State Key Laboratory of Bioactive Substance and Function of Natural Medicines Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100050, China
b.
Department of Dermatology, Vagelos College of Physicians and Surgeons, Columbia University Irving Medical Center, New York 10032, United States
ttzhang@imm.ac.cn (T. Zhang). 1 These authors contributed equally to this work.
Received Date:
02 December 2024 Accepted Date:
17 February 2025 Revised Date:
14 February 2025 Available Online:
15 December 2025
Abstract:
Interleukin-1 receptor-associated kinase 4 (IRAK4) is a key kinase downstream of the interleukin-1 receptor (IL-1R) and Toll-like receptors (TLRs) signaling pathway, whose overexpression and hyperactivation have been associated with several inflammatory diseases or cancer. Therefore, targeting IRAK4 has emerged as a promising therapeutic strategy. A range of potent and selective IRAK4 inhibitors and degraders based on draggability have been designed and developed. This article provides a comprehensive summary of the IRAK4 inhibitors and degraders that have been developed and discusses the challenges and opportunities for research in this area.
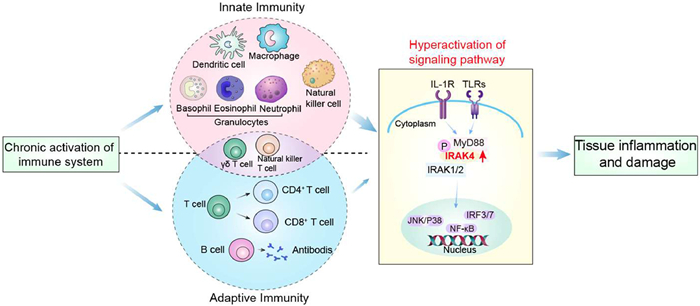
The immune response defends the body against pathogens, consisting of two branches: Innate and adaptive immunity [1,2]. The innate immunity quickly and non-specifically detects exogenous and endogenous pathogens via pattern recognition receptors (PRRs), triggering pro-inflammatory factors from various immune cells [3–5]. In contrast, adaptive immunity, which involves T and B cells, is slower and targets specific antigens [6,7].
Toll-like receptors (TLRs) are transmembrane PRRs essential for innate immunity, and interleukin-1 receptor (IL-1R), sharing similar structural domains, and functions in the same pathway [8]. Interleukin-1 receptor-associated kinase 4 (IRAK4), a key serine–threonine kinase in this pathway, is essential for activating the mitogen-activated protein kinases (MAPKs), nuclear factor-κ-gene binding (NF-κB), and interferon regulatory factor 3/7 (IRF3/7) pathways through its kinase activity and scaffold function [9]. However, the phosphorylation of IRAK4 can lead to hyperactivation of these signaling pathways, potentially resulting in development and progression of inflammatory diseases or cancers. This central role in the immune response makes IRAK4 becoming an important potential drug target in inflammatory and autoimmune diseases, as well as tumors (Fig. 1) [10].
Figure 1
Figure 1.
Chronic activation of the immune system involves a wide range of innate and adaptive immune cells. TLRs/IL-1R signaling pathway mediated the innate immunity. Overexpressed IRAK4 leads to excessive signaling pathway activation, potentially causing several diseases.
Advancements in IRAK4-based drug development have focused on small molecular inhibitors and degraders. Although prevalent, inhibitors that compete with ATP for the active site of IRAK4 do not affect its scaffolding function, which prompts the development of protein hydrolysis-targeted chimeras (PROTACs) as degraders. Up to now, although there are no drugs targeting IRAK4 on the market, IRAK4 inhibitors or PROTAC degraders currently in the clinic trials have shown promising outcomes [11].
To date, there are few review articles on the research & development of IRAK4 inhibitors or PROTAC degraders. However, systematic summary and discussion of the limitation and advantages of IRAK4 inhibitors or degraders in the design is helpful to promote the progression of research & development and avoid duplication problem. This review highlights advancements in the design and synthesis of inhibitors or PROTACs of IRAK4, and provides a comprehensive summary of the structures and activities of reported inhibitors and degraders from 2012 to the present. By analyzing structure-activity relationships (SAR) and discussing the challenges and prospects of this kind of IRAK4 agents, we aim to provide valuable insights and novel strategies for the future development & research of IRAK4 inhibitors or PROTACs.
2.
TLRs/IL-1R signaling pathway
The innate immune system provides defense by identifying exogenous and endogenous molecular patterns via PRRs, with TLRs being key transmembrane PRRs that have attracted significant biomedical interest [12]. TLRs activate signaling pathways by recognizing pathogen-associated molecular patterns (PAMPs), e.g., viruses, bacteria, to release inflammatory factors. Ten TLRs have been reported in humans [12–14]. The IL-1R shares a signaling pathway with TLRs due to similar structural domains [15,16] and activates downstream pathways by detecting damage-related molecular patterns (DAMPs), e.g., damaged cells [17].
Upon activation of TLRs/IL-1R by PAMPs and DAMPs, TIR domains recruit the adapter protein myeloid differentiation primary response protein 88 (MyD88) [18]. This leads to the recruitment of IRAK4, a crucial serine–threonine kinase [19], which then undergoes dimerizes, trans-autophosphorylation, and becomes activated [20].
Following IRAK4 activation, IRAK1 and IRAK2 are recruited to form the myddosome complex [20,21]. Within this complex, IRAK4 phosphorylates and activates IRAK1, which subsequently dissociates and interacts with the E3 ubiquitin-protein ligase TNF receptor-associated factor 6 (TRAF6). Activated TRAF6 then triggers two parallel pathways through the activation of the inhibitor of κB kinase (IKK) complex and MAPKs [22]. The IKK complex activates the downstream NF-κB pathway [23], and the MAPK activates the downstream c-Jun NH2-terminal kinase (JNK)/P38 pathway. Both cascades promote the release of inflammatory factors (IL-1, IL-6, tumor necrosis factor-α (TNF-α), etc.). These signaling cascades are collectively referred to as the MyD88-dependent signaling pathway [24–26]. In addition, TLR4 participates in a MyD88-independent signaling pathway through its scaffold function. In this pathway, the downstream junctional protein TIR-domain-containing adapter-inducing IFN-β (TRIF) of intracellular TLR3 can interacts with the downstream TRIF-associated connector molecule (TRAM) of TLR4, This interaction leads to the activation of the TLR4/IRAK4/NF-κB pathway through the scaffold function of IRAK4 (Fig. 2) [27–30].
Figure 2
Figure 2.
IL-1R/TLRs signaling pathway. Membrane IL-1R/TLRs activate the JNK/P38 and NF-κB pathways via MyD88-dependent signaling, while intracellular TLRs activate the IRF3/7 pathway via MyD88-independent signaling.
3.
IRAK4 and its role in IL-1R/TLRs signaling pathway
3.1
IRAKs family
The IRAK family belongs to the serine–threonine kinase family, which is crucial in the IL-1R/TLRs signaling pathway and includes IRAK1, IRAK2, IRAKM (IRAK3), and IRAK4 [31–33]. While IRAK1, IRAK2, and IRAK4 are broadly expressed in immune cells, IRAKM is characteristically expressed in macrophages and monocytes [34]. IRAK4 and IRAK1 have catalytic kinase activity due to aspartic acid residue [35]. Initially, IRAK2 was classified as a pseudokinase due to the substitution of a conserved aspartic acid residue with an asparagine residue. However, subsequent studies revealed that the presence of Lys213 within its kinase domain confers catalytic activity to IRAK2. In contrast, IRAKM is the sole member of the IRAK family that functions as a pseudo kinase [36]. All four kinases play key roles in the signaling pathway, including positive regulation of IRAK1, 2, and 4 and negative regulation of IRAKM [37].
3.2
The role and structure of IRAK4
IRAK4 is regarded as the "master kinase" within its family due to its pivotal role in cellular signaling processes [38]. Primarily, IRAK4 serves as the phosphorylation initiator in the IL-1R/TLRs signaling pathway, where its autophosphorylation triggers the activation of downstream proteins [39]. Additionally, IRAK4 facilitates the recruitment of other kinases to assemble the myddosome complex, thereby directly activating the downstream pathway through its scaffolding function. IRAK4's dual capacity as both a kinase and scaffold protein underscore its central role in cellular signaling transduction [28,40].
IRAK4 possesses a molecular weight of 52 kDa and is composed of 460 amino acids, featuring an N-terminal death domain (DD), a ProST domain, and a central kinase domain (KD). The structural characterization of IRAK4 was initially described by Kuglstatter et al. [41]. The active site of IRAK4 is located at the designated kinase folding site, which includes both the N-terminus and C-terminus. The hinge region of the kinase consists of Val263, Tyr264, and Met265 residues, which play a crucial role in the structure-based design of kinase inhibitors [43,44]. Tyr262, serving as a distinctive gatekeeper residue within the IRAK family, establishes a hydrogen bond with Glu233 located on helix α-C. This specific interaction maintains an active conformation that is critical for the structure-based design of selective inhibitors. Furthermore, the active site of the kinase contains a universally present catalytic Lys213, a highly conserved feature essential for kinase activity (Fig. 3) [42].
Figure 3
Figure 3.
(a) IRAKs family consists of 4 members and IRAK4 contains an N-terminal DD, a ProST domain, and a central KD (PDB ID: 2NRU). (b) The first structure of IRAK4, in the active site of IRAK4, Val263, Tyr264, and Met265 form the hinge region of the kinase, Tyr262 is a unique gatekeeper residue, and there is a ubiquitous catalytic Lys213 in the active site of the kinase (PDB ID: 2NRU). Visualized in PyMOL.
Recent studies have indicated that targeting IRAK4 inhibition is relatively safe. The mutations identified to date are primarily associated with reduced IRAK4 levels, leading to recurrent infections during childhood. However, these infections tend to diminish with age and are not typically observed after the age of 14 [45,46].
However, aberrant activation of IRAK4 plays a significant role in the pathogenesis of diseases. Dysregulation in the IRAK4 and IL-1R/TLRs signaling pathways facilitates the accumulation of various inflammatory cells and the infiltration of inflammatory factors into infected and damaged tissues, culminating in the onset of autoinflammatory/immune diseases [43,47]. Furthermore, these signaling pathway abnormalities are intricately associated with tumorigenesis, characterized by substantial infiltration of inflammatory factors within tumor tissues [48].
4.1
Autoinflammatory/immunity diseases
Recent studies have implicated TLRs in the pathogenesis of psoriasis. The topical application of imiquimod, a TLR-7 agonist, has been shown to induce psoriasis-like dermatological manifestations [49,50]. Furthermore, significant mitigation of the disease has been observed with the use of IRAK4 inhibitors [51]. Rheumatoid arthritis (RA), an autoimmune disease, has been reported with the involvement of the TLRs-IRAK4-NF-κB signaling pathway in its pathogenesis [52]. In collagen-induced arthritis (CIA) mouse models, targeted inhibition of IRAK4 has been demonstrated to attenuate disease symptoms [53]. Targeting or inhibiting IRAK4 presents a potential therapeutic strategy for chronic arthritis. In systemic lupus erythematosus (SLE), a systemic autoimmune disease [54], elevated TLR2, 7, and 9 have been observed in patients' peripheral blood mononuclear cells (PBMCs) [55,56]. Murphy et al. were the first to demonstrate that aberrant activation of IRAK4 contributes to the onset and progression of lupus in a genetically susceptible mouse model [57]. The significant efficacy of BMS-986, 126 underscores the potential of targeted IRAK4 inhibition as an alternative treatment strategy for lupus [58]. In addition, IRAK4 is involved in the pathogenesis of diseases such as inflammatory bowel disease and multiple sclerosis.
4.2
Malignant tumors
Activated B-cell-like diffuse large B-cell lymphoma (ABC-DLBCL) represents a category of malignant tumors characterized by clinically aggressive and complex phenotypes. Research indicates that the L265P mutation of MyD88 is present in approximately 30% of patients with ABC-DLBCL. IRAK4, a critical downstream kinase of MyD88, is integral to the majority of its biological functions and thus presents a potential alternative therapeutic target [59]. Furthermore, in chronic lymphocytic leukemia (CLL), where MyD88 mutations occur in approximately 3% of cases, disease remission has been achieved through the use of IRAK4 inhibitors, such as ND-2110 and ND-2158 [60].
Myelodysplastic syndromes (MDS) are a heterogeneous group of myeloid clonal disorders originating from hematopoietic stem cells (HSCs) and are characterized by ineffective hematopoiesis. High-risk MDS has the potential to progress into acute myeloid leukemia (AML) [61,62]. Evidence indicates that TLRs are expressed on HSCs and that dysregulated TLRs signaling is implicated in the pathogenesis of MDS. The administration of IRAK4 inhibitors, such as R289, has been shown to induce disease remission [20,63].
Beyond its role in hematological malignancies, IRAK4 is also implicated in the pathogenesis of solid tumors [43]. Specifically, the IRAK4-mediated NF-κB signaling pathway influences stromal fibrosis and cell viability, among other factors, in pancreatic ductal adenocarcinoma (PDAC) [64]. The application of an IRAK4 inhibitor, such as CA-4948, has been shown to suppress the activation of the NF-κB signaling pathway in tumor cells, thereby reducing inflammatory signaling and mitigating disease symptoms.
5.
The mechanism of IRAK4 inhibitors and degraders
Initially, IRAK4 serves as the phosphorylation initiation point for the IL-1R/TLRs signaling pathway and regulates the subsequent phosphorylation cascade [32]. Within the kinase's active site, specific residues are present: Met265, Tyr264, and Val263 constitute the hinge region of the active pocket; Lys213 functions as the catalytic residue; and the distinctive gatekeeper residue, Tyr262, can form a hydrogen-bonding interaction with Glu233 on the α-C helix, thereby maintaining IRAK4's active conformation [41,44]. The presence of residues in the active site is instrumental in facilitating the design of small-molecular inhibitors targeting IRAK4 [44].
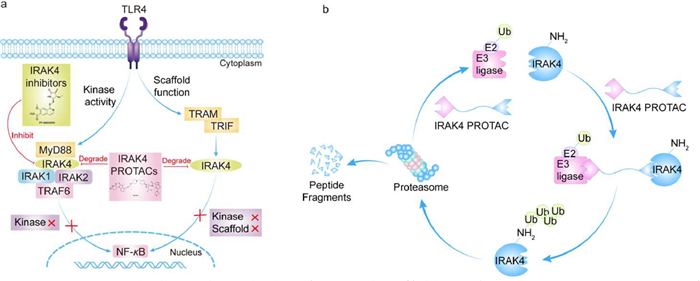
Furthermore, research has identified that IRAK4 serves a scaffolding role within the IL-1R/TLRs signaling pathway. While small molecule inhibitors influence the phosphorylation process, they do not impede the formation of the myddosome complex, which continues to activate the TLR4-NF-κB signaling pathway [28]. PROTACs have been developed to achieve comprehensive inhibition of the IL-1R/TLRs pathway. These PROTACs are a class of degraders characterized as heterodimeric bifunctional molecules, comprising three components: an E3 ubiquitin ligase-binding moiety, a linker, and a protein of interest (POI)-binding moiety. PROTACs are capable of forming a triadic complex with the E3 ubiquitin ligase, the POI, and target proteins, which subsequently undergo ubiquitination and degradation [65]. Therefore, the degradation of IRAK4 effectively inhibits both its kinase activity and scaffolding functions, resulting in a potent immunosuppressive effect. This dual inhibition leads to the comprehensive suppression of the IL-1R/TLRs signaling pathway, thereby demonstrating significant anti-inflammatory and anti-tumor activities (Fig. 4) [66].
Figure 4
Figure 4.
(a) IRAK4 dual capacity as both a kinase and scaffold protein. (b) IRAK4 PROTACs can form a ternary complex between the E3 ubiquitin ligase and the target protein, followed by ubiquitination tagging of the protein and eventual degradation by the proteasome system, achieving complete inhibition of the signaling pathway.
In recent years, a diverse array of drugs targeting IRAK4 has been reported [67]. Based on their structural features and mechanisms of action, these drugs can be primarily categorized into two distinct groups: small molecular inhibitors and PROTACs (Table S1 in Supporting information).
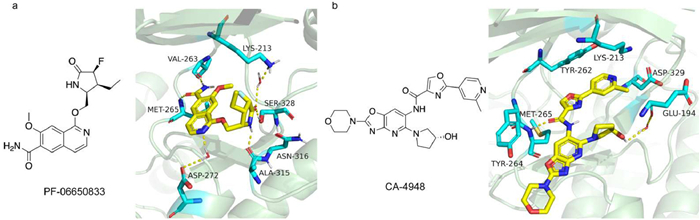
Among the small molecular inhibitors targeting autoinflammatory and immune diseases, Pfizer's PF-06650833 (Zimlovisertib) was the first to reach the clinical stage, exhibiting a half maximal inhibitory concentration (IC50) of 2.4 nmol/L for IRAK4 inhibition. Fig. 5a illustrates the co-crystalline structure of PF-06650833. Katherine et al. concluded that the incorporation of the lactam moiety, the introduction of a fluorine atom, and the presence of high-energy water significantly enhanced the activity of PF-06650833. Furthermore, the conformation of the compound was found to be highly consistent with that of the enzyme in its bound state [68]. PF-06650833 is currently in phase 2 for the treatment of rheumatoid arthritis [69], and has also been investigated in the clinic for the treatment of coronavirus disease 2019 (COVID-19) [70]. However, its phase 2 study in hidradenitis suppurativa (HS) failure led to its suspension from the IRAK4 pipeline. Gilead's GS-5718 (Edecesertib) is in phase 2 for the treatment of RA and cutaneous lupus erythematosus (CLE) [71,72]. Bayer's BAY-1834845 (Zabedosertib) and BAY-1830839 are primarily for the treatment of skin inflammatory diseases [51], and BAY-1834845 is in phase 2 for the treatment of atopic dermatitis (AD). EVO-101 is in phase 2 trials as a cream formulation for the treatment of AD and eczema [73]. BMS-986126 is the first study to explore IRAK4 inhibitor in a lupus erythematosus mouse model. However, its activation of the TRIF-dependent pathway in human PBMCs was found to potentially result in no inhibitory effect [58]. AS2444697 is currently in phase 1 for the treatment of chronic kidney disease (CKD) [74].
Figure 5
Figure 5.
(a) X-ray structure of PF-06650833 with IRAK4 (PDB: 5UIU. (b) X-ray structure of CA-4948 with IRAK4 (PDB: 7C2V). Visualized in PyMOL.
CA-4948 is the pioneering small molecule inhibitor targeting malignant tumors, with Fig. 5b illustrating its co-crystalline structure. CA-4948 does not involve interactions between Lys213 and Tyr264; instead, it forms a hydrogen bond with Glu329 mediated by water, and its terminal pyridine interacts with Asp329 [75]. CA-4948 is currently in phase 2, showing significant efficacy in the treatment of MDS/AML and synergistic effects in combination with Bruton's tyrosine kinase (BTK) inhibitors [76]. However, as a dual IRAK4/FLT3 inhibitor, the safety of CA-4948 may need careful consideration. ND-2158 and ND-2110 are mainly used for the treatment of MyD88-mutated ABC-DLBCL and CLL. However, their associated depletion of effector CD8+ T cells leads to moderate negative feedback on the disease, suggesting potential benefits from combining them with drugs that enhance T-cell function [53,60,77]. R835 [78,79] and R829 are Rigel's IRAK1/4 dual inhibitors, and R289 is a prodrug of R835 for the treatment of inflammatory diseases and MDS [80].
Significant advancements have been achieved in the development of PROTACs. Notably, KT-474 represents the first landmark IRAK4-targeting PROTAC, currently undergoing Phase 2 for the treatment of AD, HS, and RA, and it has demonstrated a favorable safety profile [66,81–83]. Additionally, LT-002–158 has emerged as the world's second PROTAC aimed at addressing inflammatory diseases [84]. Furthermore, KT-413 is presently in Phase 1 for the treatment of ABC-DLBCL [85].
7.
The recent development of IRAK4 inhibitors and PROTACs
The advancement of pharmacological agents targeting IRAK4 has progressed significantly, garnering attention from various countries and organizations regarding the therapeutic potential of IRAK4 as a drug target. While no drugs are currently available on the market, several candidates have exhibited substantial efficacy in relevant diseases, underscoring IRAK4's promise as a therapeutic target. This review aims to present a categorized summary of the inhibitors and degraders reported since 2012.
7.1
IRAK4 small molecular inhibitors
7.1.1
IRAK4 inhibitors reported by Astrazeneca
In 2017, pyrrolopyrimidine-based IRAK4 inhibitors were introduced. Compound 1 effectively inhibited the NF-κB pathway and ABC-DLBCL cells in a dose-dependent manner and showed synergistic effects with a BTK inhibitor in an ABC-DLBCL mouse model. In SAR, the binding conformation of compound 1 with IRAK4 closely matched the crystal structure, and adding morpholine improved its permeability. The morpholine's interaction with Met129 and Asp272, forming a strong interaction, is crucial for enhanced activity [86].
However, due to the poor permeability and high efflux of compound 1 observed in vivo, pyrrolizotriazine IRAK4 inhibitor 2 was developed with enhanced permeability and reduced efflux by excluding the formal HBD, making the structure more lipophilic [87].
A batch of quinazoline inhibitors were developed by high-throughput screening, which aimed at enhancing the permeability and bioavailability of lead compounds. The compound 3, featuring a morpholine moiety at the C4 position, exhibited improved physicochemical properties. This compound demonstrated a high bioavailability of 73%, a low plasma clearance rate (CL) of 22 mL min−1 kg−1, and an acceptable half-life of 1.3 h in rat models. Furthermore, compound 3, when administered orally, showed a dose-dependent inhibition of IL-6 in a TLR-7-driven inflammation model [88].
Using the quinazoline nucleus as a foundation, a series of IRAK4 inhibitors based on a 5-aza quinazoline framework were developed, as illustrated in Fig. 6. Compound 5 emerged as a potent and selective IRAK4 inhibitor with a moderate level of inhibition on IRAK1 activity. Importantly, when combined with ibrutinib, it demonstrated significant effects in an in vivo xenograft model [89]. To improve the in vitro human clearance of compound 5, compound 6 was developed, which exhibited low intrinsic clearance in human liver microsomes (HLM) and human hepatocytes (HH). However, the bioavailability of compound 6 in rodents was lower than expected [90].
Figure 6
Figure 6.
IRAK4 inhibitors by AstraZeneca. IRAK4 enz IC50 and IRAK4 cell IC50 were obtained by fluorescence resonance energy transfer (FRET) assay and flow cytometry, respectively.
In addition, a class of benzopyrrole IRAK4 inhibitors were disclosed, such as compounds 7 and 8, which inhibited IRAK4 with IC50 of 0.1 nmol/L and less than 0.03 nmol/L, respectively, with significant inhibition on kinase activity (Fig. 6) [91].
7.1.2
IRAK4 inhibitors reported by Aurigene Discovery Technologies Limited
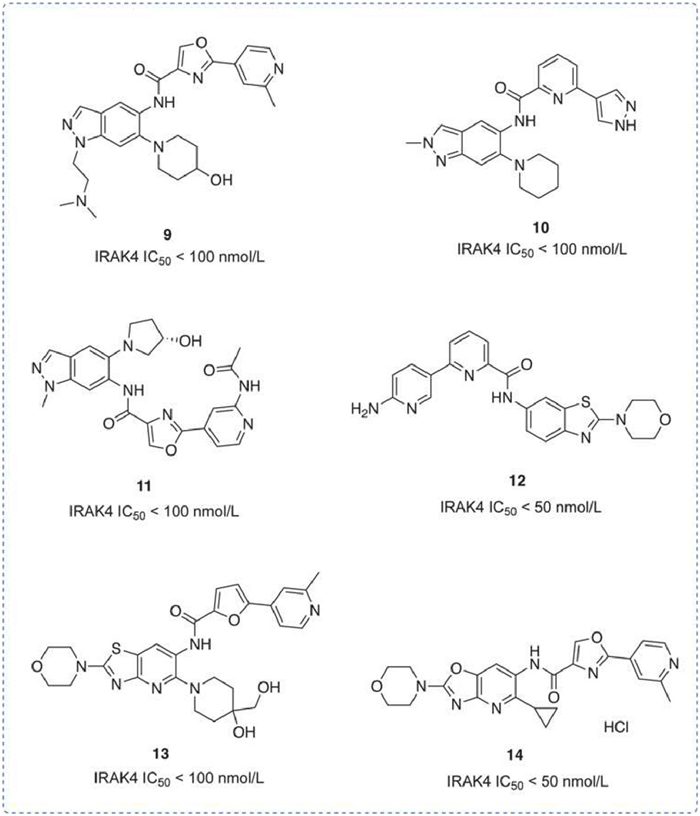
In recent years, the IRAK4 inhibitors developed by Aurigene have primarily utilized the parent ring of indazole, benzothiazole, and benzoxazole. Compounds 9 [92], 10 [93], and 11 [94] all featuring indazole as the parent ring, differ by substitutions at the 1-position, 5-position, and 6-position of the indazole moiety, respectively, with each showing an IC50 for IRAK4 inhibition less than 100 nmol/L. No additional data have been presented. Compounds 12 [95] and 13 [96] that utilize benzothiazole as the parent ring and morpholine moiety attached at position 2 maintained IRAK4 inhibitory activity (IC50 < 100 nmol/L). In the lipopolysaccharide (LPS)-induced inflammation model, compound 12 reduced TNF-α by 53% at a dose of 30 mg/kg. Compound 14 [97], a hydrochloride salt designed to enhance solubility, inhibits IRAK4 with an IC50 of < 50 nmol/L (Fig. 7).
Figure 7
Figure 7.
IRAK4 inhibitors by Aurigene Discovery Technologies Limited. IRAK4 IC50 was obtained by time-resolved fluorescence resonance energy transfer (TR-FRET) assay.
7.1.3
IRAK4 inhibitors reported by Bristol-Myers Squibb (BMS)
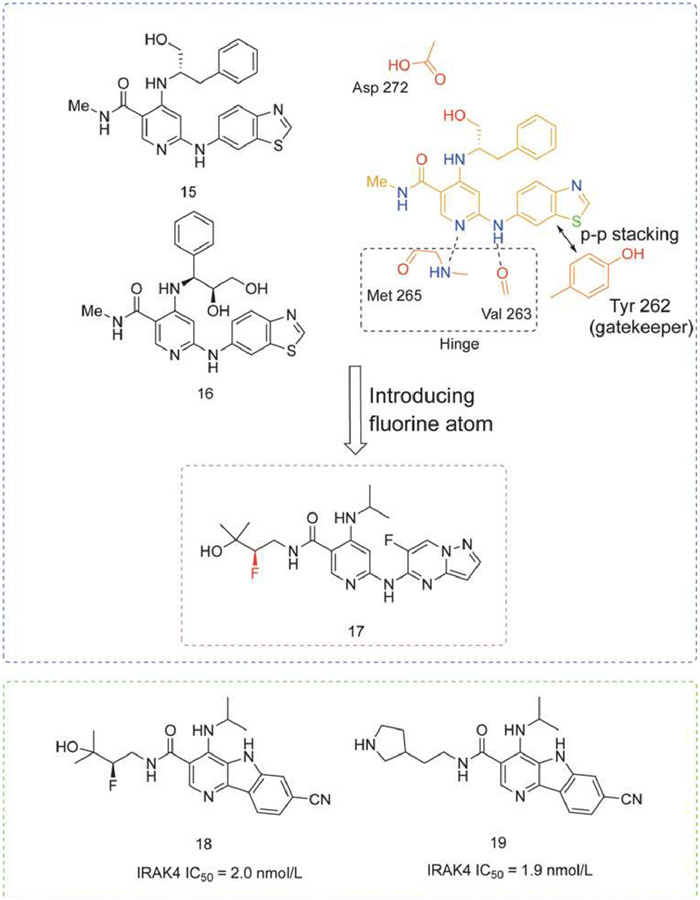
A series of highly potent and selective IRAK4 inhibitors derived from JAK3 inhibitors were reported, whose "flipped" binding mode to IRAK4 included a classical hinge region binding and an apparent π-π stacking interaction with a unique tyrosine gatekeeper. Example structures of compounds 15 and 16 are shown in Fig. 8 [98].
Figure 8
Figure 8.
IRAK4 inhibitors by Bristol-Myers Squibb. IRAK4 IC50 was obtained by TR-FRET assay.
However, due to the poor permeability of 15 and 16 to caco-2 cells, compound 17 was obtained by lengthening the amide side chain to occupy the front pocket and introducing a fluorine atom, which had a significant PD effect in an lipoteichoic acid (LTA)-induced acute inflammation model with inhibition of > 90% at a dose of 10 mg/kg. It showed a dose-dependent effect in a short-term model of psoriasis in mice [99].
In addition, BMS disclosed 82 novel tricyclic heteroaryl compounds as IRAK4 inhibitors in a patent aimed at treating inflammatory diseases, autoimmune diseases, and cancer. These compounds underwent IRAK4 kinase inhibition assays and these demonstrated significant inhibitory effects on IRAK4, exemplified as compounds 18 and 19 (Fig. 8) [100].
7.1.4
IRAK4 inhibitors reported by Bayer
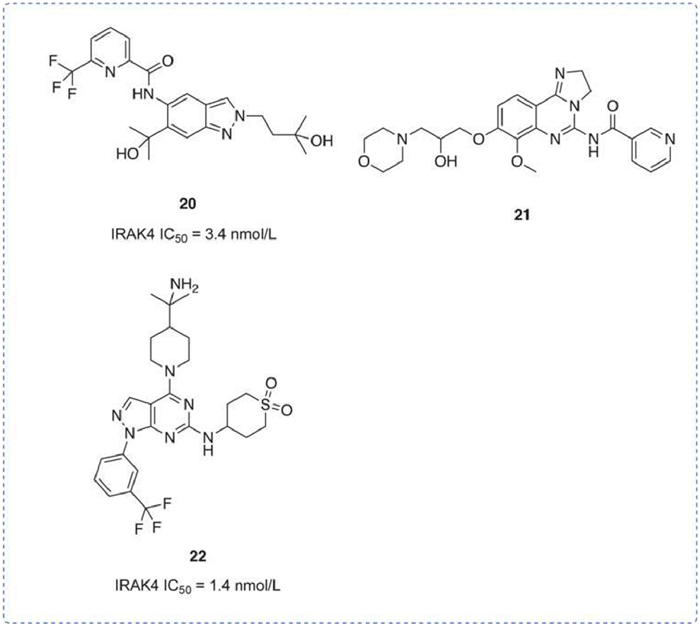
A series of indazole inhibitors, pyrimidinopyrazole inhibitors, and tricyclic thickened and analogous were disclosed. The patent WO2016174183 [101] disclosed 21 indazoles, and compound 20 had an IRAK4 IC50 of 3.4 nmol/L. In vitro, this compound also showed an IC50 for TNF-α of 0.2 µmol/L in THP-1 cells and less than 1 µmol/L in LPS-stimulated human PBMCs. In the Xenograft model, it was found that 20 in combination with ibrutinib, a BTK inhibitor, had a synergistic effect on tumor inhibition. Patent WO2017157792 [102] disclosed 103 tricyclic thickened and analogous compounds, the data showed that the combination of IRAK4 inhibitors and PI3 K inhibitors could synergistically inhibit tumor growth, e.g., compound 21. Patent WO2021064077 [103] disclosed 282 pyrimidine pyrazoles, such as compound 22, which had an IRAK4 IC50 of 1.4 nmol/L. In addition, compound 22 achieved a maximal bioavailability (Fmax) of 81% in humans, with a plasma CL of 0.25 L h−1 kg−1 (Fig. 9).
Figure 9
Figure 9.
IRAK4 inhibitors by Bayer. IRAK4 IC50 was obtained by TR-FRET assay.
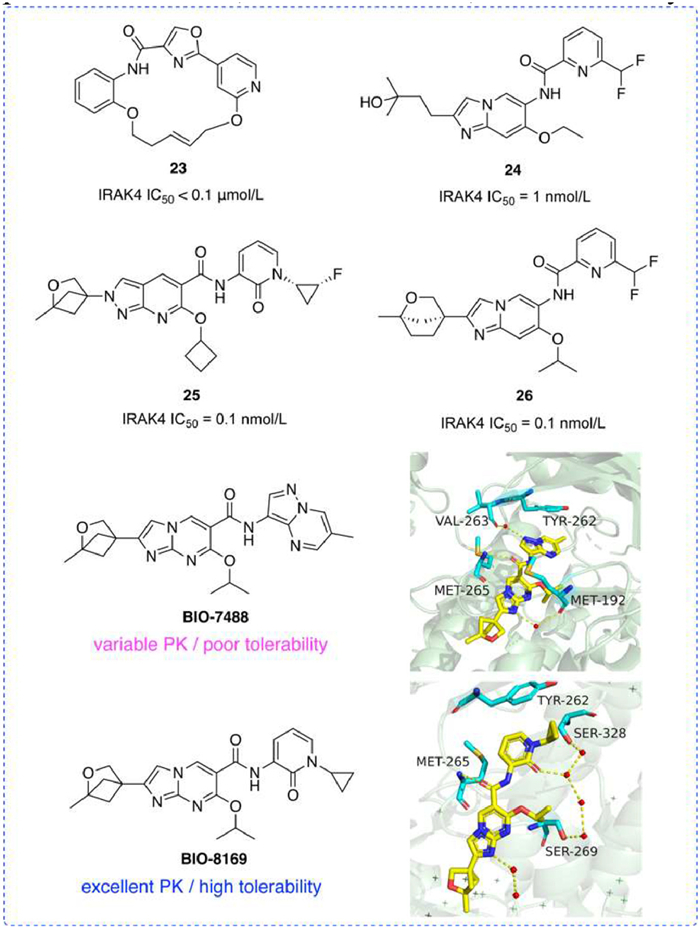
In 2014, Biogen disclosed a series of novel macrocyclic IRAK4 inhibitors that maintained inhibition of IRAK4, such as compound 23 [104]. Compounds 24 [105] and 26 [106], both based on an imidazolopyridine parent ring, demonstrated increased efficacy in inhibition. The C2 and C6 positions of 26 were connected to the bridging ring fragment and isopropoxide, respectively, which showed a 10-fold increase in inhibitory activity over 24. Additionally, compound 25 [107], another imidazopyridine IRAK4 inhibitor, exhibited an exceptionally low IC50 of 0.1 nmol/L.
Moreover, Biogen has reported on BIO-7488, a selective imidazopyrimidine IRAK4 inhibitor characterized by neuropermeability. The introduction of an oxygen atom to the parent ring and a bicyclic ether reduces oxidative metabolism. An amide bond enables BIO-7488 to exert a significant inhibitory effect on inflammation within the peripheral and central nervous systems, demonstrating excellent bioavailability and low clearance [108]. However, due to the low solubility, pyridone fragments were added to enhance solubility, leading to the development of the imidazopyrimidine analog BIO-8169, which is brain permeable, has excellent pharmacokinetics, is well-tolerated, and effectively alleviates EAE symptoms (Fig. 10) [109].
Figure 10
Figure 10.
IRAK4 inhibitors by Biogen. IRAK4 IC50 was obtained by TR-FRET assay.
7.1.6
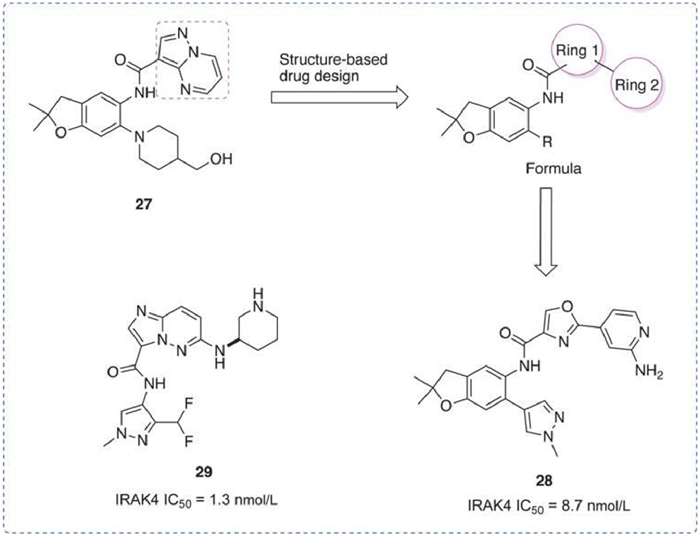
IRAK4 inhibitors reported by China Pharmaceutical University
A series of 2,3-dihydro benzofuran-based IRAK4 inhibitors were reported [110]. Benzofuran 27 was used as a lead to optimize binding with the gatekeeper Tyr262 of IRAK4. Compound 28, the most advanced compound in these compounds, showed potent IRAK4 inhibitory activity (IRAK4 IC50 = 8.7 nmol/L), good kinase selectivity, and high antiproliferative activity against MyD88 L265P DLBCL cell line (OCI-LY10 IC50 = 0.248 µmol/L). In combination with the BTK inhibitor ibrutinib, 28 exhibited enhanced apoptosis induction and antiproliferative capacity.
A series of imidazo[1,2-b]pyridazines potent IRAK4 inhibitors was also reported [111]. Compound 29 exhibited remarkable inhibitory activity (IC50 of 1.3 nmol/L for IRAK4) and preferable kinase selectivity. In addition, compound 29 in combination with the BTK inhibitor ibrutinib synergistically reduced the survival of TMD-8 cells. These results suggested that 29 may be a promising IRAK4 inhibitor for the treatment of mutant MyD88 DLBCL (Fig. 11).
Figure 11
Figure 11.
IRAK4 inhibitors by China Pharmaceutical University. IRAK4 IC50 was obtained by Z'-LYTE™ kinase assay.
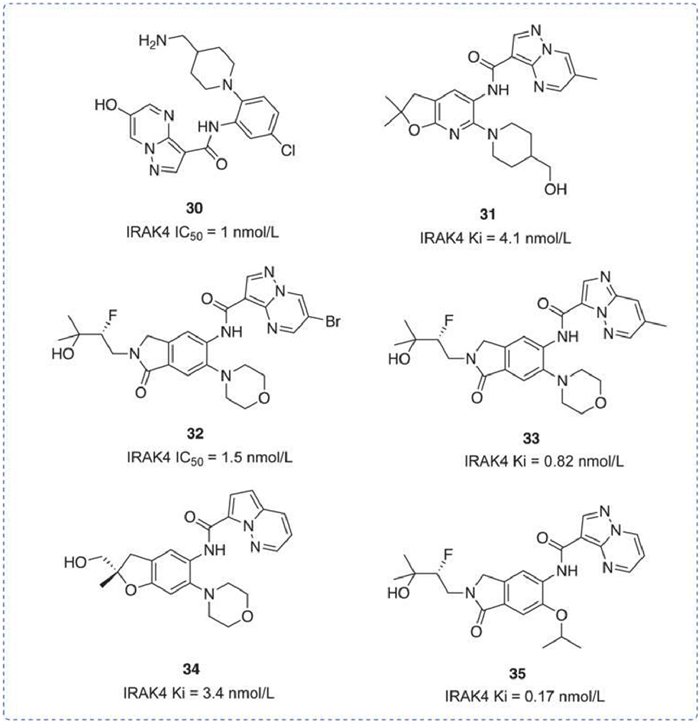
A series of pyrazolopyrimidines inhibitors were disclosed recently, including compound 30 under patent WO2012007375 [112], which features a terminal hydroxyl group and a terminal amino group. It exhibited an IRAK4 IC50 of 1 nmol/L, with no further data disclosed. Compound 31 under WO2018083085 [113] and compound 34 under WO2018234343 [114] are structurally similar and exhibit comparable inhibitory effects on IRAK4 (Ki is 4.1 nmol/L and 3.4 nmol/L respectively), despite having different substituents. No additional data was disclosed. Compounds 32 [115], 33 [116], and 35 [117], with the novel structural fragment cyclolactam, also showed no decrease in activity. No other data was disclosed (Fig. 12).
Figure 12
Figure 12.
IRAK4 inhibitors by Roche. IRAK4 IC50 was obtained by transcreener-fluorescence polarization (FP).
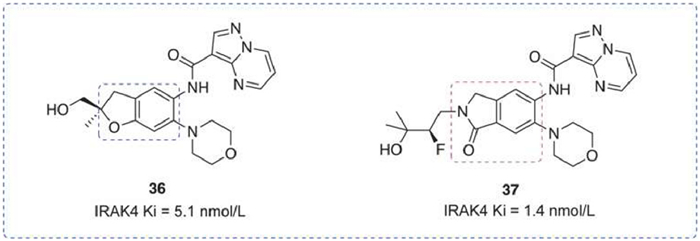
A series of dihydrobenzofuran analogs have been published. The compounds maintained excellent potency and selectivity while also exhibiting significant drug-like properties [118]. Compound 36 had low clearance and inhibited the production of IL-6 and TNF-α in an inflammation-mediated PK/PD mouse model. However, inhibition of multiple disease-associated cytokines was inconsistent. In response, Rajapaksa et al. identified a novel endocannabinoid IRAK4 inhibitor using the human whole blood assay [119]. Compound 37 not only maintained excellent biochemical potency and selectivity but also showed remarkable inhibition in human whole blood (HWB) assay (Fig. 13).
Figure 13
Figure 13.
IRAK4 inhibitors by Genentech. IRAK4 IC50 was obtained by Z'-LYTE™ kinase assay.
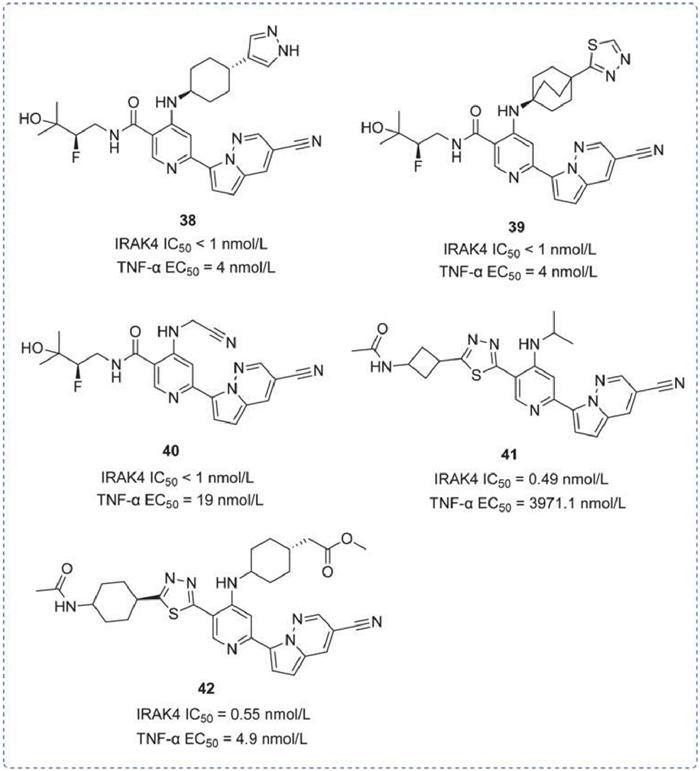
A series of pyridine-based IRAK4 inhibitors was reported, characterized by a pyridine ring with an attached pyrrolo[1,2-b]pyridazine. 75 pyridine-based compounds were disclosed in WO2020036979 [120], and compounds 38 and 39 had an IC50 for inhibition of IRAK4 of less than 1 nmol/L. In WO2020014468 [121], 10 pyridinyl compounds were disclosed, including compound 40, which demonstrated an IC50 for IRAK4 of less than 1 nmol/L and 19 nmol/L for TNF-α inhibition. The hepatic metabolic stability assay revealed an IC50 of 0.13 L h−1 kg−1 for compound 40. In addition, with an hERG IC50 greater than 10 µmol/L, compound 40 is likely safe for the heart. Under patent WO2020036986 [122], 343 IRAK4 inhibitors were disclosed, including compounds 41 and 42. These compounds are distinguished by the difference in the moiety attached at position 4, resulting in a significant variance in TNF-α inhibition, despite similar IRAK4 kinase inhibitory activity (0.49 and 0.55 nmol/L, respectively) (Fig. 14).
Figure 14
Figure 14.
IRAK4 inhibitors by Gilead. IRAK4 IC50 and TNF-α EC50 were obtained by TR-FRET immunoassay.
7.1.10
IRAK4 inhibitors reported by Merck Sharp & Dohme Corp
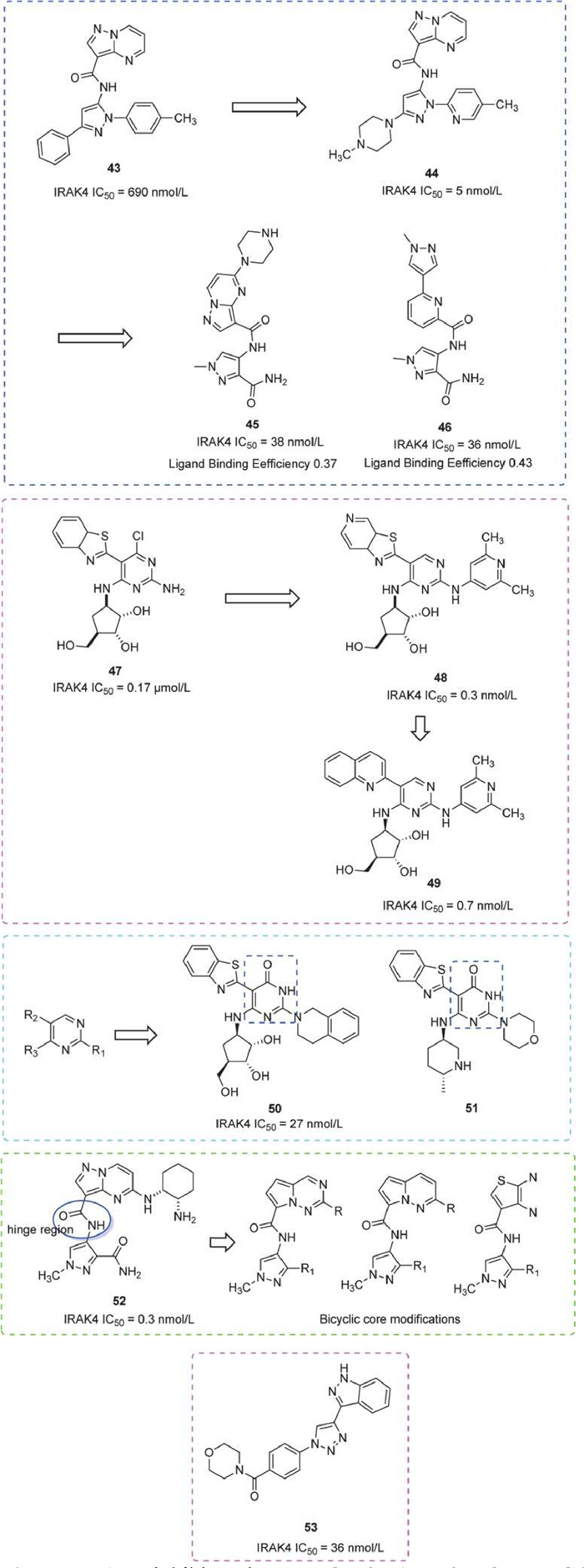
In 2015, a series of pyrazole inhibitors were reported. The lead compound was optimized by introducing pyridine at the N1 position to obtain Piperazine 44, a potent kinase inhibitor with high water solubility, cellular activity, and high selectivity for IRAK4. Additionally, Piperazine 44 reduced the production of pro-inflammatory cytokines and was orally effective in antibody-induced arthritic disease models [123]. Alternatively, replacing the pyrazolopyrimidine fragment with a pyridine bis-aromatic ring system, as seen in compound 46, can enhance cell permeability and reduce off-target effects 46, meanwhile maintaining comparable IRAK4 inhibitory activity to 45. Both compounds had desirable ligand efficiencies and acceptable off-target activities [124].
A range of novel diaminopyrimidine compounds were also identified. Compound 47, as a lead compound, exhibited prominent activity when the chlorine substituent was removed from the pyrimidine C-4 position. The C-6 carbon ribose was critical for IRAK4 inhibition. The introduction of the heteroaryl group at the C-5 position, notably azabenzothiazole resulted in enhanced activity. Amino heteroaryl groups were highly favored at the C-2 position. For instance, compound 48 exhibited nanomolar inhibition of IRAK4 (IRAK4 IC50 = 5 nmol/L) [125]. However, due to poor kinase selectivity, substituents at both the C-2 position and C-5 position of the pyrimidine nucleus were explored, to obtain compound 49, with an IC50 of 0.7 nmol/L for IRAK4 and 55 nmol/L for THP-1 cells stimulated by the TLR4 agonist LPS [126].
Further, a series of IRAK4 inhibitors by replacing the pyrimidine nucleus with a pyrimidinone nucleus were synthesized. Compound 50 demonstrated remarkable inhibitory activity (IC50 = 27 nmol/L) and excellent kinase selectivity, while 51 exhibited strong IRAK4 activity (IC50 = 93 nmol/L) and favorable bioavailability (F = 42%) in rats [127].
In addition, compound 52, as a lead compound, whose amide moiety forms a hydrogen bond with the hinge region of the IRAK4. A PK/PD response was observed in a dose-dependent manner in the R848-induced rat model. To address poor permeability and bioavailability, the investigators optimized the molecule by modifying substituent groups or scaffolds [128], and they also used the cLogD as an index to discover highly permeable IRAK4 inhibitors with superior activity and kinase selectivity [129].
Early in 2012, Merck Serono disclosed 299 imidazolyl triazole derivatives IRAK4 inhibitors, demonstrating kinase inhibition of IRAK4 and IRAK1 in this series. They also observed inhibition of IL-8 release by these compounds in THP-1 cells. In an in vivo mouse LPS-induced validation model, compound 53 achieved reductions of TNF-α and IL-6 by 97% and 83%, respectively (Fig. 15) [130].
Figure 15
Figure 15.
IRAK4 inhibitors by MERCK SHARP & DOHME CORP and Merck Serono. IRAK4 IC50 was obtained by FP assay.
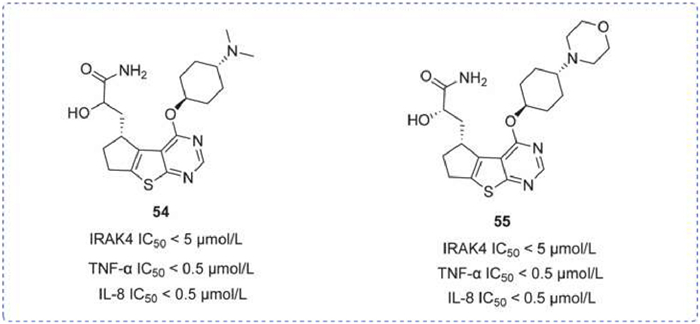
A novel series of thiazolopyrimidine-based IRAK4 inhibitors was introduced in 2015, notably compounds 54 and 55. These compounds each exhibited an IRAK4 IC50 of less than 1 µmol/L, though further detailed activity data were not provided. Both compounds were also tested in vitro for their inhibition of TNF-α and IL-8 secretion, demonstrating effective IC50 values of less than 0.25 µmol/L (Fig. 16) [131].
Figure 16
Figure 16.
IRAK4 inhibitors by Nimbus. IRAK4 IC50 obtained by FRET assay. IL-8 IC50 and TNF-α IC50 were obtained by homogeneous time-resolved fluorescence (HTRF) assay.
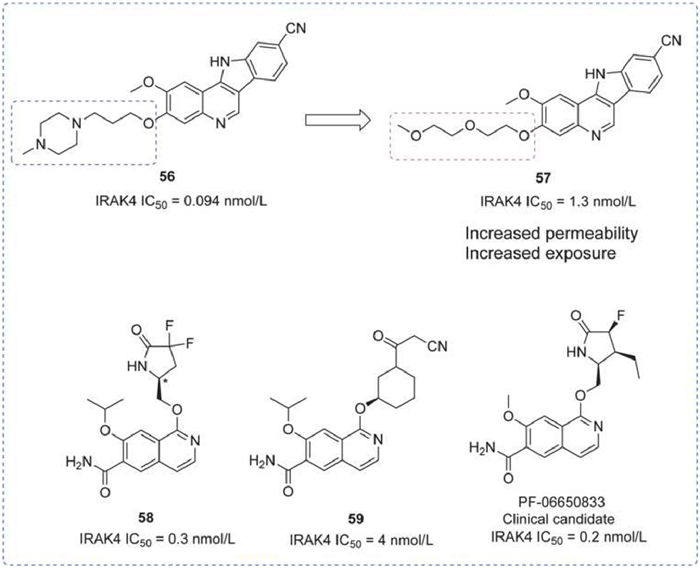
A series of indolo[2,3-c]quinoline-based IRAK4 inhibitors were reported, including compounds 56 and 57 [132]. Compound 56 was characterized by a favorable pharmacological profile (microsomal stability, TPSA, solubility, and ClogP). The removal of the ionizable solubilizing moiety from compound 56 led to increased exposure for compound 57in vivo, likely due to enhanced permeability. Both compounds effectively inhibited LPS-induced TNF-α in vivo mouse models, marking the first published experiment where small molecular inhibition of the IRAK4 pathway mimicked the phenotype of IRAK4 knockout mice.
Following in 2015 and 2017, a series of quinoline or isoquinoline-structured IRAK4 inhibitors were disclosed, for instance, compounds 58 and 59. These compounds demonstrated significant IRAK4 inhibitory activity, with PF-06650833, a clinical candidate emerging from this series (Fig. 17) [133,134].
Figure 17
Figure 17.
IRAK4 inhibitors by Pfizer. IRAK4 IC50 was obtained by DELFIA assay.
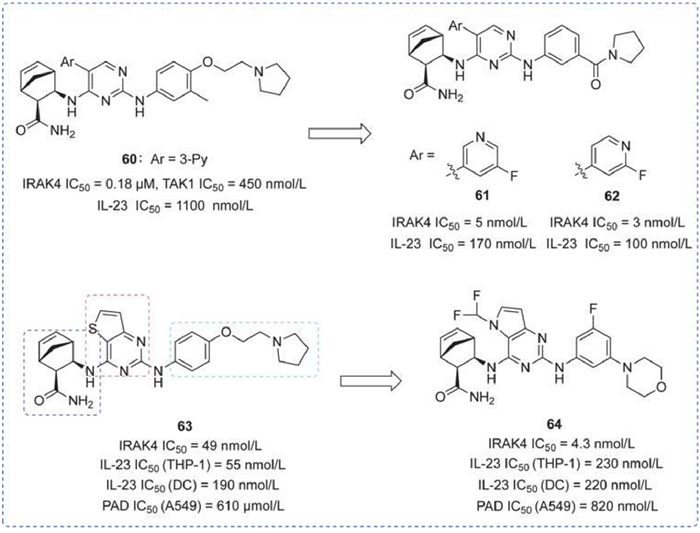
A range of potent 5-aryl pyrimidine inhibitors was identified from an HTS hit to lead optimization, especially by modifying the substituent at pyrimidine 5-position to reduce selectivity for transforming growth factor-β activated kinase 1 (TAK1). Compounds 61 and 62, showed significant inhibitory activity and selectivity for IRAK4, and both showed moderate potency in inhibiting IL-6 production in acute animal model studies [135].
In addition, bicyclic pyrimidines as potent inhibitors have also been recognized. Addressing the redundancy of promiscuous activity (PAD) properties in autoimmune diseases, the authors focused on reducing PAD activity to enhance specificity for treating autoimmune diseases. By altering the three fragments of compound 63, the obtained compound 64 showed excellent efficacy in inhibiting IL-1β-induced IL-6 production in mouse model and rat arthritis disease model studies. This achievement lays the groundwork for further development within this series (Fig. 18) [136].
Figure 18
Figure 18.
IRAK4 inhibitors by Rigel. IC50 of IRAK4 and TAK1 was obtained by ADP-Glo assay. IL-23 IC50 was obtained by ELISA assay. PAD IC50 was obtained by proliferation, apoptosis, and DNA damage assays.
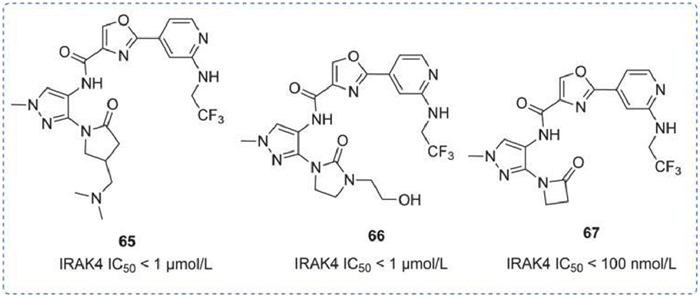
A sequence of pyrazole-based IRAK4 inhibitors was reported in 2015 and 2018. Compounds 65 and 66, differ by the fragment attached at the 2-position, both exhibited IC50 of less than 1 µmol/L. In an in vivo R848-induced inflammation model, compound 66 demonstrated a dose-dependent inhibition of TNF-α, achieving significant suppression at an oral dose of 10 mg/kg. In the non-collagenous domain of α3 chain of type IV collagen (NCI)-induced nephritis model, compound 66 showed significant alleviation of proteinuria as well as improved rat body weight after 35 days of treatment compared to the control group [137]. Compound 67, consistent with the preceding, featured a cyclic lactam structure with an IC50 of 100 nmol/L. No additional data was disclosed (Fig. 19) [138].
Figure 19
Figure 19.
IRAK4 inhibitors by Takeda. IRAK4 IC50 was obtained by lanthanide chelate excite (LANCE) assay.
7.2.1
IRAK4 PROTACs reported by Hangzhou Medical College
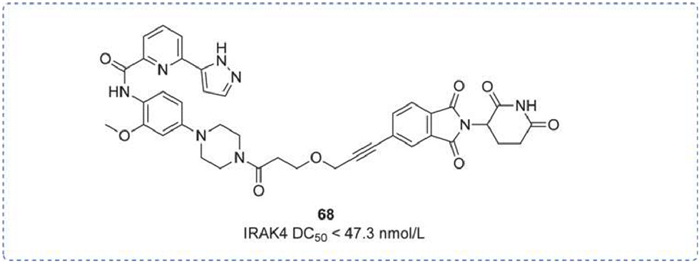
The scaffolding function of IRAK4 contributed to the development of IRAK4 protein hydrolysis-targeted chimeras. Liang's group publicly reported a class of potent PROTAC molecules, among which the most potent one, LC-MI-3 (68), could efficiently degrade intracellular IRAK4, with a degradation concentration 50 (DC50) of 47.3 nmol/L, and achieve complete inhibition of the downstream NF-κΒ pathway. Additionally, LC-MI-3 could significantly inhibit LPS and E. coli-induced skin inflammation models in vivo, suggesting its potential as a candidate IRAK4 drug (Fig. 20) [139].
Figure 20
Figure 20.
IRAK4 PROTAC by Hangzhou Medical College. IRAK4 DC50 was obtained by Western blot assay.
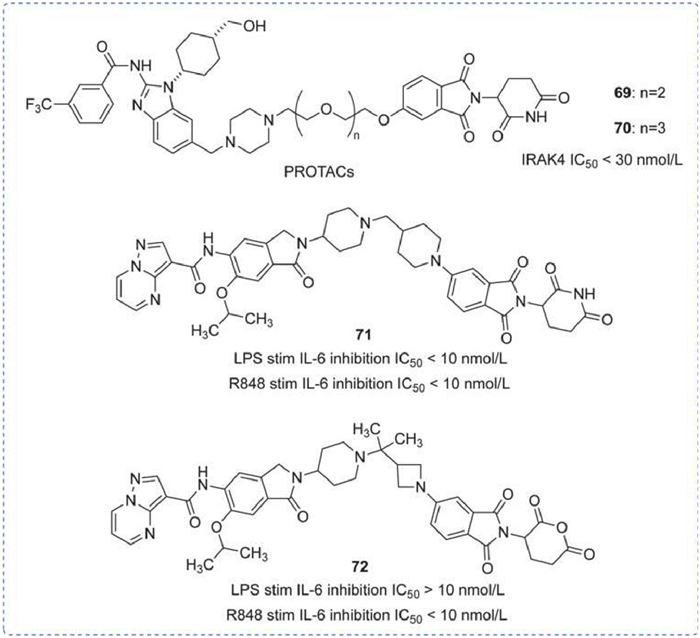
A range of degraders targeting IRAK4 had been disclosed by Arvinas, which acted by degrading ubiquitin-tagged p53 via the ubiquitin-dependent 26 s proteasome system (UPS) to achieve therapeutic effects on tumors. As shown in Fig. 21, compounds 69 and 70 consisted of a small molecular fragment, a linker, and a fragment bound to the E3 ubiquitin ligase. The distinction between 69 and 70 was their linker, and both compounds inhibited IRAK4 with an IC50 < 30 nmol/L [140,141].
Figure 21
Figure 21.
IRAK4 PROTAC by Arvinas. IRAK4 DC50 was obtained by Western blot assay. IL-6 IC50 was obtained by enzyme-linked immunosorbent assay (ELISA).
Subsequently, bifunctional degraders with ITM-LNK-CLM structure for cancer treatment were further described. As shown in Fig. 21, compounds 71 and 72 exhibited a wide range of pharmacological activities related to the degradation/inhibition of IRAK4. In particular, compound 72 has shown the most significant inhibitory effect on IRAK4 in the OCI-LY10 xenograft model or the C.B.17 SCID model, indicating its potential to treat or prevent diseases or disorders caused by target protein-mediated cellular signaling (Fig. 21) [142,143].
7.2.3
IRAK4 PROTACs reported by Kymera
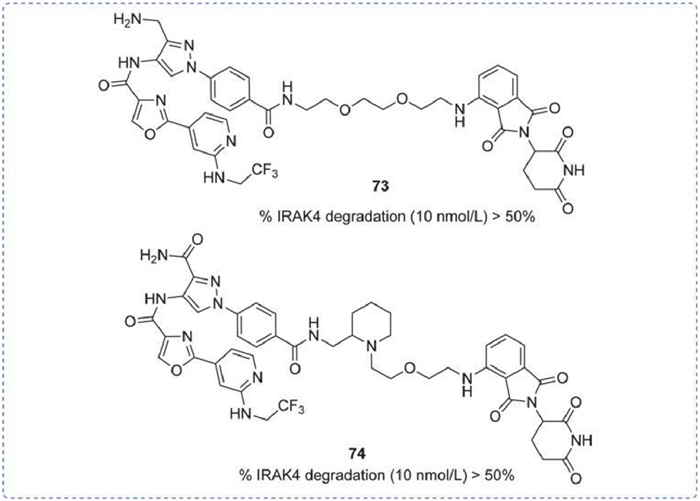
A batch of IRAK4 degraders was disclosed in 2019, which utilized pyrazole as the nucleus. The IRAK4 degradation rates of 73 and 74 were both > 50% at a dose of 10 nmol/L, with the main difference being in their linker structures (Fig. 22) [144].
Figure 22
Figure 22.
IRAK4 PROTAC by Kymera. % IRAK4 degradation was obtained by PBMCs degradation assay.
Chinese Academy of Sciences Shanghai Institute of Materia Medica reported a PROTAC 76, which effectively degraded IRAK4 in OCI-LY10 and TMD-8 cells in a concentration and time-dependent manner. Furthermore, compound 75 effectively blocked the IRAK4-NF-κB signaling pathway and exhibited significant advantages in inhibiting the growth of MyD88 L265P mutant cell lines, suggesting a potential treatment strategy for MyD88-driven B-cell lymphoma [145].
Additionally, a PROTAC molecule designed with PF-06650833 was reported. Compound 76 had a DC50 of 151 nmol/L in PBMCs, and its induced IRAK4 degradation resulted in the inhibition of multiple cytokines in PBMCs and a decrease in clearance in human and rat liver microsomes. However, the degrader did not inhibit the release of IL-6 and TNF-α in IL-β-stimulated fibroblasts (Fig. 23) [146].
Figure 23
Figure 23.
IRAK4 PROTAC by Shanghai Institute of Materia Medica & GlaxoSmithKline Medicines Research Centre. IRAK4 IC50 of OCI-LY10 and TMD-8 was obtained by cell counting kit-8 (CCK-8) assay. PBMCs DC50 was obtained by Western blot assay.
Small molecular IRAK4 inhibitors modulate downstream protein phosphorylation by competing for the active site, thereby inhibiting the signaling pathway and alleviating disease symptoms. Structural analysis of reported inhibitors has revealed three key characteristics affecting efficacy: firstly, amide fragments are crucial for enhancing inhibitor activity through interactions with the hinge region; secondly, aromatic fragments facilitate π-π interactions with the gatekeeper residue Tyr262; and thirdly, fluorine atoms improve the drug-like. These small molecule inhibitors continue to serve as the backbone of drug development and maintain their central position in the pharmaceutical industry.
Although IRAK4 candidates are promising, several challenges remain. Pfizer's PF-06650833, a promising candidate for RA treatment in Phase 2, was ultimately discontinued due to insufficient therapeutic efficacy in HS layouts, reasons for this may involve miscellaneous biological mechanisms and uncertainty in clinical trials [147]. Similarly concerning, Curis' pioneering IRAK4-targeting drug, CA-4948, exhibits substantial off-target FLT3 inhibition target, which may explain its adverse effect [148]. Indeed, CA-4948 had dose-limiting toxicity in rhabdomyolysis, and what is more, the FDA terminated a clinical study in leukemia due to patient death. Moreover, the high homology between IRAK4 and IRAK1 kinase domains emphasizes the criticality of developing selective drug design, an issue that demands immediate attention in future research.
There is also a critical limitation that IRAK4 acts as a scaffolding protein, and small molecule inhibitors are unable to fully suppress the IL-1R/TLRs pathway. PROTACs have emerged as a novel approach. PROTACs degrade IRAK4, achieving dual inhibition of both its kinase and scaffolding functions. The design of PROTACs involves: the careful selection of small molecules with the desired activities as one terminus; then, the linker, a pivotal component, determines PROTAC activity, with its flexibility, rigidity, and length contributing to its efficacy [149]; lastly, despite the identification of over 600 E3 ligase species, only a select few, including CRBN, VHL, MDM2, have been utilized for PROTAC development [150]. While the current landscape of IRAK4-targeting PROTACs remains relatively sparse, the promising theories underpinning their mechanism and the remarkable clinical successes reported thus far suggest a promising future. For example, BGB-45, 035, a degrader developed through the Cellular Degradation Targeted Complex (CDAC) platform of Beigene, achieves deeper degradation of IRAK4 [151]. GS-6791 (NX-0479) demonstrated remarkable efficacy in vivo and in vitro as a candidate in preclinical studies at the recent ACR Convergence 2025 [152].
PROTACs, while offering numerous benefits, also present limitations. They have been developed beyond "Lipinski's rule", such as molecular weight, number of H-bond acceptors and donors, and ClogP value [153]. The high molecular weight and substantial topological polar surface area (TPSA) of PROTACs contribute to poor water solubility and diminished cell permeability, which characteristics can adversely impact pharmacokinetics (PK) and bioavailability. Moreover, the non-selective expression of E3 ubiquitin ligases may raise off-target effects [154]. KT-474, a seminal PROTAC in IRAK4 research, demonstrated unexpected cardiac effects (mild QT interval prolongation) during clinical assessment, likely due to off-target inhibition of the hERG K+ channel [82].
To address these challenges, it is imperative to strategically reduce the molecular weight of PROTACs, which can optimize water solubility and metabolic stability, cell permeability can be improved through the strategic formation of intramolecular H-bonds and the incorporation of flexible linkers. Furthermore, the discovery of novel E3 ligands is crucial for enhancing oral bioavailability. Lastly, the exploration of new E3 ubiquitin ligases is essential to diversify the scope of PROTAC applications and reduce reliance on existing ligases [153,155].
9.
Summary
The IL-1R/TLRs signaling pathway holds a pivotal position within the innate immune system, highlighting its importance in various pathological processes. IRAK4, as the key kinase in this intricate signaling cascade, emerges as a compelling target for therapeutic intervention. By strategically inhibiting or degrading IRAK4, notable benefits have been observed in mitigating symptoms of numerous autoimmune, inflammatory disorders, and tumorous conditions. Through an exhaustive review and analysis of the documented inhibitors and PROTACs targeting IRAK4, this work suggests that focusing on IRAK4 could pave the way for novel therapeutic strategies to tackle an even broader spectrum of diseases in the foreseeable future.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the National Natural Science Foundation of China (Nos. 82293684, 82293680), the National Key R & D Program of China (No. 2020YFA0908004), CAMS Innovation Fund for Medical Science of China (No. 2022-I2M-1-014).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.110963.
[1]
I.M.O. Viana, S. Roussel, J. Defrêne, et al., Acta Pharm. Sin. B 11 (2021) 852–870. doi: 10.1016/j.apsb.2021.02.022
G. Teng, T. Fung, A. Mocciaro, et al., Gilead Sciences, Inc., https://www.nurixtx.com/wp-content/uploads/2024/11/IRAK4-Degrader-GS-6791-Inhibits-TLR-and-IL-1R-Driven-Inflammatory-Signaling-and-AmelioratesDisease-in-a-Preclinical-Arthritis-Model.pdf.
J. Ge, C.Y. Hsieh, M. Fang, H. Sun, T. Hou, Trends Pharmacol. Sci. 45 (2024) 1162–1174. doi: 10.1016/j.tips.2024.10.006
Figure 1
Chronic activation of the immune system involves a wide range of innate and adaptive immune cells. TLRs/IL-1R signaling pathway mediated the innate immunity. Overexpressed IRAK4 leads to excessive signaling pathway activation, potentially causing several diseases.
Figure 2
IL-1R/TLRs signaling pathway. Membrane IL-1R/TLRs activate the JNK/P38 and NF-κB pathways via MyD88-dependent signaling, while intracellular TLRs activate the IRF3/7 pathway via MyD88-independent signaling.
Figure 3
(a) IRAKs family consists of 4 members and IRAK4 contains an N-terminal DD, a ProST domain, and a central KD (PDB ID: 2NRU). (b) The first structure of IRAK4, in the active site of IRAK4, Val263, Tyr264, and Met265 form the hinge region of the kinase, Tyr262 is a unique gatekeeper residue, and there is a ubiquitous catalytic Lys213 in the active site of the kinase (PDB ID: 2NRU). Visualized in PyMOL.
Figure 4
(a) IRAK4 dual capacity as both a kinase and scaffold protein. (b) IRAK4 PROTACs can form a ternary complex between the E3 ubiquitin ligase and the target protein, followed by ubiquitination tagging of the protein and eventual degradation by the proteasome system, achieving complete inhibition of the signaling pathway.
Figure 6
IRAK4 inhibitors by AstraZeneca. IRAK4 enz IC50 and IRAK4 cell IC50 were obtained by fluorescence resonance energy transfer (FRET) assay and flow cytometry, respectively.
Figure 7
IRAK4 inhibitors by Aurigene Discovery Technologies Limited. IRAK4 IC50 was obtained by time-resolved fluorescence resonance energy transfer (TR-FRET) assay.
Figure 16
IRAK4 inhibitors by Nimbus. IRAK4 IC50 obtained by FRET assay. IL-8 IC50 and TNF-α IC50 were obtained by homogeneous time-resolved fluorescence (HTRF) assay.
Figure 18
IRAK4 inhibitors by Rigel. IC50 of IRAK4 and TAK1 was obtained by ADP-Glo assay. IL-23 IC50 was obtained by ELISA assay. PAD IC50 was obtained by proliferation, apoptosis, and DNA damage assays.
Figure 23
IRAK4 PROTAC by Shanghai Institute of Materia Medica & GlaxoSmithKline Medicines Research Centre. IRAK4 IC50 of OCI-LY10 and TMD-8 was obtained by cell counting kit-8 (CCK-8) assay. PBMCs DC50 was obtained by Western blot assay.

 DownLoad:
DownLoad:





 下载:
下载:
























