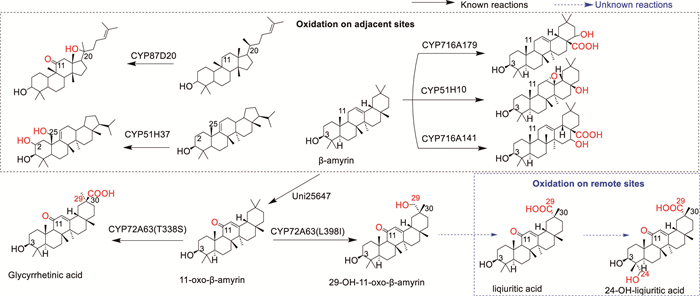
Scheme 1.
The diagram of different oxidation type of triterpenoids.
Microenvironment accessibility enables rare oxidation type of triterpenoids by plant P450
Wentao Sun , Lin Xiang , Runming Wang , Meilan Huang , Bo Lv , Youcai Hu , Chun Li

Natural products (NPs) have long been a valuable source for the development of drugs, with over 60% of pharmaceuticals being related to NPs. Expanding the structural diversity of NPs remains a pivotal strategy in uncovering novel pharmacological activities [1,2]. Plant triterpenoids possess remarkable pharmaceutical activities, among which glycyrrhetinic acid (GA) from licorice root was famous for its excellent antimicrobial, enzyme inhibitory, anti-inflammatory, analgesic, and antiviral effects [3-6]. Notably, the C-3-OH and C-30-COOH functional groups make GA very attractive lead structures for medicinal scientists, as these functionalities enable the generation of further chemical diversity to enhance pharmacological effects. One of its analogues, liquiritic acid, has been found to stimulate the release of adrenocorticotropic hormone [7] and has demonstrated high binding affinities with all severe acute respiratory syndrome coronavirus 2 (SARS‑CoV‑2) proteins, including non-structural protein 12 (NSP12), main protease, spike glycoprotein, RNA-dependent RNA polymerase, and multi-domain NSP3 [8]. The C-3-OH and C-29-COOH functional groups make liquiritic acid a platform compound expected to stimulate medicinal chemist to expand the structure and activity diversity of GA based new drug. Moreover, introducing additional OH groups to liquiritic acid could greatly expand the chemical space for bioactive derivatives. Despite its significance, no enzymes have been identified to synthesize or tailor on liquiritic acid, and its biosynthesis in microbes remains enigmatic.
Catalytic C-H bond activation is a fundamental process mediated by cytochrome P450 (P450) enzymes, playing a crucial role in the modification of plant triterpenoids [9,10]. Notable examples include CYP716, CYP90, and CYP72, which exhibit diverse tailoring capabilities [11-16]. Among these, some P450s has been identified as a dual-site oxidizer. For instance, CYP716A141 demonstrates weak oxidation at C-16 and C-28 of β-amyrin to generate cochalic acid [12]. However, current knowledge of dual-site oxidizing P450 enzymes is limited to neighboring sites, and the existence of P450s capable of remote oxidation, such as the iterative oxidation of C-29 and C-24 on 11-oxo-β-amyrin, remains elusive (Scheme 1). Hence, investigating the mechanistic underpinnings of remote oxidation are of paramount importance for the development of novel derivatives.
We here attempted to elucidate the underlying principles governing the activity, selective and remote sites oxidation within triterpenoid molecules by P450 enzymes. As the future applications in microbial cell factories and the plant P450s are bound to the endoplasmic reticulum membrane which are frequently challenging to express as recombinant proteins in vitro, the three-dimensional structure was built by AlaphFold2 [17] and the in vivo test was conducted using yeast platform carrying β-amyrin synthase (bAS) from Glycyrrhiza glabra, P450 reductase GuCPR1 and Uni25647 from Glycyrrhiza uralensis [10,18].
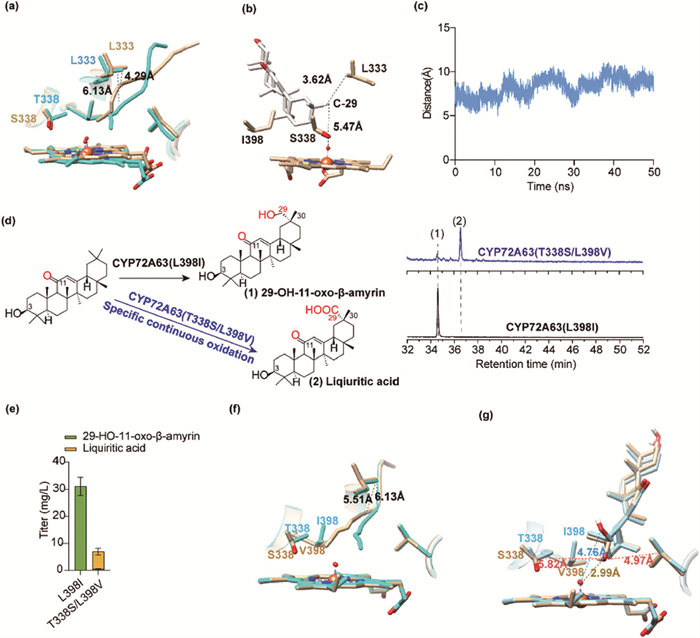
As the mutant CYP72A63 (L398I) specifically produces 29-OH-11-oxo-β-amyrin (Figs. S1–S11 and Table S1 in Supporting information), and CYP72A63 (T338S) can continuously oxidize 11-oxo-β-amyrin to glycyrrhetinic acid (Scheme 1) [9]. Consequently, we introduced the T338S mutation into the L398I variant to enable the conversion of 11-oxo-β-amyrin to liquiritic acid. However, this approach led to sharply decreased oxidation activity (Fig. S11). Compare to threonine, the reduced steric hindrance of serine significantly remodeled the substrate tunnel, which became biased toward L333 with a shorter distance of 4.29 Å, compared to 6.13 Å in CYP72A63 (L398I) (Fig. 1a). As a result, the C-29 of initial substrate11-oxo-β-amyrin leaned to the L333 with a distance of 3.62 Å and away from the FeⅣ=O moiety with a greater distance of 5.47 Å in the molecular docking analysis (Fig. 1b). The molecular dynamics (MD) stimulation further revealed an improper distance between C-29 and the FeⅣ=O moiety with an average distance of 8.28 ± 1.07 Å and a minimum distance of 4.95 Å (Fig. 1c), which was unsuitable for oxidation. Despite efforts to optimize the active site microenvironment for improved activity, the restricted substrate accessibility to this tailored environment led to reduced enzymatic performance. This highlights the critical importance of ensuring microenvironment accessibility in successful enzyme engineering. To enhance microenvironment accessibility, we introduced mutations at the 398th residue using amino acids with reduced steric hindrance (including T/V/A/D/C) to guide the substrate toward S338, a site with lower hydrophobicity. Building on the highly active CYP72A63 (T338S) variant, we aimed to explore oxidation possibilities for 29-OH-11-oxo-β-amyrin. Notably, the mutant CYP72A63 (T338S/L398V) demonstrated exceptional performance in producing a new compound (2) with a remarkable dominance (Fig. 1d), which was identified as liquiritic acid (Figs. S12–S20, Table S2 in Supporting information), constituting 91.66% of the collective oxidation products derived from 11-oxo-β-amyrin and a production of 6.40 ± 1.23 mg/L (Figs. 1d and e). Compared to CYP72A63 (L398I), the introduction of T338S and L398V mutant resulted in a reduction of steric hindrance at positions 398 and 338. Consequently, the substrate tunnel underwent a noticeable deviation away from L333 with a longer distance of 5.51 Å and extend toward S338 (Figs. 1f and g). As a result, the hydrophilic hydroxyl group at position C-29 of 29-OH-β-amyrin obtained a conformation reorientating to the FeⅣ=O moiety with a distance of 2.99 Å rather than the conformation depart from the FeⅣ=O moiety with a distance of 4.76 Å in CYP72A63 (L398I) (Fig. 1g), this conformational change made the 29-OH-11-oxo-β-amyrin more favorable to be oxidized in CYP72A63 (T338S/L398V). However, the C-29 hydroxyl group remained closer to the hydrophobic L149 residue (4.97 Å) rather than the more hydrophilic S338 (5.82 Å). This suggests that engineering the microenvironment around the 149th residue could further improve catalytic performance. Additionally, the introduction of a carboxyl group at C-29 of 11-oxo-β-amyrin would alter the hydrophobic distribution of the triterpenoid skeleton. As a result, modifications to the active site microenvironment could potentially enable a novel oxidation pathway for 29-OH-11-oxo-β-amyrin, leading to the production of new derivatives.
Notably, residue 149 also plays a pivotal role in influencing the regioselectivity between C-30 and C-29 [9,19], underscoring the complexity of this mutational strategy. To simultaneously refine the hydrophobic microenvironment, enhance substrate accessibility, and minimize impact on regioselectivity, threonine, aspartic acid, and glutamine were identified as promising candidates for mutation at the 149th position. This selection was based on their lower hydrophobicity and steric hindrance, coupled with geometric shapes akin to leucine (Fig. S21 in Supporting information). Additionally, isoleucine was also considered as a mutant to refine the tunnel architecture and adjust the orientation towards the hydrophilic microenvironment formed by T338. The catalytic activity of all mutants was meticulously evaluated by quantifying liquiritic acid production in the engineered strains. Significantly, both L149I/L398V and L149T/L398V mutants exhibited substantial conversion of 11-oxo-β-amyrin to liquiritic acid (Fig. 2a and Fig. S22 in Supporting information), with L149T/L398V demonstrating superior efficiency. Impressively, the production of liquiritic acid increased up to 25.58 ± 2.37 mg/L representing a notable 4-fold increase compared to the CYP72A63 (T338S/L398V) (Fig. 2b).
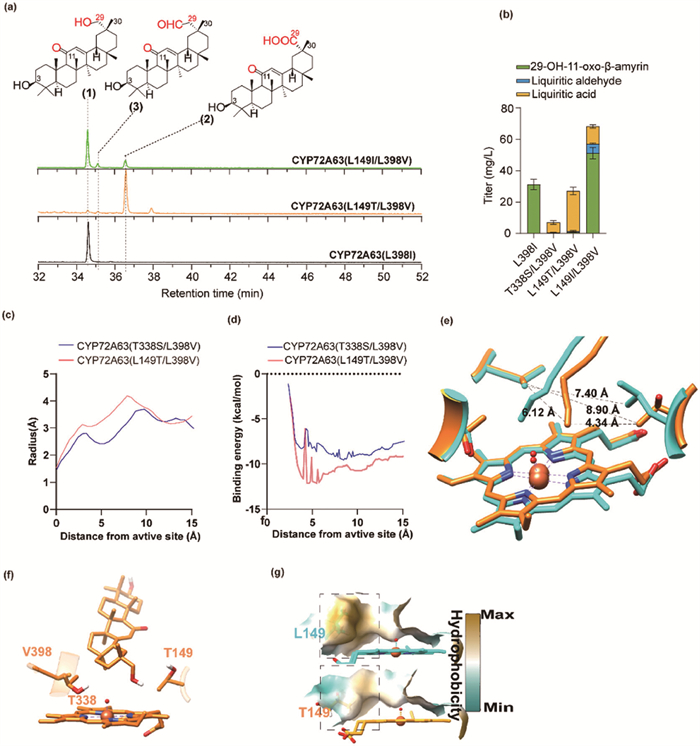
The mutant L149T reduced the hydrophobicity and expanded the substrate tunnel radius near the active site (Fig. 2c). While the enlarged tunnel could facilitate substrate accommodation, the diminished hydrophobicity may hinder the binding of the hydrophobic 11-oxo-β-amyrin. Binding process analysis via CaverDock revealed pronounced fluctuations in binding energy for 11-oxo-β-amyrin within CYP72A63 (L149T/L398V) (Fig. 2d). Nonetheless, the overall binding energy of 11-oxo-β-amyrin in CYP72A63 (L149T/L398V) was lower compared to CYP72A63 (T338S/L398V) (Fig. 2d), suggesting enhanced binding affinity and catalytic activity toward 11-oxo-β-amyrin.
Furthermore, the reduced steric hindrance of threonine instigated a reorientation of the tunnel towards T149, accompanied by a decrease in hydrophobicity (Fig. 2e). This structural modification concurrently fine-tuned the microenvironment and enhanced substrate accessibility to this microenvironment. Consequently, 29-OH-11-oxo-β-amyrin adopted an orientation favoring the more hydrophilic T149, with its C-29 hydroxyl directed towards FeⅣ=O (Figs. 2f and g). The binding free energy (ΔGbinding) for 29-OH-11-oxo-β-amyrin was lower than that in CYP72A63 (T338S/L398V), indicating a heightened substrate affinity (Table 1). Consequently, CYP72A63 (L149T/L398V) exhibited significantly improved performance.
 DownLoad:
CSV
DownLoad:
CSV
| CYP72A63 mutant | ΔGbinding (kcal/mol) |
| T338S/L398V | −38.27 ± 0.029 |
| L149T/L398V | −40.94 ± 0.035 |
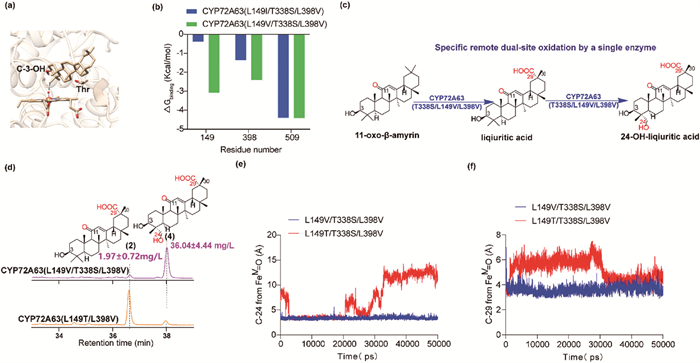
Of note, although mutant CYP72A63 (L149I/L398V) demonstrated lower production of 10.87 ± 0.99 mg/L liquiritic acid accounted for only 15.95% of the total oxidized products derived from 11-oxo-β-amyrin, the overall oxidized product yield was 2.4-fold higher compared to the L149T/L398V mutant (Fig. 2b and Fig. S23 in Supporting information). Mutant L149I caused the tunnel to shift closer to L398 and T338 (Fig. 3a). Consequently, the tunnel extended towards T338 with a relatively hydrophilic microenvironment that facilitated the orientation of the C-29 hydroxyl group towards FeⅣ=O. The distance from C-29 on 29-OH-11-oxo-β-amyrin to T338S and FeⅣ=O significantly decreased from 4.90 Å to 3.97 Å and 4.76 Å to 3.02 Å (Fig. 3b). However, the substrate binding energy of 29-OH-11-oxo-β-amyrin near the entrance of the substrate tunnel was much higher than that of 11-oxo-β-amyrin suggesting better substrate selectivity of 11-oxo-β-amyrin than 29-OH-11-oxo-β-amyrin (Fig. 3c). Hence, the L149I/L398V mutant led to limited continuous oxidation ability (Fig. 2a).

As illustrated in Fig. 3d, the establishment of a hydrophilic environment surrounding the C-29 hydroxyl group was principally facilitated by the presence of T338. The T338S mutant could downregulate the hydrophobicity of the proximal active pocket, thereby rendering it more compatible with the binding of 29-OH-11-oxo-β-amyrin, as evidenced by a lower ΔGbinding value of −44.60 ± 0.032 kcal/mol compared with both CYP72A63 (L149I/L398V) and CYP72A63 (L149T/L398V) (Table 2). Additionally, tunnel analysis of the mutant CYP72A63 (L149I/T338S/L398V) indicated a downregulation of hydrophobicity at the tunnel entrance compared with CYP72A63 (L149I/L398V) (Fig. S24 in Supporting information). This led to a convergence in the substrate binding energy between 29-OH-11-oxo-β-amyrin and 11-oxo-β-amyrin near the entrance of the substrate tunnel with similar substrate binding energy (Fig. 3e), affirming a favorable compatibility for the binding of both substrates. Consequently, the triple mutant, CYP72A63 (L149I/T338S/L398V), was constructed and demonstrated remarkable performance, yielding 33.49 ± 1.71 mg/L of liquiritic acid (Fig. 3f) and exhibiting further enhanced continuous iterative oxidation ability with no residual 29-OH-11-oxo-β-amyrin (Fig. 3g).
DownLoad:
CSV
| CYP72A63 mutant | ΔGbinding (kcal/mol) | |
| Empty Cell | 11-oxo-β-amyrin | 29-OH-11-oxo-β-amyrin |
| L149I/T338S/L398V | −37.99 ± 0.035 | −44.60 ± 0.032 |
| L149T/L398V | −35.91 ± 0.90 | −40.94 ± 0.035 |
| L149I/L398V | −36.72 ± 0.033 | −42.83 ± 0.033 |
Remarkably, mutants L149T/L398V and L149I/T338S/L398V exhibited the production of an extra derivative (4) (Fig. 3g), which was identified as 24-OH-liquiritic acid, featuring a hydroxyl group on the C-4 methyl positioned on the same side as the C-29 carboxyl group of liquiritic acid, with titers reaching 3.19 ± 0.42 and 4.05 ± 0.88 mg/L, respectively (Figs. S25–S33 and Table S3 in Supporting information). This observation indicates the successful realization of a unique oxidation pattern for triterpenoids, characterized by remote dual-site oxidation, specifically selective oxidation at C-29 and C-24. To the best of our knowledge, such a phenomenon has not been reported previously. Comprehending the underlying mechanism of this process holds significant importance as it lays the ground-work for the controlled diversification of triterpenoid types and the elucidation of P450 oxidation modes in shaping plant triterpenoids.
To elucidate the mechanism underlying C-24 oxidation, we performed a detailed analysis of the active site in the reported C-24 hydroxylase of β-amyrin, CYP93E1–9, to identify key factors responsible for this regioselectivity. A comparative active pocket analysis with CYP72A63 mutants, which do not achieve C-24 oxidation, revealed the presence of a hydrophilic threonine residue in place of leucine at the corresponding site (Fig. S34 in Supporting information). This substitution favored the binding of the hydrophilic hydroxyl group on the C-3 tail of the β-amyrin skeleton, thereby promoting oxidation at C-3 tail rather than at the hydrophobic C-30 tail (Fig. 4a). These findings suggested that the oxidation at the more hydrophilic C-3 tail of the β-amyrin skeleton is also driven by substrate microenvironment accessibility within the active site.
Therefore, achieving a balanced microenvironmental accessibility in the active site for 11-oxo-β-amyrin, 29-OH-11-oxo-β-amyrin, and liquiritic acid, each with distinct hydrophobic properties, was crucial for facilitating remote dual-site oxidation of 11-oxo-β-amyrin to produce 24-OH-liquiritic acid using a single P450 enzyme. Given the superior binding affinity of CYP72A63 (L149I/T338S/L398V) for 11-oxo-β-amyrin and its intermediate 29-OH-11-oxo-β-amyrin, as well as their enhanced oxidative susceptibility, this variant was selected for further engineering efforts. This was aimed at achieving the rare remote dual-site oxidation, producing 24-OH-liquiritic acid with minimal accumulation of precursors, thereby significantly streamlining the downstream purification process.
The substrate tunnel analysis showed that residues I149, V398, and L509 exerted the most significant influence on the microenvironment within the proximal active site, thereby dictating substrate orientation (Fig. S24). To distinguish the primary contributor among these residues, MM/GBSA binding free energy decomposition was performed for liquiritic acid. The analysis revealed that I149 exhibited substantially higher binding free energy compared to both V398 and L509 (Fig. 4b), identifying I149 as the principal factor limiting the binding of liquiritic acid.
Consequently, site 149 was targeted for refining microenvironmental accessibility. Threonine, aspartic acid, valine and glutamine can be chosen due to their lower hydrophobicity while minimizing significant alterations in the geometric shape of the active pocket, avoiding the influence on regioselectivity as much as possible. Since Asp and Gln exhibited inhibitory effects on enzymatic activity, threonine and valine were chosen as replacements for I149. Notably, the triple mutant L149V/T338S/L398V showcased remarkable proficiency in the generation of 24-OH-liquiritic acid through the selectively sequential oxidation of C-29 and C-24 of 11-oxo-β-amyrin, achieving a noteworthy titer of 36.04 ± 4.44 mg/L and an impressive selectivity ratio of 94.83% (Figs. 4c and d).
In mutant CYP72A63 (L149V/T338S/L398V), the positioning of C-24 of liquiritic acid in close proximity to FeⅣ=O, maintained a mean distance of 3.38 Å over 50 ns of MD simulations (Fig. 4e and Fig. S35 in Supporting information), laid a robust foundation for the targeted synthesis of 24-OH-liquiritic acid. The MM/GBSA binding free energy decomposition showed that the key residues, V149, V398 and L509, all displayed low binding free energy for liquiritic acid. Total binding free energy analysis revealed that, in comparison with both CYP72A63 (L149T/L398V) and CYP72A63 (L149I/T338S/L398V), this mutant exhibited a lower binding free energy for the initial substrate 11-oxo-β-amyrin and the two intermediates, namely, 29-OH-11-oxo-β-amyrin and liquiritic acid (Table 3). This indicates a higher affinity of these substrates for oxidation. Moreover, the mutant displayed a more analogous binding free energy between 29-OH-11-oxo-β-amyrin and liquiritic acid compared to CYP72A63 (L149T/L398V) and CYP72A63 (L149I/T338S/L398V), implying more balanced substrate affinity between them (Table 3). Consequently, the mutant L149V/T338S/L398V demonstrated specific and continuous oxidation of 11-oxo-β-amyrin on remote sites, leading to the specific production of 24-OH-liquiritic acid.
DownLoad:
CSV
| CYP72A63 mutant | ΔGbinding (kcal/mol) | ||
| Empty Cell | 11-oxo-β-amyrin | 29-OH-11-oxo-β-amyrin | Liquiritic acid |
| L149T/L398V | −35.91 ± 0.90 | −40.94 ± 0.035 | −38.59 ± 0.038 |
| L149I/T338S/L398V | −37.99 ± 0.035 | −44.60 ± 0.032 | −34.97 ± 0.031 |
| L149V/T338S/L398V | −38.23 ± 0.035 | −45.58 ± 0.037 | −43.99 ± 0.033 |
However, CYP72A63 (L149T/T338S/L398V) exhibited a marked reduction in its capacity for continuous oxidation, failing to produce 24-OH-liquiritic acid while accumulating substantial amounts of 29-OH-11-oxo-β-amyrin (Fig. S36 in Supporting information). Compared to CYP72A63 (L149V/T338S/L398V) and CYP72A63 (L149T/L398V), this mutant significantly enlarged the active pocket (Fig. S37 in Supporting information) and weakened the hydrophobic interactions between the enzyme and the triterpenoid scaffold due to the incorporation of smaller, hydrophilic residues. These changes likely disrupted substrate orientation. Consequently, the distance between C-29 of 29-OH-11-oxo-β-amyrin and FeⅣ=O displayed significant fluctuations and was longer than in CYP72A63 (L149V/T338S/L398V) (Fig. 4f). Similarly, the C-24 of liquiritic acid was positioned far from FeⅣ=O (Fig. 4e). These structural alterations impaired oxidation at 29-OH-11-oxo-β-amyrin and abolished catalytic activity toward 24-OH-liquiritic acid formation.
Conversely, introducing bulkier residues (F, Y, H, R, W, K, E, N) at position 149 in the CYP72A63 (L149I/T338S/L398V) background led to enzyme inactivation, underscoring the critical role of proper steric hindrance at this site in maintaining activity. Furthermore, despite having similar steric hindrance to leucine, the introduction of Q and D, which significantly reduce hydrophobicity, also led to enzyme inactivation. These findings highlight that both the geometry and the hydrophobic microenvironment at position 149 are essential for sustaining the enzymatic activity and determining the regioselectivity.
These findings underscore the critical importance of optimizing microenvironmental accessibility, not only in modulating catalytic selectivity but also as a fundamental basis for the development of novel oxidation reactions. By analyzing and identifying the key factors affecting microenvironmental accessibility, we can effectively streamline the scope of the mutagenesis library.
In summary, by creating the microenvironmental accessibility for substrates within a plant P450 enzyme, we have achieved precise control over oxidation selectivity and enhanced catalytic activity though a minor mutagenesis library, enabling the specific production of rare licorice triterpenoids. Notably, this work introduces a novel oxidation model characterized by remote dual-site oxidation mediated by a single enzyme. The implications of this research extend to establishing a robust framework for modulating P450 catalytic properties, highlighting the potential of heterologous biosynthesis for generating natural products and related compounds within a broader chemical space.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Wentao Sun: Validation, Investigation, Methodology, Funding acquisition. Lin Xiang: Validation, Investigation, Data curation. Runming Wang: Investigation, Formal analysis. Meilan Huang: Visualization, Software, Formal analysis. Bo Lv: Methodology, Conceptualization. Youcai Hu: Visualization, Formal analysis, Data curation. Chun Li: Visualization, Supervision, Methodology, Funding acquisition, Formal analysis, Data curation, Conceptualization.
This project was supported by grants from the National Natural Science Foundation of China (Nos. 22108154, 22138006, 32171430).
Supplementary material associated with this article can be found, in the online version, at doi:
L.J. Kim, M. Ohashi, Z. Zhang, et al., Nat. Chem. Biol. 17 (2021) 872–877. doi: 10.1038/s41589-021-00834-2
R. Fasan, ACS Catal. 2 (2012) 647–666. doi: 10.1021/cs300001x
R. Pompei, O. Flore, M.A. Marccialis, et al., Nature 281 (1979) 689. doi: 10.1038/281689a0
Y. Kiso, O. Kato, H. Hikino, Plant. Med. 51 (1985) 50–52. doi: 10.1055/s-2007-969391
H.Y. Park, S.H. Park, H.K. Yoon, et al., Arch. Pharm. Res. 27 (2004) 57. doi: 10.1007/BF02980047
H. Okamoto, D. Yoshida, Y. Saito, et al., Cancer Lett. 21 (1983) 29–35. doi: 10.1016/0304-3835(83)90079-4
H.Y. Lee, D.Y. Jung, H.K. Ha, et al., BMB Rep. 40 (2007) 979–985. doi: 10.5483/BMBRep.2007.40.6.979
C. Joshi, A. Chaudhari, C. Joshi, et al., J. Biomol. Struct. Dyn. 40 (2022) 8405–8419. doi: 10.1080/07391102.2021.1922095
W. Sun, H. Xue, H. Liu, et al., ACS Catal. 10 (2020) 4253–4260. doi: 10.1021/acscatal.0c00128
M. Zhu, C. Wang, W. Sun, et al., Metab. Eng. 45 (2018) 43–50. doi: 10.1016/j.ymben.2017.11.009
T. Tanaka, T. Koyano, T. Kowithayakorn, et al., J. Nat. Prod. 64 (2001) 1243–1245. doi: 10.1021/np010196w
K. Tamura, Y. Teranishi, S. Ueda, et al., Plant Cell. Physiol. 58 (2017) 874–884. doi: 10.1093/pcp/pcx043
S. Ghosh, Front. Plant Sci. 8 (2017) 1886. doi: 10.3389/fpls.2017.01886
C.C. Hansen, D.R. Nelson, B.L. Moller, et al., Mol. Plant 14 (2021) 1244–1265. doi: 10.1016/j.molp.2021.06.028
E. Biazzi, M. Carelli, A. Tava, et al., Mol. Plant 8 (2015) 1493–1506. doi: 10.1016/j.molp.2015.06.003
Y. Peng, Y. Wang, T.J. Chen, et al., Chin. Chem. Lett. 35 (2024) 108818. doi: 10.1016/j.cclet.2023.108818
M. Baek, F. DiMaio, I. Anishchenko, et al., Science 373 (2021) 871–876. doi: 10.1126/science.abj8754
W. Sun, S. Wan, C. Liu, et al., Acta. Pharm. Sin. B 14 (2024) 4134–4148. doi: 10.1016/j.apsb.2024.04.032
M.Z. Fanani, E.O. Fukushima, S. Sawai, et al., Front. Plant Sci. 10 (2019) 1520. doi: 10.3389/fpls.2019.01520

Figure 1 Continuous oxidation of 11-oxo-β-amyrin for controllable synthesis of liquiritic acid. (a) The influence of the substrate tunnel by introducing mutant T338S, the line in the enzyme indicating the central line of the substrate tunnel, CYP72A63 (L398I) (cyan) and CYP72A63 (T338S/L398I) (tan). (b) The orientation of 29-OH-11-oxo-β-amyrin in the active pocket of CYP72A63 (T338S/L398I). (c) The distance between C-29 and FeIV=O moiety of CYP72A63 (T338S/L398I) during 50 ns molecular dynamics (MD) stimulation. (d) Total ion chromatogram (TIC) of the product spectra from yeast strains expressing CYP72A63 (L398I) and CYP72A63 (T338S/L398V), analyzed by GC–MS, (1) 29-OH-11-oxo-β-amyrin, (2) liqiuritic acid. (e) Triterpenoid production of yeast strains expressing different CYP72A63 mutants. Data are presented as mean ± standard deviation (SD) (n = 3 biologically independent samples). (f) The impact of introducing the T338S and L398V mutations on the substrate tunnel was demonstrated, including the visualization of the central line of the tunnel. CYP72A63 (L398I) is shown in cyan, and CYP72A63 (T338S/L398V) in tan. (g) The 338th, 398th and 149th residue had significant influence of substrate binding.

Figure 2 Restoring the enzyme’s activity for liquiritic acid production. (a) TIC of the product spectra from yeast strains expressing different mutants, analyzed by GC–MS. (1) 29-OH-11-oxo-β-amyrin, (2) liqiuritic acid, (3) liqiuritic aldehyde. (b) Triterpenoid production of engineered stains harboring different CYP72A63 mutants. Data are presented as mean ± SD (n = 3 biologically independent samples). (c) The radius of the substrate tunnel in mutant CYP72A63 (T338S/L398V) and CYP72A63 (L149T/L398V). (d) The binding process analysis of 11-oxo-β-amyrin in mutant CYP72A63 (T338S/L398V) and CYP72A63 (L149T/L398V). (e) The center line of the substate tunnel of mutant CYP72A63 (T338S/L398V) (blue) and CYP72A63 (L149T/L398V) (orange). (f) The docking results of mutant CYP72A63 (L149T/L398V) with 29-OH-11-oxo-β-amyrin. (g) The hydrophobicity in the active pocket of the mutants CYP72A63 (T338S/L398V) and CYP72A63 (L149T/L398V).

Figure 3 Enhancing the enzyme’s activity for liquiritic acid production. (a). The center line of the substate tunnel of mutant CYP72A63 (L398I) (green) and CYP72A63 (L149I/L398V) (tan) (b) The docking result of mutant CYP72A63 (L149I/L398V) (left) and CYP72A63 (L398I) (right) with 29-OH-11-oxo-β-amyrin. (c) The substrate binding energy (calculated in Autodock Vina) of 11-oxo-β-amyrin (E1) and 29-OH-11-oxo-β-amyrin (E2) near the entrance of the substrate tunnel of CYP72A63 (L149I/L398V). (d) The microenvironment of C-29-OH in the active pocket of CYP72A63 (L149I/L398V). (e) The substrate binding energy (calculated in Autodock Vina) of 11-oxo-β-amyrin (E1) and 29-OH-11-oxo-β-amyrin (E2) near the entrance of the substrate tunnel of CYP72A63 (L149I/T338S/L398V). (f) The production of liquiritic acid of engineered strains harboring CYP72A63 (L149I/T338S/L398V) and CYP72A63 (L149T/L398V). Data are presented as mean ± SD (n = 3 biologically independent samples). (g) TIC of the product spectra from yeast strains expressing CYP72A63 (L149I/T338S/L398V) and CYP72A63 (L149I/L398V) analyzed by GC–MS. (1) 29-OH-11-oxo-β-amyrin, (2) liqiuritic acid, (3) liqiuritic aldehyde, (4) 24-OH-liqiuritic acid.

Figure 4 Remote dual-site oxidation of 11-oxo-β-amyrin to produce 24-OH-liquiritic acid. (a) The C-24 of β-amyrin was orientated to FeⅣ=O for oxidation in CYP93E1. (b) MM/GBSA binding free energy decomposition for the key residues, I149, V398 and L509. (c) The specific remote dual-site oxidation on triterpenoids. (d) TIC of the product spectra from yeast strains expressing CYP72A63 (L149V/T338S/L398V) (violet) and CYP72A63 (L149T/L398V) (orange) analyzed by GC–MS, (2) liqiuritic acid, (4) 24-OH-liqiuritic acid. (e) The distance between C-24 of liquiritic acid and FeⅣ=O in mutants CYP72A63 (L149V/T338S/L398V) and CYP72A63 (L149T/T338S/L398V) during a 50 ns MD simulation. (f) The distance between C-29 of 29-OH-11-oxo-β-amyrin and FeⅣ=O in mutants CYP72A63 (L149V/T338S/L398V) and CYP72A63 (L149T/T338S/L398V) during a 50 ns MD simulation.
Table 1. The binding free energy of 29-OH-11-oxo-β-amyrin calculated by MM/GBSA.
| CYP72A63 mutant | ΔGbinding (kcal/mol) |
| T338S/L398V | −38.27 ± 0.029 |
| L149T/L398V | −40.94 ± 0.035 |
 下载: 导出CSV
下载: 导出CSV
Table 2. The binding free energy of 11-oxo-β-amyrin and 29-OH-11-oxo-β-amyrin calculated by MM/GBSA.
| CYP72A63 mutant | ΔGbinding (kcal/mol) | |
| Empty Cell | 11-oxo-β-amyrin | 29-OH-11-oxo-β-amyrin |
| L149I/T338S/L398V | −37.99 ± 0.035 | −44.60 ± 0.032 |
| L149T/L398V | −35.91 ± 0.90 | −40.94 ± 0.035 |
| L149I/L398V | −36.72 ± 0.033 | −42.83 ± 0.033 |
下载: 导出CSV
Table 3. The binding free energy of 11-oxo-β-amyrin, 29-OH-11-oxo-β-amyrin and liquiritic acid calculated by MM/GBSA.
| CYP72A63 mutant | ΔGbinding (kcal/mol) | ||
| Empty Cell | 11-oxo-β-amyrin | 29-OH-11-oxo-β-amyrin | Liquiritic acid |
| L149T/L398V | −35.91 ± 0.90 | −40.94 ± 0.035 | −38.59 ± 0.038 |
| L149I/T338S/L398V | −37.99 ± 0.035 | −44.60 ± 0.032 | −34.97 ± 0.031 |
| L149V/T338S/L398V | −38.23 ± 0.035 | −45.58 ± 0.037 | −43.99 ± 0.033 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
