative strategies all focus on therapeutic interventions targeting STING.
2.2
STING and immunity
STING pathway can be initiated by the host immune system to defense against infections by various pathogens including DNA virus, RNA virus, retrovirus, bacteria and parasites, etc. These pathogens contain DNA or generate DNA in their life cycles [20]. A typical case is that the STING signaling in inflammatory diseases. The pathogenesis of inflammatory diseases is sometimes associated with the STING mutations and constitutive activation [21-24]. Chronic activation of the STING pathway can also result in the onset of autoimmune diseases, a certain kind of inflammatory diseases, broadly speaking [24,25]. Both type Ⅰ IFN and self-nucleic acids are recognized as pivotal contributors to the pathogenesis of systemic autoimmune diseases [26].
The aforementioned STING-associated immune responses might be in effect in anticancer therapy. Due to unstable genomes, cancer cells easily encounter chromosome mis-segregation during mitosis. This process results in the formation of micronuclei, which are characterized by easy rupture and release of dsDNA into the cytoplasm [27-29]. Oxidative stress and mitochondrial dysfunction may cause cancer cells to release mitochondrial DNA (mtDNA) into the cytoplasm [30]. Both DNA fragments may directly initiate the STING signaling in cancer cells. Activated STING triggers the type Ⅰ IFN production, as well as some proinflammatory cytokines, chemokines, proteases, and even growth factors [31,32]. This cascade produces immunostimulatory factors that can either inhibit tumor growth in a cancer cell-autonomous manner or recruit immune cells to fight against the tumor [31,33]. Another alternative path is that tumor DNA may translocate into the cytoplasm of dendritic cells (DCs) and macrophages to activate STING-IRF3-induced IFN signaling, which enhances the presentation of tumor antigen on DCs and thereby cross-triggers antitumor immunity in CD8+ T cells [34,35]. Instead of transferring self-DNA, tumor cells can secrete cGAMP into the extracellular space, which then enters host immune cells via the folate transporter protein SLC19A1 [36]. Tumor-derived cGAMP then activates the host STING-IRF3 pathway, which in turn induces NK-mediated tumor killing [37].
However, although favorable antitumor immunity in most scenarios, STING sometimes exhibits inhibitory effects on the immune system. It was reported that cancer cells treated with STING agonist exhibited a marked increase in programmed cell death ligand 1 (PD-L1) expression and proinflammatory cytokines [38]. Oddly, STING-dependent DNA sensing may shape an immune-suppressive tumor microenvironment [39,40]. Chromosomal instability can lead to the activation of the STING pathway and downstream noncanonical NF-κB signaling, which unexpectedly drove tumor metastasis [41]. These findings demonstrate the dual function of STING pathway in tumor immunity.
2.3
Emerged STING activators
Activators to initiate the STING signaling have continually been identified. These activators can be broadly classified into two categories: those that activate STING via the cGAS-STING pathway and those that bind directly to STING protein. Moreover, a STING mimic can activate the STING downstream pathways in the absence of endogenous STING, which is also involved in this section.
2.3.1
dsDNA that activates the cGAS-STING
DNA mis-segregation during cell division generates micronuclei that consist of chromatin surrounded by nuclear membrane. Following the rupture of the micronucleus membrane, exposed chromatin DNA can be recognized by cGAS and thus activates the cGAS-STING pathway. In addition to replication stress and genomic instability, micronuclei can also occur after exposing to genotoxic stress, ionizing radiation, or the induction of chromosome segregation errors. However, the micronuclei formed as a result of genotoxic insults contain histone-bound self-DNA, which inhibits the binding of cGAS and therefore cannot trigger the cGAS-STING pathway [42]. Apart from the micronuclei DNA, the ectopic mtDNA is also a stimulator of inflammatory response [43,44]. In certain pathological conditions, e.g., genotoxic stress, oxidative stress, viral infection, high levels of proinflammatory factors, and mitochondrial dysfunction, mtDNA can be released and activates the cGAS-STING pathway [45]. RNA virus infection may result in cellular damage and cell death, which can lead to the release of cellular DNA [46]. DNA intermediates are produced during reverse transcription process that occurs in retroviruses [46]. Summarily, DNA that initiates cGAS-STING pathway can be endogenous from the nucleus and mitochondria or is exogenous from viruses or bacteria.
2.3.2
STING agonists
2′3′-cGAMP is a natural agonist of STING, and yet unfavorable pharmacological properties curb the application of this natural CDN. The focus thus shifts to explore synthetic CDNs and small molecules as STING agonists. In this section, natural CDNs, synthetic CDNs, and other small molecules are introduced as STING agonists, respectively.
(1) Natural CDN
2′3′-cGAMP, as typical natural CDN, has been widely demonstrated to work as endogenous STING agonist in mammalian cells. Other natural CDNs include cyclic dimeric GMP (c-di-GMP), cyclic dimeric AMP (c-di-AMP), and 3′3′-cGAMP, which can activate STING signaling in prokaryotic cells [47]. However, natural CDNs are susceptible to hydrolysis by phosphodiesterase, suggesting them as suboptimal drug candidates [48].
(2) Synthetic CDN
Synthetic CDNs aim to improve STING efficiency. Some CDN analogues were synthesized with nonhydrolyzable characteristics. Li et al. reported a hydrolysis-resistant bisphosphothioate analog of 2′3′-cGAMP, 2′3′-cGsAsMP, which demonstrated a comparable binding ability for human STING (hSTING) and an increase in the potency of IFN-β secretion [49]. ML RR-S2 CDA, also known as ADU-S100, is another a widely used synthetic CDN. ADU-S100 can resist the degradation by phosphodiesterase and thus effectively generate CD8+ T cell priming initiated by tumor [50]. Additionally, a macrocycle-bridged STING agonist (MBSA) derivative of ADU-S100 was demonstrated to exhibit prolonged antitumor efficacy in murine tumor models [51].
(3) Small molecule STING agonists
Flavone acetic acid (FAA) has been demonstrated to possess antitumor properties and has also been identified as a murine STING (mSTING) agonist [52]. Due to its unsuccessful application in animal models, a series of analogous compounds were developed for use as STING agonists. For instance, 5,6-dimethylxanthenone-4-acetic acid (DMXAA) is a drug that destroys tumor blood vessels and causes hemorrhagic necrosis of tumor tissues. Furthermore, it has been demonstrated to activate mSTING and initiate innate immune responses [53,54]. However, the inability of the DMXAA to bind to hSTING represents a significant obstacle to its potential clinical application. 10-Carboxymethyl-9-acridanone (CMA) is another similar derivative that has also been identified as an exclusive mSTING agonist [55-57]. Zhang et al. identified α-Mangostin, a DMXAA analog with xanthone skeleton, as a ligand that binds to STING protein and activates STING pathway [56-58].
After analyzing the structure of STING agonists, Zhao et al. demonstrated that nitrogen-containing heterocyclic structures are essential for binding with the homodimeric STING protein complex and forming hydrogen bond interactions in the binding pocket [59]. The same specific structural characteristic was also identified in linked dimeric amidobenzimidazoles (diABZIs) and SR-717, two synthetic STING agonists. The diABZIs were created by linking two symmetry-related ABZI-based compounds, thereby enhancing the binding affinity to STING and cellular function [60]. The diABZIs was observed to elicit robust antitumor activity, resulting in complete and enduring regression of tumors. SR-717 is another small molecule STING agonist identified through a series of phenotypic screens related to the STING pathway and subsequent structure-function analysis [61]. Intraperitoneal administration of SR-717 has been demonstrated to exhibit antitumor activity and prolong overall survival in B16F10 tumor models [61]. Meanwhile, SR-717 also inhibited lung metastasis of melanoma in mouse models. Although diABZIs and SR-717 have similar structure with cGAMP, diABZIs bind to STING through an open lid confirmation, while SR-717 induces a closed lid conformation [62].
Pan et al. identified benzothiophene oxobutanoic acid (MSA-2) as a non-nucleotide-based STING agonist through the use of phenotypic screening [63]. Two molecules of MSA-2 interact with each other through their aromatic cores, resulting in predimerization before binding to STING [62]. The administration of MSA-2 in the colorectal cancer mouse model demonstrated a dose-dependent antitumor activity. Moreover, MSA-2 also elicited long-term antitumor immunity and a synergistic effect when combined with anti-PD-1 antibodies.
Finally, 4-(2‑chloro-6-fluorobenzyl)-N-(furan-2-ylmethyl)−3-oxo-3,4-dihydro-2H-benzo[b][1,4]thiazine-6 carboxamide, denoted G10, is a special small molecule STING agonist that was identified through high-throughput screening [64]. The mechanism of some small molecule STING agonists is based on the indirect binding to STING. G10 could activate IRF3 and IRF3-dependent transcriptional activity via a STING-associated pathway, rather than directly binding to STING protein [65].
2.3.3
STING mimic
Despite that CDNs and small molecule STING agonists have continually been reported, we should realize that these agents could not work without STING protein. Unfortunately, in some tumor cases, epigenetic silencing or missense mutations inhibit the STING expression [66]. To activate STING pathway in those special cases, Wang et al. designed a universal STING activation strategy (uniSTING) independent of endogenous STING expression. They genetically fused a highly stable tetramerization motif with the non-membrane-bound domain of STING [67]. Interestingly, uniSTING has been shown to imitate the cGAMP-induced oligomerization of endogenous STING, which results in the constitutive activation of tumor-control IRF3/IFN-I pathways, but not tumor-progression NF-κB pathways. Furthermore, delivering mRNA encoding STING mimic with the lipid nanoparticles (LNP) resulted in potent antitumor efficacy across metastatic tumor models. Collectively, uniSTING addressed current limits of STING agonists, particularly in the context of STING-deficient tumors.
2.3.4
Metal ions
Manganese (Mn2+) was identified as a naturally occurring and inducible potentiator for STING activation [68-70]. The binding capacity of cGAS for dsDNA is enhanced by Mn2+, and vice versa. Meanwhile, cGAMP-STING binding affinity seems could be augmented by Mn2+ [68]. Nevertheless, another study demonstrated that rather than affecting the affinity of STING binding, Mn2+ actually enhanced the activity of STING agonists [71]. Notably, not just positively coupling with dsDNA-dependent activation, Mn2+ could also activate cGAS without dsDNA [72]. To examine the downstream signal of STING, Mn2+ further induced the TBK1 and p65 phosphorylation in a STING-independent manner [71]. Mn2+ could also lead to IRF3 phosphorylation in combination with STING agonists, which amplified the STING signaling cascade.
Free zinc (Zn2+) has been demonstrated to enhance the enzymatic activity of cGAS [73]. Zn2+ stabilizes the cGAS–DNA complex by binding to cGAS and increases the cGAMP generation. Designed zinc cyclic di-AMP nanoparticles successfully suppressed tumor via endothelial STING activation [74]. In addition, magnesium (Mg2+) seems served as the catalytic cofactor of cGAS to catalyze the formation of cGAMP [75].
2.4
STING inhibitors
Some bioactive molecules have been reported to inhibit the STING signaling through diverse mechanisms. It was reported that signal transducing adapter molecule 1 (STAM) functions as a transporter that directly interacts with STING, facilitating the transport of STING into EVs. This process served as a mechanism for the degradation of STING and the subsequent negative regulation of STING signaling [76]. Furthermore, the STING pathway can be suppressed by inhibiting the upstream signaling of STING. Ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1), which express on the surface of tumor cell membrane, can degrade extracellular cGAMP, thereby inhibiting the STING pathway [77]. Some microRNAs (miRNAs) were also reported to curb the STING signal [78,79]. Apart from naturally occurring proteins and miRNAs, some emerged STING antagonist compounds have been comprehensively summarized elsewhere [26].
3.
Overview of EVs
3.1
Classification and biogenesis of EVs
The EVs are lipid bilayer-closed particles, released by cellular organisms, and cannot replicate on their own [80,81]. Currently, there is no standardized criteria for the classification of EVs. The EVs can be roughly classified into two types: exosomes and ectosomes, based on their biogenesis [3]. Exosomes are of endosomal origin, formed within multivesicular bodies (MVBs), and are released into the extracellular space after fusing with cell membrane [82,83]. Ectosomes are plasma membrane-derived EVs. One representative of ectosomes is the microvesicle, which is formed by the outward budding and fission of the plasma membrane at the cell surface [84]. Furthermore, EVs can be classified according to their dimensions. Small EVs (such as exosomes), which approximately range in size from 50 nm to 150 nm, are the most prevalent type of EVs in biological fluids. Microvesicles are representative medium EVs with an approximate diameter of 200–800 nm. Large EVs, e.g., apoptotic bodies, constitute the least abundant population of EVs with a diameter of ≥1 µm. Notably, extracellular particles such as exomeres and supermeres should not be considered as EVs as they lack the defining characteristics of a lipid bilayer.
It is worth noting that bacteria are also capable of producing those vesicles. Bacterial EVs (BEVs) are spherical vesicles derived from the bacterial membrane of both Gram-positive and Gram-negative bacteria. They are bilayered lipid membrane nanostructures with a size range of 20–400 nm [85]. BEVs contain numerous biological contents found within the parent bacterium but in a non-replicative form [86]. Based on this characteristic, BEVs express pathogen-associated molecular patterns (PAMP) that are recognized by host pattern recognition receptors (PRR) both extracellularly and intercellularly [86]. This process results in the activation of host immunity. Compared with bacteria, BEVs are non-infectious and exhibit a superior safety profile, which suggests their potential therapeutic effects.
3.2
Sources of extracellular vesicles
EVs can be segregated from multiple body fluids including blood, urine, and cerebrospinal fluid. However, some proteins may co-isolate with EVs, thereby rendering these body fluid-derived EVs as potential candidates for diagnostic purposes rather than therapeutics [81]. Moreover, EVs can be isolated from intestinal lumen contents. These intestinal EVs contain gut microbial DNA, which has the capacity to trigger the STING signaling [87,88]. As delineated in Section 3.1, bacteria constitute an additional substantial source of EVs. Host bacteria or bacteria from non-target species are optimal EVs sources.
All types of cells release EVs, and their features are highly determined by their source. The source could be immune cells. T cell-derived EVs have been demonstrated to effectively participate in the immunomodulation of autoimmune and infectious diseases, or STING activation by relying on surface-associated DNA [89-91]. As the antigen presenting cells (APCs), macrophages/DCs-derived EVs are used in immunotherapy as well as drug delivery [92-94]. Macrophage-derived EVs are known to express major histocompatibility complex (MHC) class I and MHC class II, which preferentially induce Th1-type immune response that directs T cells to attack cancer cells [92]. The antigen-presenting molecules enriched in DC-derived EVs contribute the antitumor therapy [95]. To examine the drug delivery performance, EVs derived from these two types of cells can easily cross biological barriers such as blood-brain barrier (BBB) [96-98]. Macrophage-derived EVs loaded with chemotherapeutics seems preferentially accumulate in cancer cells, suggesting their tumor-targeting ability [99]. Natural killer (NK) cells constitute a distinctive population of innate lymphoid cells that possess intrinsic capabilities to identify and eliminate virally infected cells and tumor cells [100]. NK cell-derived EVs are considered to contain the same cytotoxic proteins, cytotoxic receptors, and cytokines as NK cells themselves. Moreover, they can directly eliminate tumor cells and engage in the communication between immune cells in the tumor microenvironment, which serves as the biological basis for drug delivery candidates [101].
Among the current EV-based immunotherapeutic approaches, EVs from mesenchymal stem cells (MSCs) occupy a leading position [3]. MSCs are adult stem cells which can differentiate into both mesenchymal and non-mesenchymal cells [102]. MSCs can be used for cell therapy due to their easy isolation properties and specialized biological functions. MSC-derived EVs have similar function to MSCs, and are considered as potential alternatives to MSCs in the context of drug delivery [103,104]. However, it was reported that MSC-derived EVs exhibited immunosuppressive effects by suppressing NK cell activity and IFN-γ production [105]. MSC-derived EVs may not be the good vehicles for the STING activation.
Red blood cells (RBCs) represent the most abundant cell type in the human body and can be obtained in a safe and straightforward manner [106]. Furthermore, RBCs are devoid of both nuclear and mitochondrial DNA, rendering them an ideal vehicle for drug delivery [107]. RBC-derived EVs have been observed to exhibit characteristics similar to those of their cellular origins. These EVs have been demonstrated to possess the capacity to target tumors and to facilitate the delivery of therapeutic RNAs [108,109]. Platelets are anucleate cells derived from megakaryocytes and capable of maintaining hemostasis and vascular integrity [110]. Moreover, platelets play a pivotal role in cancer progression, both directly through interactions with cancer cells and indirectly via long-range interactions mediated by platelet releasates [111]. Although platelets are unable to traverse tissue barriers, platelet-derived EVs are capable of entering the lymphatic system, bone marrow, and synovial fluid [112]. The number of platelet-derived EVs is considerable, representing the majority of EVs in the bloodstream. Platelet-derived EVs are also involved in the modulation of tumor progression. It was reported that platelet-derived microparticles infiltrated solid tumors and transferred miRNAs to suppress tumor growth [113], different from the conventional perspective that platelet-derived EVs are tumor-supportive [111]. The dual role of platelet-derived EVs in tumor progression is attributable to the different cargoes they carry. Therefore, it is imperative to take the contents of platelet-derived EVs into consideration when utilizing them.
Tumor-derived EVs are reported to carry tumor antigens, costimulatory molecules, and DNA fragments that are similar to those of their parental cells. This results in the elicitation of a potent T cell-dependent antitumor immune response [114,115]. Meanwhile, tumor-derived EVs show tumor cell-specific targeting by exploiting the intrinsic homo-adhesion properties of the membrane antigen [116]. Several studies have been conducted to investigate the potential of tumor-derived EVs for the treatment of cancer [117,118]. A recent study reported that tumor cell-derived small EVs could act as a defense system against nanoparticle tumor delivery by binding and trafficking nanoparticles to liver Kupffer cells for degradation [119]. However, it's worth noting that tumor-derived EVs can facilitate cancer progression through a number of mechanisms, including enhanced cell proliferation and evasion of apoptosis, induction of angiogenesis, metabolic reprogramming, increased invasive and disseminating capacity, and avoidance of immune surveillance [120]. Safety issue remains a big concern for the application of this type of EVs.
4.
Mechanisms linking STING pathway and EVs
The complex interactions of STING pathway and EVs highly associate with the immune regulation and intercellular communication. The STING pathway affects the EVs biogenesis and cargo selection. In turn, EVs have the capacity to transfer certain components that function in the regulation of the STING pathway, such as dsDNA, 2′3′-cGAMP, and ENPP1. In this section, the primary focus is on the mechanisms that link the STING pathway and EVs.
The biogenesis of EVs involves distinct cellular pathways including the endosomal system. Activated STING triggers the production of type Ⅰ IFNs. Type I IFNs have been demonstrated to modulate the endosomal sorting complex required for transport machinery, which is essential for the biogenesis of exosomes [121]. Moreover, the STING pathway can influence EVs cargo selection. Upon activation, the activated STING protein can be packaged into EV, thereby modulating the immune response in recipient cells [122,123].
EVs contain a complex assortment of bioactive molecules, encompassing proteins, metabolites, lipids, genetic material, and organelle fragments, which differ across cell types, cell signaling states, and disease conditions [124]. It has been observed that certain components transferred by EVs can activate the STING pathway. In comparison to EVs derived from normal cells, EVs derived from tumors have been shown to contain a greater amount of DNA [125-128]. Moreover, the DNA content of EVs derived from different tumor cell lines exhibited variation. Thakur et al. reported that exosomes from lung and pancreatic cancer cell lines contained lower amounts of DNA, compared to those from melanoma and leukemia [125]. Furthermore, the DNA contents were found to be correlated with the size of the EVs. The abundance of DNA in large EVs released by Huh-7 cells was higher than that in small EVs [126]. A similar phenomenon was observed in EVs obtained from prostate cancer patient plasma [129]. Such differences may be attributed to variations in the internal DNA content rather than external DNA [126]. Furthermore, external EV-DNA was predominantly observed on small EVs, whereas internal EV-DNA was primarily found within large EVs [128].
EVs carry different DNA species, including single-stranded DNA (ssDNA), dsDNA, and mtDNA. Several studies have reported differences in the distribution and proportion of different DNA species in EVs. Whether ssDNA or dsDNA is predominant in EVs remains a controversial issue. Lázaro-Ibáñez et al. reported that the predominant form of DNA in EVs is ssDNA [130]. However, other researchers demonstrated that the majority of EV-DNA is dsDNA [125,128]. Additionally, Jiao et al. indicated that the distinction between ssDNA and dsDNA is in distribution rather than the proportion. They reported that dsDNA was mainly located within the EVs while ssDNA was on the EVs surface [126]. Interestingly, Jeppesen et al. reported that EVs did not contain dsDNA [131]. And those extracellular dsDNA and histones are secreted through an autophagy- and multivesicular endosome (MVE)-dependent, but exosome-independent mechanism. This finding is in contradiction with the conclusion of another study regarding the mechanism by which DNA enters the exosome via micronuclei [127]. Different isolation methods and cell sources of exosomes utilized in the studies may account for the discrepancies observed in their respective conclusions. Additionally, the DNA present within EVs also include endogenous DNA from the mitochondria [130,132,133]. However, some research reported that mtDNA was absent in EVs [125,134]. These discrepancies highlight the necessity to investigate the various subsets of EVs derived from different cellular sources.
The miRNAs are also identified in EVs. Cells received exosomal miRNAs may completely change the characteristics [135]. Moreover, EVs from different cellular sources contain unique miRNAs expression profiles and may also be differ from their parental cells [136]. The quantification of miRNAs within EVs is important in elucidating their biological functions. It has been investigated that the number of miRNAs molecules in exosomes is limited, suggesting that there is less than one given miRNAs per exosome [137]. Several studies have indicated that miRNAs present within EVs have the capacity to regulate the STING pathway.
Apart from nucleic acid, EVs contain a substantial number of protein molecules both inside and on their surface. Proteins largely shared by diverse EV subtypes are those associated with biogenesis pathways [124]. Typically, abundant EV proteins are parental cytosolic, cytoskeletal, heat shock, and plasma membrane proteins, as well as proteins involved in vesicle trafficking. EVs barely contain proteins from organelles of the parental cell [138]. Ni et al. identified that tumor-derived exosomes were enriched with cGAS and 2′3′-cGAMP proteins that can activate the STING pathway [139]. Moreover, following the administration of agonists or chemoradiotherapy, activated STING protein could be packaged into a type of non-canonical autophagosome. The non-canonical autophagosome was subsequently transformed into an MVB-like structure and secreted intraluminal vesicles, which were designated a new type of EV. These EVs can carry activated STING protein [140]. Additionally, during herpes simplex virus-1 (HSV-1) infection, STING can be released from infected cells in exosomes [141,142]. However, the stability of STING protein in EVs is uncertain. It was reported that STAM can transport STING into EVs, thereby serving as a mechanism for the degradation of STING [76]. Furthermore, it has been reported that EVs can also inhibit the STING pathway by carrying ENPP1. An et al. reported that exosomes derived from various tumor cells carry ENPP1, which can hydrolyze both synthetic and endogenous 2′3′-cGAMP produced by cells, thereby inhibiting the STING pathway in immune cells [143]. Moreover, T cell-derived apoptotic extracellular vesicles have been demonstrated to carry ENPP1, which can alleviate radiation enteritis [144].
In this review, we mainly focus on the immunomodulatory role of the EVs containing STING regulators. In the subsequent section, these EVs will be designated as EV@STING.
5.
EV@STING in pathogen infection
Pathogen infections trigger the generation of EV@STING. Here the STING activators in EVs could be microbial DNA, immune cell DNA, and even STING protein. Produced EV@STING affects the crosstalk between microorganism and host cells or the communication among infected and uninfected cells. This section mainly discusses the immunomodulatory role of EV@STING in infectious diseases.
5.1
EV@STING mediates the communication between microorganism and host cells
In the majority of cases, following infection, the aberrant DNA or CDNs present in the cytosol can directly activate the STING pathway. However, EVs derived from gut bacteria was reported to deliver bacterial DNA into host cells, triggering the STING pathway and priming the systemic antiviral immunity (Fig. S1a in Supporting information) [145]. Meanwhile, the DNA-containing EVs from the gut microbiota were observed to be present in the blood circulation, indicating that they may facilitate DNA delivery into host cells at distant sites. Both results suggest that gut commensals are vital for systemic immunity to respond to viral infection. Furthermore, EV plays a similar role in fungal infections. Harding et al. reported that Candida albicans (C. albicans), which is an important fungal pathogen, could secrete EVs contain microbial DNA (Fig. S1b in Supporting information). C. albicans DNA packaged in EVs triggered the STING pathway in macrophages (Figs. S1c and d in Supporting information) [146]. However, Chen et al. reported the uncanonical transit patterns of STING after fungal infection. Upon stimulation of C. albicans, STING was observed to transit alongside the ER to the phagosomes and negatively regulated the anti-fungal immune responses [147].
5.2
EV@STING mediates the communication between infected and uninfected cells
EVs also transmit signals between infected and uninfected cells, thus propagating immune responses against infection. It has been demonstrated that Plasmodium falciparum (P. falciparum)-infected RBC (iRBC)-derived EVs contain parasite non-coding RNA and parasite genomic DNA (gDNA) [148]. Subsequently, the EVs were internalized by human monocytes, resulting in the transfer of the P. falciparum DNA within EVs into the host cell cytosol, which in turn activated the STING pathway. Following the Kaposi's sarcoma-associated herpesvirus (KSHV) infection, EVs derived from infected cells carried mtDNA to activate the STING pathway in uninfected cells, thus exerting antiviral response [149]. In addition to DNA, EVs also transported STING protein to exert antiviral effect after HSV-1 infection [141,142].
However, this process does not always facilitate antibacterial defense. Nandakumar et al. demonstrated that intracellular bacterial DNA was sorted into EVs and delivered to bystander cells, thereby stimulating the STING pathway [150]. This phenomenon occurred when infected with Listeria monocytogenes (L. monocytogenes), Legionella pneumophila and Francisella tularensis. Nevertheless, EVs from L. monocytogenes-infected cells demonstrated the capacity to impede T cells proliferation and prime T cells for apoptosis. The results suggested that intracellular bacteria can exploit the foreign DNA delivered by EVs to impair antibacterial defense.
5.3
Immune cell derived EV@STING
It has been demonstrated that T cell derived EVs are effective tools for reprogramming DCs and enhancing the innate immune response against pathogens [91]. Upon contact between antigen-bearing DCs and T cells, the gDNA and mtDNA contained within the T cell derived EVs are transmitted to DCs. This process initiates the STING pathway and induces the antiviral response in DCs.
6.
EV@STING in inflammatory disease
STING signaling associates with various inflammatory diseases. When the pathway is overactivated, it can cause excessive inflammation, contributing to conditions such as autoimmune diseases, cardiovascular diseases and metabolic diseases. Inflammatory pathogenesis is generally EV@STING-involved.
6.1
EV@STING in autoimmune diseases
Both the STING pathway and EVs have been implicated in the pathogenesis of autoimmune diseases. Li et al. have demonstrated that EVs isolated from the plasma of patients with dermatomyositis contained dsDNA, which triggered the STING pathway to induce type Ⅰ IFN release and mediated proinflammatory response [151]. Inflammatory bowel disease shares some features with autoimmune diseases. In Crohn's disease, EVs could transport pathogenic mtDNA and nuclear gDNA from damaged intestinal epithelial cells to macrophages [152]. Similarly, another study demonstrated that microbial DNA-containing EVs derived from the gut microbiota activated the STING pathway, thereby stimulating inflammation in inflammatory bowel disease [88].
6.2
EV@STING in cardiomyopathy
In states of cardiomyopathy, the injured mitochondria result in significant cardiomyocyte apoptosis and the release of EVs containing mtDNA [153]. EVs intercellularly communicate between cardiomyocytes and fibroblasts. The mtDNA-containing EVs derived from cardiomyocyte have been observed to activate the STING activation and promote fibroblast proliferation [153,154].
6.3
EV@STING in metabolic disease
STING activation can exacerbate the chronic inflammation of many metabolic diseases. Luo et al. have demonstrated that the release of gut microbial DNA-containing EVs mediates the obesity-associated tissue inflammation and insulin resistance via the STING pathway [87]. EVs are also capable of transporting mitochondria, which can then mediate inflammatory responses. Gao et al. reported that EVs derived from M1 macrophages (M1-EVs) encapsulating inflammatory mitochondria penetrated pancreatic beta cells [155]. Upon fusion with the mitochondria of beta cells, this process triggered the ferroptosis pathway, leading to mitochondrial disruption and subsequent release of mtDNA into the cytoplasm. Subsequently, the released mtDNA activated the STING pathway, which ultimately resulted in apoptosis.
6.4
EV@STING in radiation-induced inflammatory disease
Tissues exposed to radiation therapy easily cause inflammation. As a radiation-sensitive organ, the intestine is susceptible to radiation enteritis and the pathogenesis is STING-involved. But the participation of EVs can negatively regulate STING signaling and alleviate the inflammatory response. Zhou et al. demonstrated that apoptotic EVs (ApoEVs) released by T cells could target the intestines of abdominal irradiated mice [144]. The ENPP1 located on the surface of the ApoEVs could hydrolyze cGAMP to inhibit the STING pathway, thereby alleviating radiation enteritis (Fig. S2 in Supporting information).
7.
EV@STING in cancer
The STING pathway plays a pivotal role in antitumor immune response through the production of type Ⅰ IFN and other pro-inflammatory cytokines, leading to the recruitment and activation of various immune cells. EVs, such as tumor-derived EVs, have intrinsic biocompatibility and natural tumor-targeting capability, rendering them the potential vectors of STING regulators. Based on these properties, EV@STING was utilized as an effective antitumor therapeutic strategy in the field of biomaterials. In this section, we collectively analyze the roles of native and engineered EV@STING in antitumor immune response. Engineered EV@STING applications in the field of cancer therapy were presented comprehensively (Table S1 in Supporting information).
7.1
Native EV@STING
7.1.1
Tumor-derived EVs
Tumor-derived EVs can function as mediators of intercellular communication in the tumor microenvironment [124]. They can transfer DNA fragments to DCs, which then proceed to produce type Ⅰ IFN through the STING pathway [156]. The inclusion of a fraction of dsDNA in tumor microvesicles (TMVs) is regulated by the cycling of ARF6 GTP/GDP and occurs together with cGAS [157]. The transfer of dsDNA to recipient cells by TMVs is able to change the biological performance of the recipient cells. Meanwhile, tumor-derived exosomes, representing another type of EVs, could activate STING pathway in tumor-infiltrated T cells by exosomal TGF-β, cGAS, and 2′3′-cGAMP. This consequently may cause the expansion of iTregs in cervical cancer [139]. The findings suggest that tumor-derived EVs may potentially be utilized as a tumor vaccine due to their ability to present innate DNA signals.
Tumor-derived EVs influence the activation and differentiation of tumor-associated macrophages (TAMs) via the STING pathway. After taken up by macrophages, Ma et al. demonstrated that tumor-derived EVs induced M2-type TAMs through the STING pathway, promoting tumor growth, metastasis and cancer stem cell development [158]. Furthermore, EVs released by triple negative breast cancer (TNBC) enabled monocytes to differentiate to be distinct macrophage subsets [159]. The addition of colony-stimulating factor-1 (CSF-1) led to the differentiation of proinflammatory macrophage subset in TNBC EVs. Furthermore, tumor-derived EVs have been demonstrated to induce macrophages to release a key chemokine C-C motif ligand 2 (CCL2) [160]. CCL2 can attract monocytes to the site of vaccine injection, thereby enhancing the process of endocytosis of antigen. Mechanically, tumor-derived EVs can activate the STING signaling through the release of DNA fragments, which then induce monocytes to upregulate the expression of IRF4, which is a key factor in the differentiation of monocytes into monocyte-derived DCs (moDCs). moDCs are capable of presenting tumor antigens to T cells, thereby eliciting a robust antitumor immune response. Moreover, monocytes that have internalized tumor-derived EVs gain the capacity to treat tumors.
Unexpectedly, tumor-derived EVs could also inhibit the STING signaling. An et al. reported that ENPP1 was presented in various tumor-derived exosomes (Fig. S3a in Supporting information) [143]. As shown in Fig. S3b (Supporting information), ENPP1 localized on the surface of exosomes can hydrolyze 2′3′-cGAMP bound to LL-37 (an effective transporter of 2′3′-cGAMP). To isolate exosomes from human lung and breast cancer tissue, those tumor-derived EVs contained high level of ENPP1, which was observed to inhibit the T cell infiltration. Moreover, EVs participate in the STING degradation. STAM engages in direct interaction with the STING oligomer, subsequently facilitating its transportation to EVs, where it undergoes degradation in the recipient cells (Figs. S3c–e in Supporting information). The transfer of activated STING oligomers into exosomes by STAM represents an additional mechanism through which STING degradation and the suppression of the innate immune response can occur [76]. This biological process may serve to prevent the overactivation of innate immunity.
It has been demonstrated that miRNAs in EVs also regulate the STING pathway. In PARPi-resistant TNBC cell lines, Bustos et al. demonstrated that miR-181a downregulated the STING protein and the downstream proinflammatory cytokines to enhance PARPi resistance [78]. EVs derived from PARPi-resistant TNBC cell lines work as the vehicle to transfer miR-181a to PARPi-nonresistant parental cells, impairing STING and aggravating PARPi resistance. Hypoxia was observed to enhance the release of EVs from glioblastoma (GBM) cells and upregulate the expression of miR‑25/93 in both cells and in EV cargos [79]. The transfer of miR-25/93-containing EVs to macrophages resulted in the impairment of the STING pathway, leading to a reduction in type Ⅰ IFN expression and secretion by macrophages.
In addition to EVs produced during tumor progression, both chemotherapy and radiotherapy can eliminate significant number of tumor cells, resulting in the production of tumor-derived EVs [158]. Chemotherapy is a major antitumor therapeutic strategy. Following the administration of antitumor agent topotecan (TPT), breast cancer cells have been observed to induce the secretion of danger-associated molecular patterns (DAMPs), which in turn could trigger the activation of DCs and the subsequent production of cytokines. Cancer cells treated with TPT could release DNA containing exosomes, which activated DCs through a STING-dependent pathway [161]. Radiotherapy also plays a significant role for the treatment of cancer. Diamond et al. reported that tumor-derived exosomes (TEX) produced by irradiated mouse breast cancer cells (RT-TEX) transferred dsDNA to DCs and stimulated DC upregulation of costimulatory molecules and STING-dependent activation of type Ⅰ IFN [162]. Hematopoietic stem cell transplantation (HSCT) is a treatment for leukemia. In mouse models of myeloid leukemia, EVs were observed to be constitutively secreted from myeloid leukemia cells during HSCT. These EVs transferred dsDNA to donor cells, which resulted in the activation of the STING pathway, thereby reducing leukemia cell viability through the generation of reactive oxygen species (ROS) [163].
7.1.2
EVs purified from other cells
Apart from tumor-derived EVs, EVs secreted by other cell types can also affect the tumor immunity. Activated CD4+ T cells secrete EVs (i.e., T-EVs). These T-EVs can sensitize macrophages to elevate STING activation by virtue of the self-contained IFN-γ rather than self-contained DNA or cGAMP [164]. Liu et al. purified the exosome-like particles from Artemisia annua (A. annua), which were termed as artemisia-derived nanovesicles (ADNVs) [165]. The researchers identified ADNVs facilitated the internalization of plant-derived mtDNA by TAMs and induced the STING activation and macrophage polarization. ADNVs also reportedly inhibited tumor growth and boosted antitumor immunity in a mouse lung cancer model. EV donators could be diverse and exhibit distinct modulation in antitumor therapeutic treatment.
7.1.3
Rafeesome-derived EVs
The newly identified organelle, rafeesome, is formed through the fusion of RAB22A-mediated non-canonical autophagosome with an early endosome [140]. Relying on rafeesome to intercellularly transfer activated STING has become a novel strategy for the development of STING-targeted therapies (Fig. S4a in Supporting information). Here the rafeesome, which exhibits an MVB-like structure, is designated to fuse with the plasma membrane and to release the inner vesicles of non-canonical autophagosomes. The new type of extracellular vesicle is defined as RAB22A-induced extracellular vesicles (R-EVs) (Fig. S4b in Supporting information). The R-EVs serve as the carrier of activated STING, thereby inducing antitumor immunity in recipient cells (Figs. S4c–e in Supporting information). Subsequently, Guo et al. utilized MSCs to produce R-EVs containing activated STING [166]. It was reported that induced pluripotent stem cell (iPSC)-derived MSCs can generate R-EVs with a size and formation mechanism that is comparable to that of R-EVs produced by cancer cells. This cell-free strategy also represents a potential therapeutic approach for enhancing antitumor immunity.
7.2
Engineered EV@STING
7.2.1
Normal cell-derived EVs
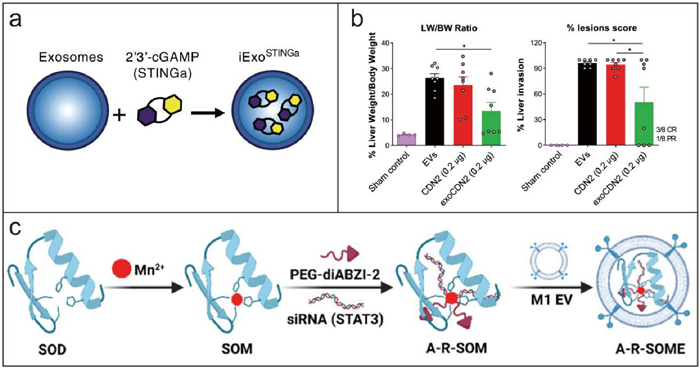
Previous studies have highlighted the immense potential of EVs as drug-delivery systems. McAndrews et al. engineered exosomes derived from HEK293T cells to deliver the cGAMP (iExoSTINGa), which can selectively target the STING pathway in APCs (Fig. 3a) [167]. Moreover, better than STINGa alone, iExoSTINGa highly efficiently deliver STINGa into DCs, which led to an enhanced accumulation of activated CD8+ T cells and an augmented antitumor immune response. Jang et al. utilized HEK293-derived EVs to load potent CDN agonists and obtained the engineered exoSTING [168,169]. In this research, PTGFRN-overexpressing EVs were employed due to their safety and the absence of endogenous immune modulatory effects. The exoSTING loaded by CDN1 (ML RR-S2, a 2′−3′ CDN) and CDN2 (cAIM(PS)2 Difluor, a 3′−3′ CDN) enhanced local Th1 responses and the recruitment of CD8+ T cells, thereby generating systemic antitumor immunity. Compared with free CDN, exoSTING treatment apparently relieved tumor burden (Fig. 3b). Meanwhile, exoSTING at therapeutically active doses did not induce excessive inflammatory response, thereby expanding the therapeutic window.
Figure 3
Figure 3.
Engineered EV@STING derived from normal cells. (a) Schematic representation of iExoSTINGa generation. Reproduced with permission [167]. Copyright 2021, American Society for Biochemistry and Molecular Biology. (b) 15 days after administration of exoSTING, liver weight (LW) over body weight (BW) was calculated and% lesions scores were evaluated. Reproduced with permission [168]. Copyright 2021, Springer Nature. (c) Schematic illustration of the A-R-SOME preparation. Reproduced with permission [169]. Copyright 2024, American Chemical Society.
EVs derived from macrophages have been demonstrated to possess immunomodulatory properties [93,170]. Yang et al. filled hollow MnO2 radiosensitizer and metformin (Met) into macrophage-derived EVs and got multi-functional Met@HMnER [171]. The Met@HMnER effectively improved the hypoxic TME through two mechanisms: hollow MnO2-catalyzed O2 generation and Met-associated cellular respiratory depression. These two mechanisms expressed a remarkable synergistic sensitization effect on radiotherapy. Meanwhile, the combination of HMn and EVs maintained the biological barrier permeability while exhibiting a high-drug loading capacity. Moreover, the modulation of immunosuppressive TME was also accomplished by the innate immune enhancement of NK cells via the activation of the STING pathway with Mn2+. In order to remodel the immunosuppressive microenvironment in malignant pleural effusions (MPE), Li et al. designed a nanoplatform, designated A-R-SOME, through a synergistic integration of enzyme-dynamic therapy (EDT) and metalloimmunotherapy [169]. A-R-SOME comprises M1 macrophage derived EVs (M1-EVs) and loaded cargos of manganese-based superoxide dismutase (SOD) enzyme, diABZI-2, and signal transducer and an activator of transcription 3 (STAT3) small interfering RNA (Fig. 3c). Endogenous ROS within tumors induced immunogenic cell death via the process of EDT. Mn2+ and diABZI-2 acted synergistically to activate the STING pathway. Given that STING activity in human cancer cells is inhibited by the JAK2/STAT3 pathway, it can be posited that the suppression of the STAT3 pathway may serve to enhance the activity of STING agonists [172]. Moreover, inhibiting STAT3 can also reverse the immunosuppressive microenvironment of MPE. M1-EVs have limitations such as inadequate cargo encapsulation. Therefore, Guo et al. developed hybrid nanovesicles (LEV) by fusing M1-EVs with liposomes, thereby enabling EVs to exhibit a tunable composition and additional features [173]. The liposomes were loaded with reversine (REV) and SR780Fe (REV@SR780Fe@Lip). REV is an antitumor agent which can activate the STING pathway in breast cancer [174]. SR780 is an amphiphilic photosensitizer developed by the researchers. Once bound to Fe3+ (SR780Fe), the compound is inactivated. However, upon the release of Fe3+ in mildly acidic conditions, the compound undergoes a transformation, becoming activated. To integrate M1-EVs withREV@SR780Fe@Lip, formed hybrid nanovesicles after the modification of RS17 peptide inhibited CD47–signal regulatory protein α (SIRPα) signaling and enhanced macrophage engulfment. These tumor-targeting hybrid nanovesicles achieved a combination of photodynamic therapy, ferroptosis, and STING pathway activation, which enhanced antitumor efficacy through synergistic effects.
7.2.2
Tumor-derived EVs
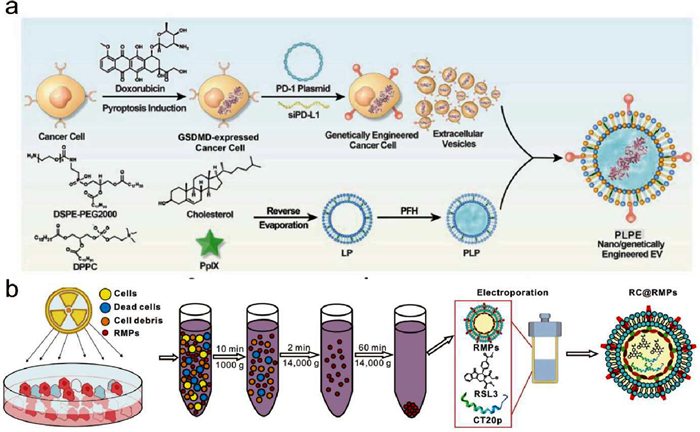
The properties of tumor-derived EVs presented in Section 3.2 render them potential drug-delivery candidates for targeted therapy. Zhou et al. fused tumor-derived EVs with sonosensitive nanoliposomes, which gathered functional groups of surface-presented programmed death 1 (PD-1), sonosensitive protoporphyrin IX (PpIX), gasdermin D (GSDMD) protein, and phase change perfluorohexane (PFH) (Fig. 4a) [175,176]. This nanovesicles-sensitized US-controlled immunoengineering therapy of tumor (NUITT) tactics exhibited both sonosensitive activity and protein transport functions for performing chemical, physical, and biological catalysis and inducing PANoptotic cell death pathways. PANoptotic cell death could induce tumor DNA release, which may subsequently activate the STING pathway and elicit an immunological antitumor response.
Figure 4
Figure 4.
Engineered EV@STING derived from tumor cells. (a) Schematic illustration of the design and synthetic route of nano/genetically engineered EVs. Reproduced with permission [175]. Copyright 2024, Wiley. (b) Schematic illustration of RC@RMP preparation. Reproduced with permission [176]. Copyright 2024, BioMed Central.
According to previous investigations, radiated tumor cell-derived microparticles (RMPs) are the primary mediators of radiation therapy-induced bystander effects, which also exhibit unique and broad therapeutic antitumor effects [177]. RMPs could induce ferroptosis of tumor cells and the reprogramming of M2 macrophages, which may promote type Ⅰ IFN expression through the STING pathway. Given that RMPs originate from the tumor cells, they are inherently capable of targeting tumors. Therefore, Deng et al. designed a nanoplatform wherein the ferroptosis inducer RSL-3 was loaded into RMP [176]. The apoptosis inducer CT20 peptide (CT20p) was incorporated into the RMPs to enhance the ferroptosis (Fig. 4b). The results showed that RMPs encapsulating RSL-3 and CT20p (RC@RMPs) retained the characteristics of RMPs and effectively targeted prostate tumor cells. Furthermore, RC@RMPs activated DCs and M1 polarization of macrophages, enhancing both adaptive immunity via CD8+ T cells and innate immunity via STING pathway, which effectively eliminated prostate cancer cells.
7.2.3
Hybrid EVs
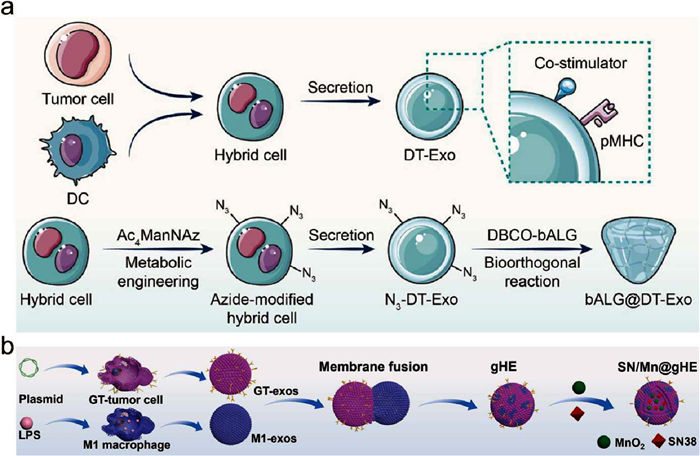
In comparison to EVs derived from a single cell type, chimeric EVs from at least two cell types retain the parental cell-specific signatures. Given this mechanism, Bao et al. developed a novel immunotherapeutic platform, DT-Exo-STING, which employs a DC-tumor chimeric exosome (DT-Exo) and a co-delivery of cGAMP [178]. The exosome-assisted delivery strategy has the potential to enhance the efficacy of exogenous STING agonists. In addition, DT-Exo could load antigen-derived pMHC and costimulatory molecules owing to the characteristics of the parental cells. Thus, DT-Exo-STING evoked a potent immune response, allowing for the near-complete eradication of GBM foci. Subsequently, the research team upgraded the DT-Exo-STING (Fig. 5a). They reported an in situ formed immunotherapeutic gel that can act as an artificial lymph node structure within the GBM tumor tissues after surgery [179]. In addition to leveraging the characteristics of DT-Exos, the azide-modified DT-Exos were obtained through metabolic glycoengineering. For the purpose of postoperative application, the exosome-crosslinked gel was formed by the bio-orthogonal reaction of azidegroup-modified exosomes with alkyne-modified alginate polymers. Unlike free chimeric exosomes that are rapidly removed, this exosome-cross-linked gel structure guaranteed a direct and long-acting antitumor T cell response. Meanwhile, this exosome-containing gel platform exhibited considerable potential for enhancing the efficacy of STING agonists.
Figure 5
Figure 5.
Engineered EV@STING generated from hybrid cells. (a) Schematic illustration of bALG@DT-Exo gels preparation. Reproduced with permission [179]. Copyright 2024, American Chemical Society. (b) Schematic diagram of SN/Mn@gHE preparation. Reproduced with permission [180]. Copyright 2023, Elsevier.
Hybrid EVs could be diverse. Cheng et al. designed and synthesized genetically engineered hybrid exosomes (gHE) by fusing exosomes derived from M1 macrophages (M1-exos) with exosomes derived from genetically engineered CD47-overexpressed tumor cells (GT-exos). It was reported that gHE ameliorated antitumor immunity through polarizing TAMs and inhibiting the "do not eat me" signal from tumor cells [180]. The hybrid exosomes were further encapsulated with MnO2 nanoparticle and DNA damage chemotherapy drug SN38, designated as SN/Mn@gHE (Fig. 5b). SN/Mn@gHE exhibited an efficient capacity to evade immune clearance through the disguise of CD47 and repolarize TAMs to the M1 phenotype. Meanwhile, it could induce immunogenic cell death (ICD) through the chemotherapeutic effect of SN38, which subsequently activates the STING pathway, and promotes DCs maturation, CTL infiltration, and NK cell recruitment.
7.2.4
Apoptotic bodies
Besides exosomes and microparticles, apoptotic bodies (ABs) represent another category of EVs [124]. ABs are membrane-enclosed particles derived from cell apoptosis [181]. They are distinguished as being leakproof and having tumor-specific antigen constituents, which makes them suitable for use as vectors in immunotherapy. Bao et al. exploited a nanovaccine platform to induce Fenton reaction and STING activation in cancer cells, thus generating tumor cell-derived ABs [182]. The experimental nanovaccines were the membrane fusogenic lipid-coated nanoparticles that contained Fe2+ and cGAMP molecules in the core. To facilitate the delivery of the nanovaccine payload into the cytoplasm, the researchers induced apoptosis in cancer cells. This process resulted in the generation of ABs that were loaded with cGAMP and subsequently engulfed by APCs. The findings of this study indicate that ABs might be a suitable candidate in antitumor therapy.
7.2.5
Bacteria-derived EV (BEV)
The role of tumor commensal bacteria in the etiology and progression of tumor has drawn increased attentions recently. Previous research demonstrated that modulating the microbiome could have impact on the efficacy of cancer treatment, thus suggesting intratumor bacteria as a potential therapeutic target [183-185]. Inspired by the phenomenon that patients with breast cancer exhibited a prevalence of commensal bacteria, Zhang et al. developed an activatable biointerface comprising commensal BEVs encapsulated in a responsive nanocloak, with the objective of enhancing immunoreactivity against intratumoral bacteria and breast cancer [186]. They employed MnO2 to program the immunointerface of Bacteroides fragilis-derived BEVs and achieved nanocloaked BEV (cBEV) with pH-responsive behavior. It was demonstrated that cBEV has heightened immunogenicity through intercellular responsive immunogenicity, which facilitates DCs maturation through the activation of the STING pathway.
7.2.6
Cellular membrane nanovesicles
In view of the low yield of cell-derived EVs for drug delivery purposes, alternative solutions have been proposed. Cancer cell membrane-coated exosome-mimetics are cost-effective. This innovative drug delivery system can effectively target tumors, exhibit high biocompatibility, and demonstrate effective drug loading [187]. Guo et al. developed exosome-mimetics to load REV and Doxorubicin (DOX), also known as EM@REV@DOX [188]. The accumulation of EM@REV@DOX at the tumor site was facilitated by the homing targeting effect, which allows for the release of REV and DOX. This leads to DNA damage, which activates the STING pathway and promotes DCs maturation, macrophage polarization, and T cells activation.
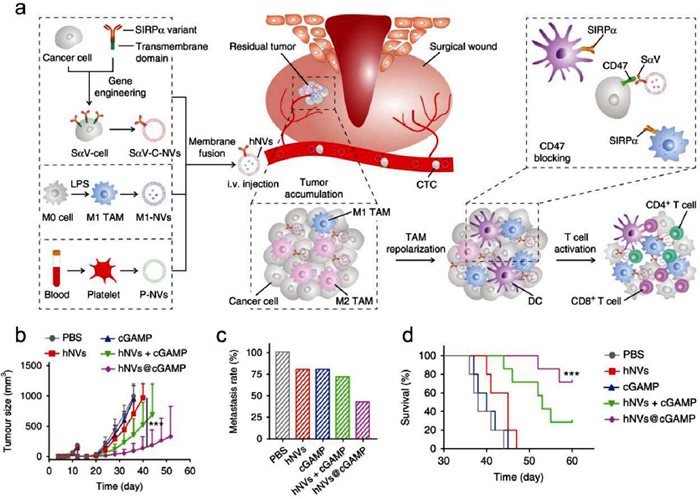
Hybrid cellular membrane nanovesicles (hNVs) have emerged as promising alternatives to EVs [189-191]. hNVs contain proteins and lipids derived from the parental cells, and thus inherit a variety of characteristics from them [192]. For instance, hNVs were reported by Rao et al. to amplify immune responses against post-surgery cancer recurrence and metastasis [193]. The hNVs comprised three categories: platelet-derived NVs (P-NVs), M1 macrophage-derived NVs (M1-NVs), and cancer cell-derived NVs overexpressing high-affinity SIRPα variants (SαV-C-NVs) (Fig. 6a). The nanoparticles were observed to accumulate efficiently at surgical wound sites, interact with circulating tumor cells (CTCs), promote M1 phenotype repolarization, and block the CD47-SIRPα interaction. This resulted in enhanced macrophage phagocytosis of cancer cells, augmented antitumor T cell immunity, and a reduction in the side effects associated with systemic infusion. Subsequently, hNVs@cGAMP was developed by loading cGAMP into the hNVs, indicating that hNVs enhanced the cGAMP delivery, and exhibited better antitumor effect in TNBC models (Figs. 6b–d). These findings suggest that cellular membrane nanovesicles may serve as a potential alternative to EVs.
Figure 6
Figure 6.
hNVs@cGAMP for cancer therapy. (a) Schematic illustration of the preparation and antitumor mechanism of the hNVs. (b) Tumor growth kinetics in different groups. (c) Metastasis rates after indicated treatments. (d) Survival curves for different treatment groups. Reproduced with permission [193]. Copyright 2020, Springer Nature.
EV-based therapeutics possess cell-free novelty. However, there are some challenges in clinical translation. The major challenges are isolation and characterization methods, which were not provided in most of the EV-related clinical trials [194]. Owing to the heterogeneity of EVs, it is difficult to standardize these methods. For the purpose of clinical applications, the method capable of isolating large amounts of EVs is required. Ultracentrifugation, which is widely used in the laboratory, yet is not suitable for large-scale production. Tangential flow filtration (TFF) and size-exclusion chromatography (SEC) can isolate EVs from larger volumes of cell culture media, potentially, thus facilitating their broad clinical application [2]. EVs transport bioactive molecules that may influence the immune microenvironment. However, there is a potential risk of unwanted immunogenicity associated with these intrinsic contents. The utilization of cell sources with safety profiles and low immunogenicity, such as stem cells, may prove an effective solution to this concern. Moreover, novel techniques to measure protein or nucleic acid contents in single EVs may provide insights into the mechanisms underlying the therapeutic potential of EVs. Additionally, issues such as reproducibility, loading strategy and storage condition are obstacles for the clinical translation of EV-based therapeutics. In addition to the aforementioned challenges associated with EV-based therapeutics, more limitations still exist. Firstly, there is an incomplete understanding of the mechanisms linking the STING pathway and EVs. This lack of comprehensive knowledge can hinder the optimization and predictability of therapeutic outcomes. Secondly, there are potential safety concerns related to the EV@STING, e.g., the risk of unintended immune activation or inflammatory responses. Addressing these limitations requires our deep understanding on the interactions between the STING pathway and EVs.
Over the past few decades, a number of EV-based clinical trials have emerged. In 2005, two clinical trials of EV-based antitumor therapies were initiated [195,196]. DC-derived exosomes loaded with tumor antigen were utilized to treat non-small cell lung cancer and melanoma, demonstrating safety and antitumor immunity in Phase Ⅰ and Phase Ⅱ clinical trials. In addition, EV-based delivery vehicles with small interfering RNAs (siRNAs) targeting KRASG12D and STAT6 antisense oligonucleotides have been demonstrated to control tumor growth [197,198]. The administration of exoSTING, an engineered EV loaded with STING agonists, resulted in localized STING pathway activation and dose-dependent immune activation in phase Ⅰ/Ⅱ clinical trial. Intratumoral administration of exoSTING was well tolerated and demonstrated tumor retention, with no evidence of systemic exposure to the STING agonist. In a subset of patients, a reduction in tumor size was observed in both injected and distal non-injected lesions. Although the exoSTING clinical trial demonstrated the advantage of local administration of exosomes, intratumoral injection is an alternative that can be achieved with conventional chemotherapy. Furthermore, there is a lack of head-to-head comparative trial with conventional drug delivery systems, such as liposomes, which consequently results in a paucity of robust evidence to substantiate the promising future of EV-based therapy in clinical applications. Apart from antitumor therapy, EVs were also employed in anti-inflammatory therapy. ILB-202 was an engineered exosome loaded with super-repressor IκBα, which inhibited the activity of NF-κB, thus inhibiting the inflammation. The anti-inflammatory effect of ILB-202 has been demonstrated in ischemia-reperfusion-induced acute kidney injury animal models [199]. Furthermore, ILB-202 exhibited favorable safety and tolerability profile in Phase Ⅰ clinical trial. So far, these EV-based clinical trials demonstrate the safety of heterologous EVs, and the clinical outcomes support the effectiveness of EV-based therapy.
In this review, distinct immunomodulatory tasks of EV@STING in pathogen infection, inflammatory diseases, and cancer have been individually summarized. Pathogen infections trigger the generation of EV@STING generated after pathogen infections is mainly responsible for the communications among microbes, infected, and uninfected cells. EV@STING usually worsen the progress of inflammatory diseases, but not for therapy. Distinctively, the proposal of utilizing STING-based EVs as new regimens for cancer therapy is promising.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the National Natural Science Foundation of China (NSFC, Nos. 32222090, 32101069 and 32171318), Faculty of Health Sciences, University of Macau, the Multi-Year Research Grant (MYRG) of University of Macau and the University of Macau Development Foundation (UMDF) (Nos. MYRG2022-00011-FHS and MYRG-GRG2023-00013-FHS-UMDF), the Science and Technology Development Fund, Macau SAR (Nos. 0002/2021/AKP, 0133/2022/A3, 0009/2022/AKP, and 0006/2023/ITP1), Ministry of Education Frontiers Science Centre for Precision Oncology, University of Macau (No. SP2023-00001-FSCPO), and Guangdong Provincial Applied Science and Technology Research and Development Program (No. 2024A1515011140).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.110990.
A. Daei Farshchi Adli, R. Jahanban-Esfahlan, K. Seidi, S. Samandari-Rad, N. Zarghami, Chem. Biol. Drug Des. 91 (2018) 996–1006. doi: 10.1111/cbdd.13166
[55]
T. Cavlar, T. Deimling, A. Ablasser, K.P. Hopfner, V. Hornung, EMBO J. 32 (2013) 1440–1450. doi: 10.1038/emboj.2013.86
Figure 1
Schematic illustration of EV@STING for immune regulation. EVs are lipid bilayer-closed particles that can be obtained from a variety of sources, including body fluids, microorganisms, and diverse cell sources. Many bioactive molecules have the capacity to regulate the STING pathway, which can be classified into activators and inhibitors. The activators of the STING pathway are comprised of dsDNA, STING agonists, metal ions, and STING mimic (i.e., uniSTING). Conversely, ENPP1, STAM, and certain specific miRNAs have an inhibitory effect on the STING pathway. The EVs, when loaded with different STING regulators, can exert immunomodulatory effects in cancer, inflammation, and infection.
Figure 2
The STING pathway. Following the binding of dsDNA, the activated cGAS convert ATP and GTP into cGAMP. cGAMP binds to the STING, which induces a conformational change in STING that allows for TBK1 binding and subsequent IRF3 and NF-κB phosphorylation. Phosphorylated IRF3 functions in conjunction with NF-κB to induce the expression of type Ⅰ IFNs and other immune-regulating molecules. Copied with permission [9]. Copyright 2016, Springer Nature.
Figure 3
Engineered EV@STING derived from normal cells. (a) Schematic representation of iExoSTINGa generation. Reproduced with permission [167]. Copyright 2021, American Society for Biochemistry and Molecular Biology. (b) 15 days after administration of exoSTING, liver weight (LW) over body weight (BW) was calculated and% lesions scores were evaluated. Reproduced with permission [168]. Copyright 2021, Springer Nature. (c) Schematic illustration of the A-R-SOME preparation. Reproduced with permission [169]. Copyright 2024, American Chemical Society.
Figure 6
hNVs@cGAMP for cancer therapy. (a) Schematic illustration of the preparation and antitumor mechanism of the hNVs. (b) Tumor growth kinetics in different groups. (c) Metastasis rates after indicated treatments. (d) Survival curves for different treatment groups. Reproduced with permission [193]. Copyright 2020, Springer Nature.

 DownLoad:
DownLoad:

 下载:
下载:







