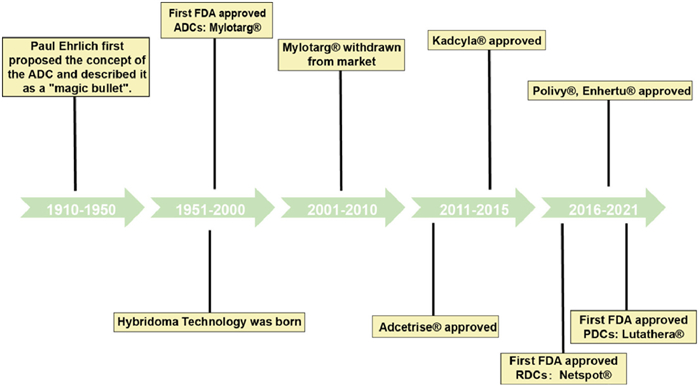
Figure 1.
The timeline of the drug conjugates development process.
Cancer-targeting drug conjugates drives a new era in precise cancer treatment
Jia Deng , Jianbin Shi , Dan Li , Xue Jiao , Jinrui Liu , Haowen Tian , Na Liu , Cong Luo , Ken-ichiro Kamei , Chutong Tian

Cancer is currently the second leading cause of premature mortality and shortened life expectancy worldwide [1]. Despite various therapeutic approaches, clinical outcomes often fall short of expectations. Surgery, chemotherapy, and radiation therapy are the most commonly employed treatments; however, surgery is only effective for solid tumors and localized lesions [2]. Although radiation therapy and chemotherapy are used to treat most types of cancer, they often cause side effects and significant damage to normal cells and tissues [3], significantly limiting their clinical efficacy. The immunotherapy represents a promising therapeutic strategy that harnesses the body's immune system to combat tumors [4]. However, its efficacy in treating solid tumors and preventing tumor recurrence and metastasis has been suboptimal [5]. To surmount these problems and increase anti-cancer efficiency, additional therapeutic strategies are urgently required.
Targeted therapies can precisely target and eliminate tumors, reducing the risk of progression and prolonging patient survival [6,7]. As a result, there has been growing interest in developing these therapies. Several targeted treatments are currently approved for cancer, including hormonal therapies, protease regulators [8], signal inhibitors, gene expression modulators [9], apoptosis inducers, immunotherapies, and antibody-drug conjugates, among others. Among these, drug conjugates are the most promising targeted therapies due to the high specificity of the targeting ligands and the high activity of the payload [10]. Drug conjugates consist of three basic components: targeting ligands, linkers, and payloads. Targeting ligands deliver drugs precisely to specific cell surfaces, while linkers allow payloads to be released accurately within the target cell. Therefore, drug conjugates are also known as "biological missiles" or "magic bullets" [11,12].
After decades of research, drug conjugates are the fastest-growing class of targeted therapies. Antibody-drug conjugates (ADCs) are the key to unlocking the door to conjugated drugs, which were first proposed in the 1950s. Monoclonal antibodies (mAbs) are commonly used as carriers for ADCs due to their targeted delivery and specific binding properties to overexpressed antigens on tumor cells. Despite their advantages, such as high target specificity and binding affinity, good stability in circulation and internalization efficiency, mAbs also have limitations including their high molecular weight, restricted distribution, low penetration ability, and potentially life-threatening side effects [13]. To overcome these limitations, alternative drug conjugates subtypes using smaller molecular weight tumor-homing carriers like peptides have been developed. Peptide-drug conjugates (PDCs) show improved efficacy and reduced side effects compared to mAbs with enhanced penetration ability, reduced immunogenicity, simpler design, and cheaper synthesis. However, PDCs development has been slow in clinical settings due to short circulatory half-life and rapid renal clearance [14]. Other types of drug conjugates such as small molecule-drug conjugates (SMDCs) [15], aptamer-drug conjugates (ApDCs), radionuclide-drug conjugates (RDCs) are being developed to enhance the efficacy of anticancer therapies, with most currently in preclinical or clinical trials [16,17]. RDCs, in particular, are characterized by the use of nuclides as payloads, distinguishing them from other drug conjugates. This approach offers greater flexibility in selecting targeting ligands, which can include antibodies, peptides, small molecules, or nucleic acids. Such versatility enables the customization of targeting ligands for a wide range of therapeutic applications.
In recent years, the range of applications and application scenarios for drug conjugates have significantly expanded, encompassing concepts such as combination therapy and precision drug delivery. Various new drug conjugates concepts have emerged in a never-ending stream, and the industry has unleashed a wave of new drug conjugates development. Therefore, it is time to conduct a comprehensive review of drug conjugates. This review is poised to provide a comprehensive summary of emerging drug conjugates for cancer-targeted therapy, with a specific focus on the ADCs, PDCs, SMDCs, ApDCs and RDCs from the point of view of components. Furthermore, we discuss the merits and demerits of two kinds of linkers. Finally, we briefly analyze the advantages, challenges, and future prospects of drug conjugates in clinical cancer therapy. This paper provides a valuable reference for the design and construction of drug conjugates-based anticancer treatments.
Targeting ligands play a pivotal role in the development of drug conjugates, serving as carriers for efficacious payloads such as small molecule drugs and enabling precise drug delivery due to their specific targeting activity. The continuous advancements in genetic engineering and conjugation technologies have led to the emergence of diverse types of targeting ligands in the field of drug conjugates, encompassing antibodies, peptides, small molecules, and aptamers, each exhibiting unique advantages. As the key component in targeted drug delivery, an ideal targeting ligand should exhibit high selectivity and affinity for the target, low immunogenicity (or no immunogenicity), functional efficacy, promote drug uptake, and possess an appropriate molecular size and others.
The concept of ADCs originated from Paul Ehrlich's "golden bullet" doctrine of monoclonal antibodies proposed in 1936, which aimed to achieve cancer cell-specific treatments by precisely targeting antigens with monoclonal antibodies. With the advancement of genetically engineered antibody production technology and new chemical ligation technologies, the concept of ADCs has gradually become a reality. ADCs combine mAbs with highly potent cytotoxic payloads through chemical linkers, achieving efficient and tumor-specific delivery of cytotoxins, reducing their toxicity to normal cells and increasing the therapeutic index [18]. As a navigation system for ADCs, ideal antibodies should possess the following characteristics: target specificity, high target binding affinity, effective internalization, low immunogenicity, low cross-reactivity, maintaining a relatively long plasma half-life, and appropriate conjugation properties. The mAbs used in ADCs are typically selected from one of four isotypes of human immunoglobulin G (IgG), which are defined by different sequences of heavy chain amino acids (IgG1, IgG2, IgG3 or IgG4) [12]. Considering the half-life, structural stability, immune function of Fc fragment and coupling convenience of different IgG types of antibodies, IgG1 (human-derived, or PEGylated) is preferred by ADCs [19]. Despite their numerous advantages, ADCs currently face several challenges that need to be addressed, including large molecular weight of antibodies, complex manufacturing process, low drug delivery efficiency, non-specific expression of target antigens in normal tissues and heterogeneity of target antigen expression in tumor cells [20-22].
PDCs, as the subsequent generation of targeted therapies succeeding ADCs, possess enhanced cellular permeability and improved drug selectivity as their core advantages. PDCs usually use linear, cyclic single-chain polypeptides as targeted peptides, with a flexible and simple structure, high disulfide bond independent stability, and low molecular weight (usually only a few kDa) [23]. Compared to mAbs as previously mentioned, peptides can offer some significant advantages over antibodies, including deep tissue permeability, low immunogenicity and toxicity, low production costs, easy introduction of structural modifications, support for drug and rational design, improved stability and activity, high selectivity and specificity, fewer drug-drug interactions and low off-target toxicity [24-26].
Peptides can be classified according to their functions: cell-penetrating peptides (CPPs), cell-targeting peptides (CTPs). CPPs refer to small, short molecular peptides that can enter cells without disrupting their membrane integrity [27]. CPPs tend to be short, positively charged sequences of alkaline amino acids, which allows them to interact well with cell membranes and effectively deliver drugs into cells utilizing endocytosis [28]. CPPs can be divided into three classes: protein derived CPPs, modified CPPs [29] and designed CPPs [30]. However, due to their low cell specificity, the application of CPPs to these types of PDCs has been limited. In contrast, CTPs serve as an ideal carrier molecule that can specifically bind to receptors overexpressed on the surface of tumor cells and exhibit similar effects to monoclonal antibodies while overcoming several limitations. One advantage of utilizing CTPs instead of monoclonal antibodies lies in their superior tissue penetration capability, enabling efficient transmembrane transport and intracellular targeting within tumor cells [31]. Additionally, CTPs offer numerous advantages in comparison to conventional monoclonal antibody therapies, encompassing cost-effectiveness, enhanced stability, and absence of immunogenicity. Consequently, CTPs have gained widespread utilization in PDCs research. Meanwhile, self-assembling peptides (SAPs), as a peptide-targeting ligand, exhibit remarkable versatility in different research fields due to the unique capability to spontaneously form well-defined structures without requiring external stimuli, making them with good biocompatibility, biodegradability and versatility [32]. Responsive peptides are polypeptide structures that undergo structural changes when external conditions change [33]. Due to the unique tumor microenvironment (TME), it has also attracted the attention of many researchers (Table S1 in Supporting information).
The steady development of peptide chemistry has made it possible to obtain PDCs relatively directly [34]. However, the utilization of peptides as targeting ligands also poses potential challenges [35], including low affinity, limited in vitro half-lives, and susceptibility to protease-mediated degradation [36,37]. With the advancement of technology, chemical and structural modifications of peptides can increase stability and improve peptide pharmacokinetics, including binding to albumin, pegylated and so on.
Small molecule-based drug delivery systems have been employed for targeted delivery of therapeutic agents to tumor sites and enhanced the clinical outcomes of anticancer drugs, offering advantages such as low immunogenicity, cost-effectiveness, and enhanced distribution and permeability in tumor tissue due to their smaller molecular size. Additionally, small molecules enable accurate drug-ligand ratios, while off-target low-molecular weight drug conjugates are commonly excreted from the body, reducing undesired toxicity in normal cells [38]. Therefore, the unique advantages of small molecules have promoted the development of SMDCs. An ideal SMDCs ligand should possess characteristics such as chemical inertness, non-immunogenicity, optimal size, high affinity for target molecules, facile synthesis and amplification processes, as well as straightforward transportation and storage procedures [38-40]. Several targets of SMDCs, such as glucose transporter 1 (GLUT1), fusion protein BCR/ABL, the folic acid, aminopeptidase N (APN), phosphatidylserine (PS), αvβ3, and the somatostatin receptor (SSTR), have been investigated in clinical trials or preclinical study of SMDCs [40-42].
The molecular composition of SMDCs exhibits similarities to that of ADCs, thus leading to a gradual evolution in the development process of SMDCs alongside the advancements in ADCs. With enhanced computational support data, cost-effective properties, and simplified characterization compared to macromolecular ligands, novel small molecule ligands have been embraced for their potential as cancer cell-targeting agents [43]. However, the rate of development of SMDCs is limited due to the difficulty in finding suitable receptors and ligands for SMDCs and the scarcity of targets and indications.
In recent years, nucleic acid drugs have become increasingly important in disease treatment, particularly for conditions not addressable by small molecule or protein drugs. An aptamer, a member of the nucleic acid drug class, is a small RNA or single-stranded DNA that adopts a specific three-dimensional structure. Utilizing base stacking, hydrogen bonding and electrostatic interaction, aptamers exhibit high affinity and selectivity towards specific target tissues [44,45]. Besides binding to targets, aptamers can also act as therapeutic agents to regulate the biological function of biomarkers.
Aptamers offer higher screening efficiency than antibodies and can be generated through various artificial screening technologies, including Systematic Evolution of Ligands by Exponential Enrichment (SELEX), Cell-SELEX, capillary electrophoresis-SELEX, and automated SELEX [44]. Nucleic acid aptamers offer an alternative targeting approach for antigens that are difficult to detect with antibodies due to low sensitivity. Their synthesis is mature and cost-effective, and their low molecular weight ensures high tissue permeability [46]. Meanwhile, nucleic acid aptamers can target various molecules (e.g., metal ions, small organic molecules, proteins, sugars, lipids and cells) [47].
Both RNA aptamers and DNA aptamers exhibit strong binding affinity for different targets. Recent studies have shown that while DNA and RNA aptamers exhibit similar efficacy, DNA aptamers offer several advantages over RNA ligands. These include the absence of transcription requirements, ease of modification [48], lower cost and time demands, and improved stability, owing to the C—H bonds at the 2′-deoxyribose position in DNA nucleotides [46]. Therefore, DNA-based aptamers have a good research prospect.
Currently, most ApDCs consist of three parts: targeting ligand, linker and payload. Interestingly, it has a unique coupling mode, non-covalent connection, with intermolecular forces such as van der Waals force, hydrogen bonding and electrostatic interaction, to achieve the direct binding of nucleic acid aptamers and drugs, without the need for linkers. For example, doxorubicin (DOX) molecules can embed nucleic acid base fragments due to its flat aromatic ring structure, directly forming the aptamer DOX complex [49-51], e.g., A10-DOX, HB5-DOX. ApDCs is a promising targeted drug delivery platform that is still in its infancy and clinical translation needs to be further advanced.
The key requirement for the linker is that it must ensure the chemical stability of the drug conjugates in the blood and allow for rapid release of the payload at the target site after internalization. In addition, the ideal linker should also possess the following characteristics: (1) Non-toxic: the linker must keep the coupling in an inactive and non-toxic state when it binds to the antibody; (2) Controlled drug release: linkers should facilitate controlled release of cytotoxic drugs upon internalization; (3) High water solubility: the linker aids in coupling reactions and prevents formation of inactive aggregates [52-54]. Drug conjugates commonly employ two types of linkers: cleavable and non-cleavable linkers, which play a pivotal role in determining the overall functionality and efficacy of these conjugates (Fig. S1 in Supporting information).
The cleavable linker can be disrupted by intracellular biomolecules or enzymes, thereby enabling targeted release of payloads at specific sites, and is extensively employed in drug conjugates. This release can lead to a "bystander effect", affecting neighboring cells not directly targeted by drug conjugates. While this can amplify the therapeutic effect, it also carries a risk: It is more likely to produce toxicity at non-target sites. Compared to non-cleavable linkers, the ability to specifically cleave at the target tissue and rapidly release the drug to reach therapeutic concentrations, while reducing toxic side effects to other tissues or organs, makes the concept of intelligent drug delivery a reality. Cleavable linkers can be divided into three main categories, including: (1) pH-sensitive linkers [15]: acetals, hemiacetals, hydrazone [55], imines and hydrazines; (2) Enzyme-sensitive linkers: matrix metalloproteinases (MMPs) [56], Val-Ala [57], Carbamate bond, Amide bond; (3) GSH concentration-sensitive linkers: disulfides [58], thioethers [59], ferrocene [60], and metalthiols [61]; reactive oxygen species (ROS)-responsive linker [62] and others.
Unlike cleavable linkers, non-cleavable linkers are able to resist degradation under physiologic conditions, providing stability and structural integrity to drug conjugates, thereby ensuring that the therapeutic payload remains securely attached until reaching the designated target. Non-cleavable linkers also contribute to prolonging circulation time in the systemic circulation, enhancing bioavailability and improving overall pharmacokinetics. Compared with the cleavable ligand, the non-cleavable ligand has the following advantages, such as good plasma stability, low off-target toxicity, wide therapeutic window and is not easy to develop drug resistance [63]. The action mechanism of non-cleavable linkers is based on the internalization of the drug complex, followed by the degradation of certain targeting molecular components in the lysosome, resulting in the release of cytotoxic drugs to kill tumor cells (e.g., thioether linkers, maleimide caproyl linkers and others). They do not release cytotoxic substances at the off-target site and therefore do not damage healthy cells [64]. However, for non-cleavable linkers, the effect of the targeting molecules must be degraded in the lysosome after the internalization of the coupled drug. Therefore, differences between the parent drug and potential coupled drug metabolites must be taken into account. Its application is limited due to the difficulty in exerting the effects of onlooker cells and the high structural requirements for payloads [65].
As the warhead of drug conjugates, the payload primarily performs the cytotoxic function. Their activity and physicochemical properties have a direct impact on the anti-tumor efficacy of conjugated drugs, including solubility, hydrophilicity, permeability and modifiability, while their mechanism of action is a critical determinant of the performance of conjugated drugs. Thus, the selection of appropriate payloads is imperative for attaining desired therapeutic outcomes [66]. An ideal payload should have the following characteristics: high cytotoxicity, sufficiently low immunogenicity, high stability [15,67], and low molecular weight [68]. At the present time, the most commonly used clinical agents can be divided into three main categories, including chemical drugs, biomacromolecule drugs and radioactive isotopes.
Among drugs conjugate, the most applied payloads are chemical drugs. These drug conjugates show strong potential in targeted therapy by delivering their payload directly to target cells. Upon internalization, the drug conjugate is degraded in lysosomes. This degradation releases the active payload, enabling it to exert its therapeutic effect and ultimately induce target cell death. In recent years, numerous chemical drugs have been extensively studied and are currently undergoing clinical trials, such as microtubule-disrupting agents maytansinoids [69], DNA damaging agents calicheamicin [70], topoisomerase inhibitors DOX [71], transcription inhibitors thailanstatin [72] and so on.
Although chemical drugs are widely used, conditions such as safety and stability limit their further expansion, leading to a focus on non-chemical drugs. Many proteins and peptides have been developed to target different active processes within tumor cells due to the clinical limitations of traditional payloads, such as poor efficacy, drug resistance, and the overactive metabolic processes of tumor cells. Compared to chemical drugs, these may exhibit higher activity, fewer side effects, and easier structural and chemical modification, including tumor necrosis factor (TNF) [73], polypeptide RNA polymerase inhibitors amatoxins [74], Bcl-xL inhibitors [75], niacinamide phosphate ribose transferase (NAMPT) [76] and others.
In addition to drug-based payloads, some non-drug payloads have also gradually caught the attention of researchers, such as radionuclides. Radionuclides are potent cytotoxic agents that emit alpha, beta, and gamma radiation, which can destroy cells through ionization. Currently, they are widely used in tumor diagnosis, treatment, and tissue imaging. However, due to their potential to damage surrounding healthy cells, radionuclides are primarily utilized in targeted therapies [77].
Based on this, the RDCs formed by linking radioactive isotopes to target ligands have gradually become a research hotspot in the field of drug conjugates [78,79]. When RDCs work inside the body, the linker arm does not need to be broken or in direct contact with the cells. It can only achieve its tumor cell-killing effect through internal radiation. In addition, it also exhibits bystander and distant effects, thereby giving it potential for use in diffuse tumors. This further enhances the stability and safety of RDCs in the body [44]. An ideal radioactive nuclide should possess the following characteristics: a half-life that strikes a balance between minimizing prolonged radiation effects after the application of radioactive drugs and allowing adequate time for production and transport; sufficient stability both in vitro and in vivo; and other relevant factors [16,80,81]. Compared to ADCs, RDCs have several advantages including high targeting specificity, lower risk of systemic toxicity, higher drug safety, good tolerance, and immune enhancement. Although RDCs are in their infancy, it can be predicted that it has a good application prospect.
Drug conjugates span multiple fields, including antibodies, small molecules, cell therapies, and targeted therapies, with growing clinical and commercial importance (Fig. 1). Currently, more than 20 drug conjugates are approved worldwide, of which 15 are ADCs (Table S2 in Supporting information). This surge mirrors trends in other drug fields, creating a competitive landscape. Consequently, research on novel drug conjugates remains active, especially on those with similar structures and mechanisms to ADCs, such as PDCs, SMDCs, ApDCs, and RDCs.
Research on ADCs dates back to 1980; however, the Food and Drug Administration (FDA) approved the ADCs for acute treatment only in 2000. Unfortunately, owing to severe toxicity concerns, this drug was withdrawn from the market in 2010. Technological revisions led to the development of first-generation ADCs that received FDA approval in 2017. After several decades of optimization of key components, there are currently 200 active drugs in clinical stages across different countries worldwide [82].
The mAbs used in previous ADCs studies were derived from mice, resulting in an immune response and production of human anti-mouse antibodies. Since the first generation of ADCs, recombinant humanized IgG4 has been used as an antibody that binds to low-toxicity cytotoxic drugs via unstable linkers, such as gemtuzumab ozogamicin and inotuzumab ozogamicin.
Calicheamicin is a potent DNA-damaging agent isolated from Micromonospora echinospora that induces toxicity in a variety of cells, limiting its application. However, its toxic effects can be minimized when incorporated into drug conjugates. Petersdorf et al. developed gemtuzumab ozogamicin (MylotargⓇ), an anti-CD33 IgG4 monoclonal antibody conjugated to calicheamicin via a cleavable hydrazone linker. This ADC had an average drug-antibody ratio (DAR) of only 1.5 and contained approximately 50% uncoupled monoclonal antibodies. Once internalized, the hydrazone bond hydrolyzes in the acidic endosome, releasing ozogamicin. Glutathione then reduces it to the active form, which binds to DNA grooves, selectively cleaving double-stranded DNA. MylotargⓇ was the first ADC approved for acute myeloid leukemia treatment and the first to be withdrawn due to severe cytotoxicity. It was later reintroduced with an optimized "dosing regimen" [83-85]. However, hydrazone bonds are prone to premature drug release into the bloodstream owing to their instability.
Kantarjian et al. developed inotuzumab ozogamicin (BesponsaⓇ), a recombinant human anti-CD22 IgG4 kappa antibody. Ozogamicin binds to inotuzumab via a cleavable linker based on hydrazone and disulfide bonds, specifically binding to the CD22 target on the cell surface for the treatment of relapsed or refractory precursor B-cell acute lymphoblastic leukemia [86,87]. Ozogamicin, a semi-synthetic acanthomycin derivative, is produced through microbial fermentation and synthetic modification. As a DNA synthesis inhibitor, it induces double-stranded DNA breaks, playing a tumor-killing role [88]. Compared to MylotargⓇ, BesponsaⓇ uses a linker with greater steric hindrance at the ortho-disulfide bond, improving stability and reducing premature release of the cytotoxic drug.
First-generation drug conjugates have advanced targeted therapies by improving efficacy, reducing systemic toxicity, and optimizing pharmacokinetics, pharmacodynamics, and in vivo distribution. However, limitations remain: (1) Insufficient drug efficacy: low blood concentrations and poor target antigen expression result in inadequate intracellular drug delivery. (2) Linker instability: hydrophobic payloads cause antibody aggregation, leading to a short half-life, rapid clearance, and poor immunogenicity. (3) High heterogeneity: random coupling of lysine and cysteine residues affects DAR, which in turn influences efficacy, pharmacokinetics, and therapeutic indices. (4) High immunogenicity: the use of murine or chimeric human antibodies increases the risk of immune reactions. Consequently, there is still a need for further enhancements in the development of first-generation ADCs.
Compared to first-generation ADCs, second-generation ADCs feature enhanced antibodies, linkers, and payloads, using chimeric human antibodies or humanized monoclonal antibodies, more stable linkers, and more potent small-molecule drugs such as brentuximab vedotin (Fig. 2A) and ado-trastuzumab emtansine.
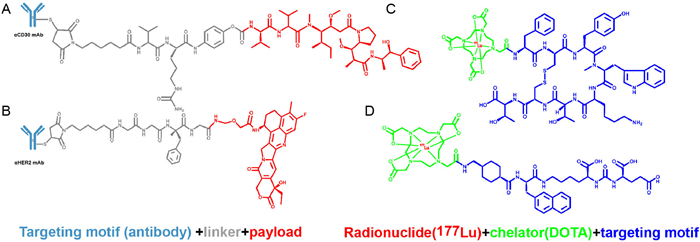
Mertansine (DM1) is a microtubulin inhibitor that is isolated from the shrub Maytenus ovatus. Compared to calicheamicin, it has higher toxicity but with more serious side effects. Verma et al. developed ado-trastuzumab emtansine (KadcylaⓇ), a HER2-targeting ADC. It was synthesized using humanized HER2 IgG1 trastuzumab and DM1 via a thioether linker (MCC), with an average DAR of 3.5 [89]. The MCC linker improves plasma stability and releases the payload after endocytosis by HER2-positive cancer cells, resulting in effective "mass killing" of HER2-positive tumor cells. It is commonly used in patients with HER2-positive breast tumors [90], although MCC's poor water solubility requires further stability improvements.
Younes et al. synthesized brentuximab vedotin (AdcetrisⓇ), coupling monomethylauristatin E (MMAE), a natural tubulin inhibitor, with brentuximab, a CD30-targeting monoclonal antibody. A protease-sensitive, cleavable citrulline-Val-CIT with a mean DAR of 4 was used [91,92]. Brentuximab vedotin is internalized by a clathrin-dependent mechanism and is transferred to endosomes and lysosomes, where the linker is hydrolyzed by cysteine proteases such as cathepsin B, which hydrolyses the linker [93]. The released free MMAE then binds to tubulin and inhibits its polymerization, leading to cell cycle arrest and cell death. In 2011, brentuximab vedotin was approved for the treatment of primary cutaneous anaplastic large-cell lymphoma (pcALCL) and CD30-expressing mycosis fungoides (MF) after systemic therapy [94].
Compared to first-generation ADCs, second-generation ADCs benefit from the IgG1 isotype, which is better suited for bioconjugation and cancer cell targeting. Using a more toxic payload while increasing water solubility and coupling efficiency allows more payload molecules to be loaded onto each monoclonal antibody without inducing antibody aggregation. Enhanced linkers offer greater plasma stability and uniform DAR distribution, improving overall clinical efficacy and safety. However, challenges remain, such as off-target toxicity. Future advancements will require site-specific coupling to optimize DAR, alongside continued improvements to mAbs and payloads.
The development of ADCs has been greatly facilitated by extensive research and the rapid development of monoclonal antibodies. Consequently, the third generation of ADCs came into being, which employ site-specific binding of small molecule drugs to monoclonal antibodies, achieving a DAR of 2 or 4. Stability and pharmacokinetic problems were greatly improved, with low rates of coupling loss and high drug activity maintained, even at low antigen levels. Examples of such ADCs include polatuzumab vedotin and fam-trastuzumab deruxtecan (T-DXd) (Fig. 2B).
DAR affects drug stability in circulation and influences both toxicity and pharmacokinetics. While earlier ADCs had DAR inconsistencies, the third generation resolved many of these issues. Dornan et al. designed and synthesized polatuzumab vedotin (PolivyⓇ), which is composed of polatuzumab, a monoclonal antibody targeting CD97b, bound to MMAE via a cleavable ligand, mc-vc-PABC, with an average DAR of 3.5 [95]. Similar to brentuximab vedotin, polatuzumab vedotin selectively binds to CD79b, undergoes endocytosis and proteolytic cleavage to release MMAE, which induces cell cycle arrest and cell death [96]. In 2019, PolivyⓇ was approved by the FDA for use in combination with bendamustine and rituximab for the treatment of r/r diffuse large B-cell lymphoma (DLBCL) in patients who have had at least two prior lines of therapy.
Further advancements were made in linker technology. Modi et al. designed and synthesized fam-trastuzumab deruxtecan (EnhertuⓇ), which is linked by a cleavable linker of trastuzumab, a humanized monoclonal antibody targeting HER2, to an exatecan derivative of a novel topoisomerase 1 inhibitor [97]. These linkers contain an enzyme-cleavable peptide (GGFG) and an aminoethyl moiety that can be released by internalized ADCs subjected to lysosomal proteases, followed by rapid hydrolysis of the aminoethyl moiety to ammonia and formaldehyde [98]. Finally, DXd is released into the cells, triggering cell death [99]. Compared to previous generations of ADCs, DXd is a more potent active form of irinotecan. Its higher potency, combined with novel tetrapeptide-based linker technology, improves plasma stability and reduces systemic toxicity. DXd's cell membrane permeability allows for a paracrine antitumor effect, a major advantage in ADC design. This linker-payload system is particularly beneficial in treating refractory HER2+ metastatic breast cancer [100].
The third generation of ADCs addresses the shortcomings of first and second generations by incorporating key optimizations: (1) Fully humanized antibodies replace chimeric ones to reduce immunogenicity. (2) Site-specific binding of small-molecule drugs to monoclonal antibodies results in ADCs with a DAR of 2 or 4, offering reduced toxicity, higher stability, improved pharmacokinetics, minimal coupling loss, and no unconjugated antibodies. (3) Unique amino acid sites and selective modifications, such as inter-chain disulfide bonds, glycosyl, and chemically selective modifications, enhance targeting precision. (4) More efficient payloads, such as PBD, microtubulins, and immunomodulators, are used. However, the third-generation connector types remain unchanged. To avoid interference with the immune system and improve circulation time, third-generation ADCs use more hydrophilic linker modifications, such as PEGylation.
Recently, PDCs have emerged as a prominent research focal point in the field of cancer treatment and have garnered extensive attention. Due to their remarkable selectivity and specificity, PDCs exhibit enhanced therapeutic efficacy. As an emerging targeted drug delivery system, PDCs offer new hope for treating various cancers and are gradually entering clinical use. Researchers are actively exploring diverse strategies to optimize the design and formulation of PDCs tailored to specific tumor types or individual patients. Currently, only three PDCs are marketed worldwide for therapeutic purposes.
Most gastroenteropancreatic neuroendocrine tumors (GEP-NETs) have relatively high levels of somatostatin receptor (SSTR) expression, and radiolabelled somatostatin analogs have high affinity for SSTR. These analogs can be internalized into cells. Their low molecular weights allow them to be rapidly cleared from the blood, making them ideal drug candidates. LutatheraⓇ (Fig. 2C), developed by Novartis, was the first peptide-targeted PDC, using radionuclides as its killing mechanism. Specifically, it uses radionuclide 177Lu combined with a somatostatin analog (octreotate) and 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) for SSTR-targeted therapy. In 2018, the FDA approved LutatheraⓇ for treating GEP-NETs, and Novartis is exploring its use in other cancers, including lung, prostate, breast, and colorectal cancers [101].
Multiple myeloma (MM) is a common hematological malignancy that is difficult to treat because of recurrence. Fortunately, aminopeptidases have emerged as promising targets. PDCs have been developed using oncopeptides, such as melphalan flufenamide (melflufen, PepaxtoⓇ), which links melphalan to para-fluoro-L-phenylalanine through simple peptide bonds. Melphalan flufenamide enters the cell via passive diffusion and is hydrolyzed by aminopeptidases in the cytoplasm to produce the hydrophilic cytotoxic alkylating agent melphalan and other active metabolites. In February 2021, the FDA approved melflufen combined with dexamethasone for the treatment of adult patients with relapsed/refractory multiple myeloma (R/R MM). However, because it showed safety risks in confirmatory studies, Oncopeptides voluntarily withdrew PepaxtoⓇ from the U.S. market in October 2021 [102].
Prostate-specific membrane antigen (PSMA) is a carboxypeptidase highly expressed in prostate cancer cells. Although existing PSMA inhibitors have good tumor-targeting properties, their pharmacokinetic effects are severely compromised by their typical low-molecular-weight membranes, limiting their application. To overcome this, Novartis developed PluvictoⓇ (PSMA-617) (Fig. 2D), an RDC that uses radionuclide 177Lu as a payload attached to a PSMA-targeting peptide fragment (Glu-NH—CO—NH-Lys) via a DOTA and Glu-UREA-LYS linker. PluvictoⓇ emits radiation over a short range, allowing precise tumor targeting while limiting damage to surrounding healthy cells. In March 2022, the FDA approved PluvictoⓇ for treating patients with PSMA-positive metastatic castration-resistant prostate cancer (mCRPC) [103].
With new scientific explorations uncovering the details of cancer cell receptor overexpression, the potential of SMDCs as targeted therapies has received widespread attention. Compared to large-molecule ligands, small-molecule ligands offer advantages such as lower cost, better computational modeling, and easier characterization. Moreover, SMDCs are highly effective, cost-efficient, and safe drug delivery systems.
Vintafolide, developed by Endocyte, is an SMDC targeting FRα, connecting folic acid (FA) and the vinca alkaloid desacetylvinblastine hydrazide (DAVLBH) by means of a peptide spacer and a reducible and spontaneous disulfide linker. Following receptor-mediated endocytosis, DAVLBH is released into the cells by cleaving the disulfide bond. Despite its potential, vintafolide's phase III clinical trial failed, halting its progress toward market approval [104].
Similar to ADCs, ApDCs were proposed for the first time in 2009. Aptamers have better stability, lower process cost, lower immunogenicity, and deeper tissue penetration owing to their small size. Despite these advantages, ApDCs development remains in its early stages, with no products currently on the market as of October 2024.
Gemcitabine is a first-line chemotherapeutic agent used for treating pancreatic cancer. However, owing to the frequent occurrence of resistance to chemotherapy, its application is limited. Aptamers, which can be effectively internalized into cancer cells by binding to target molecules with high affinity and specificity, offer a promising alternative. Park et al. developed APTA-12 as an ApDC that incorporates gemcitabine into the loop region of the DNA aptamer AS1411, which targets the cellular protein nucleolin. Once internalized, APTA-12 disrupts the stability of bcl-2 mRNA and releases gemcitabine after nuclease-mediated degradation, playing a role in DNA incorporation. In vitro experiments confirmed that it exhibited good targeting and serum stability, while in vivo experiments confirmed that it showed good tumor-inhibiting efficacy compared to controls. A Phase I clinical study is currently underway [105].
The clinical application of drug conjugates, alongside innovations in targeting ligands and payload modifications, has driven the rise of RDCs as a new therapeutic category. As an innovative form of medicine, RDCs exhibit numerous similarities and distinct differences compared with ApDCs and SMDCs. In contrast to the increasingly saturated field of ADCs, RDCs offer a novel alternative with a less competitive development landscape. The research and development of RDCs has thrived in both academia and industry, resulting in a steady increase in the number of drug candidates entering clinical trials. RDCs rank second in the number of marketed coupling drugs, but only two, both developed by Novartis, have demonstrated therapeutic efficacy: LutatheraⓇ and PluvictoⓇ. These RDCs, which use peptides as targeting ligands (making them PDCs), illustrate the potential of this approach, though the broader therapeutic potential of RDCs remains largely unexplored. As ADCs were being developed, multiple other drug conjugates also entered clinical research (Table S3 in Supporting information).
To address the limitations of conventional IgG antibodies, such as high molecular weights and low penetrability, smaller antibody formats such as single-domain antibodies (sdAbs), single-chain antibody fragments (scFvs), and minibodies have been developed. Compared with intact IgG, their faster clearance rate, higher stability, and solubility may be advantageous for reducing the number of potentially immunogenic epitopes. Moreover, their unique light chain variable domains (VL) make them promising scaffolds for ADCs development.
Ganglioside GD2, a validated clinical target overexpressed in several cancers, is a promising marker for anti-tumor therapy [106]. Combining small antibody fragments with ADCs can enhance therapeutic efficacy. Daniel et al. employed two different GD2-binding formats, minibodies and scFvs, both containing variable antibody domains identical to those of dinutuximab, conjugated to MMAE via thiol-maleimide chemistry, resulting in DAR of 2 and 1 [107], respectively (Fig. 3A). Stability, affinity, and cytotoxicity experiments showed that mini-body-based fragment-drug conjugates (FDCs) demonstrated more pronounced cytotoxic effects and stronger antigen binding than scFv-based FDCs (Figs. 3B–D). Currently, sdAbs are the smallest functional antigen-binding fragments obtained from conventional IgG, consisting only of VH or VL. Their small size, rapid clearance, good stability, high solubility, and low immunogenicity make them attractive ligands for ADC development [108,109]. Ana et al. developed a novel rabbit-derived VL sdAb-based bioconjugation linker and created DAB-SN-38, a complex molecule featuring the SN-38 drug, an ROS-responsive deazapurine linker, and a maleimide bioconjugation handle. Conjugated at Cys80, DAB-SN-38 achieved a DAR of 1 [110]. In animal models of canine non-Hodgkin's lymphoma, DAB-SN-38 showed potent cytotoxicity linked to DNA Topo I inhibition, demonstrating high efficacy against canine diffuse large B-cell lymphoma at nanomolar concentrations. This highlights the potential of rabbit-derived sdAbs as targeting moieties for ADCs.

Currently, most targeting ligands used in ADCs are mono-specific antibodies. However, monospecific antibodies are prone to defects such as poor affinity and targeting, random directions of binding sites, and poor long-term storage stability due to structural changes in the antibodies, which limits their application. To address these issues, the development of bifunctional antibodies is necessary. Bispecific antibodies possess two antigen-binding sites that allow ADCs to target two different epitopes on the same antigen or to completely target two different antigens. The benefits of bispecific antibodies over monospecific ones include: (ⅰ) Enhanced targeting by engaging two binding sites on the same antigen, potentially increasing the binding affinity and specificity of the antibody; (ⅱ) the ability to target multiple antigens simultaneously, which is advantageous for cancer and immunological therapies; and (ⅲ) the ability of bispecific antibodies to cross-link cancer cells and immune cells, such as T-cells, to facilitate targeted cell killing. For example, He et al. constructed ADCs by conjugating anti-programmed cell death-ligand 1 (PD-L1) THIOMAB to the bifunctional immunomodulator D18 via a redox-cleavable linker. The ADCs HE-S2 not only induces a potent anti-tumor immune response by blocking the programmed cell death 1 (PD-1)/PD-L1 interaction and activating the Toll-like receptor 7/8 (TLR7/8) pathway, but also upregulates its targeted PD-L1 expression via epigenetic regulation. This enhances sensitivity to PD-1/PD-L1 blockade and suggests that treatment with ADCs HE-S2 may provide greater tumor suppression than treatment with D18 combined with an anti-PD-L1 antibody. This approach represents a novel strategy for enhancing the antitumor immune response to immune checkpoint blockade (ICB) therapy [111].
Linkers used in ADCs, whether cleaved or non-cleaved, are internalized by cells to release the payload. However, off-target toxicity remains a problem. Optimizing linkers is critical for improving ADCs safety and efficacy. Ha et al. introduced a novel valine-citrulline (VCit)-based linker, glutamate-glycine-citrulline (EGCit) (Fig. 4A), which replaces valine with glutamic acid. This modification significantly enhances the stability of VCit-based linkers. In vitro experiments have demonstrated that the EGCit linker is resistant to degradation by human neutrophil proteinases. In vivo, the linker exhibited long-term stability with minimal signs of blood or liver toxicity in healthy mice. These results suggest that this kind of linker could provide excellent in vivo stability and release a traceless payload in normal tissues [112].

A phenol-containing compound is an effective agent for masking and subsequent cleavage upon activation. Various methods have been developed to utilize the phenolic group in generating self-immolative units, including the use of carbonate, carbamate, and ether linkages [113]. Wei et al. developed Ate-PPS-CA4, utilizing a phenoxysilyl linker (Fig. 4B), Atezolizumab as the targeting ligand, and CA4, a microtubule depolymerizer, as the payload. In vitro experiments have demonstrated that Ate-PPS-CA4 effectively targets PD-L1-positive cells and delivers the active payload, CA4, through self-cleavage [114]. The application of the phenolic linker provides a foundation for designing the next generation of ADCs, warranting further research.
The endogenous free radical nitric oxide (NO) is one of the smallest and most ubiquitous signaling molecules involved in many physiological processes. It plays a dual role in tumors: high concentrations inhibit tumor growth, while optimal concentrations promote tumor growth. Identifying an effective NO donor that can deliver therapeutic amounts at the target site with minimal side effects is crucial [115]. Sun et al. reported a new NO- donor, HL-2, which is covalently linked to G7mAb via a disulfide bond. This conjugate target human CD24, highly expressed in solid tumors. At high GSH concentrations, cleavage is triggered and NO is released (HN-01) [116]. The results of in vivo and in vitro experiments indicated that HN-01 retained its specific binding ability and showed more potent antitumor activity in vivo than either component, G7mAb, or HL-2, offering a promising new strategy for treating CD24+ malignancies.
The treatment of bacterial infections poses numerous challenges. As one of the most promising tools in biomedical sciences, ADCs also hold promise for addressing bacterial infections and autoimmune diseases. Johnson et al. developed an ADC by fusing an antimicrobial peptide to the C-terminal end of the VH and/or VL-chain of a monoclonal antibody, VSX, targeting the lipopolysaccharide core of Pseudomonas aeruginosa. This ADC, VSX-AMP, demonstrated specific bactericidal activity against P. aeruginosa with minimal non-specific cytotoxicity to mammalian cells in vitro and in vivo. Tvilum et al. developed a mitomycin C ADC to treat implant-associated biofilm infections caused by Staphylococcus aureus. Using a disulfide bond linker, they conjugated the S. aureus antibody to mitomycin C. This novel mechanism allows drug release without cellular entry, possibly interacting with mercaptan on the bacterial surface. The ADC showed good antibacterial activity in vitro and in vivo in an osteomyelitis model [117,118].
Glucocorticoids (GCs) are widely used to treat autoimmune and inflammatory diseases. However, systemic administration of GCs causes side effects. Budesonide is a potent GC with low oral bioavailability and extensive first-pass liver metabolism. One promising strategy to reduce its toxic side effects is to use it in targeted therapies. Han et al. modified budesonide and conjugated it to anti-prolactin receptor (PRLR) antibody using site-specific carbamate bonds and cathepsin-cleavable linkers. This anti-PRLR antibody rapidly internalizes into PRLR-expressing cells, delivering the ADC payload effectively. In vitro experiments demonstrated the safety and absence of cardiotoxicity of the modified drug. Additionally, the anti-inflammatory efficacy of the drug was superior to that of the parent drug in a mouse model [119]. When used in a targeted manner to deliver GC to cells of interest, it can achieve its biological effects in a targeted manner and reduce the side effects caused by systemic administration. Thus, the development of novel GC-ADCs for the treatment of inflammatory and autoimmune diseases is a promising therapeutic strategy.
The advancement of ADCs is heavily influenced by evolving coupling technologies that ensure the production of drugs with stable DAR. However, creating homogeneous ADCs with high DAR while avoiding increased hydrophobicity and aggregation remains a challenge. McPherson et al. designed and synthesized a modular platform capable of producing homogeneous, stable, high DAR ADCs. This system includes: (1) A maleimide-based "multi-arm linker" for drug attachment; (2) a scFv-Fc antibody; and (3) a 6-mer peptide with a Zn2+-binding cysteine that activates the maleimide reaction without forming unwanted disulfide bonds. Using this technology, they developed a novel ADC, TE-1146, incorporating lenalidomide molecules as the payload and reconfigured daratumumab as the targeting ligand capable of targeting CD38. In vitro experiments confirmed that it remained intact in the plasma and effectively entered MM cells to release the payload. Simultaneously, it had a stronger cell-killing effect than the other controls. In vivo experiments showed significant tumor volume reduction and notable tumor-inhibiting effects [73]. In addition, numerous studies on PDCS technologies have been conducted (Table S4 in Supporting information).
Although paclitaxel (PTX) is one of the most commonly used natural antitumor drugs in clinical practice, it causes many adverse effects, including arthralgia, myalgia, and myelosuppression. Matrix metalloproteinases (MMP-2), which can recognize and cleave peptides with specific sequences, have emerged as key targets for controlled drug release [120-122]. Tang et al. developed SynB3-PVGLIG-PTX (Fig. 5A), where the MMP-2-sensitive linker PVGLIG is combined with CPPs to form a dual-functional peptide with blood-brain barrier (BBB) permeability [122]. In vitro studies showed that SynB3-PVGLIG-PTX specifically inhibited the proliferation, migration, and invasion of GBM cells in vitro in the presence of MMP-2. Further in vivo investigations showed that SynB3-PVGLIG-PTX readily entered the brain of U87MG xenograft nude mice and achieved a better suppressive effect on GBM through the controlled release of PTX from SynB3-PVGLIG-PTX compared to PTX and temozolomide (Figs. 5B–H). SynB3-PVGLIG-PTX, with its specificity and permeability across the BBB, can serve as an innovative drug delivery system for GBM treatment [123]. Similar to PTX, Docetaxel (DTX) is a chemotherapeutic agent used as first-line treatment for various malignancies. However, the limited solubility of DTX hampers its efficacy and leads to significant excipient-related toxicity [124,125]. To address this, Li et al. designed a peptide drug conjugate (CTCE-DTX) by coupling two functional molecular (CXCR4) antagonist peptides with the chemotherapeutic agent DTX through a flexible linker [126]. CTCE-DTX can self-assemble into nanoparticles (CTCE-DTX NPs) in water, thus improving their solubility. In vivo and in vitro data suggested that free DTX preparations may cause severe systemic toxicity, including red blood cell hemolysis and weight loss, decreased white blood cell count, and liver function impairment. CTCE-DTX NPs, however, improved toxicity profiles and reduced metastasis risk by blocking the CXCR4 pathway.

Although 4–1BB is an important target for tumor immunotherapy, its application is limited because of its dose-dependent liver toxicity. Hinner et al. developed RPS-343, a bispecific anti-CD137/anti-HER2 protein combining an engineered IgG4 variant of trastuzumab and anticalin. Its activity was characterized using in vitro experiments and humanized mouse models. Safety was assessed using in vitro human cells and cyborg monkey toxicity assays. The results showed that the bispecific binding of HER2 and 4–1BB promoted tumor targeting by T cells, whereas PRS-343 was well-tolerated without significant toxicity. Compared to monospecific methods, it has a higher level of efficacy and lower level of peripheral toxicity [127].
As a clinical broad-spectrum antitumor drug, the application of DOX is limited by its potential cardiotoxicity. Therefore, there is an urgent need to enhance its targeting to reduce its cardiotoxicity. To address this, Zeng et al. designed a novel type of PDC supramolecular self-assembled nanofiber: baicalin-FFYEEG-ARVYIHPF and BA-NFs. The ARVYIHPF peptide targets AT1R as a ligand. As a payload, baicalin (BA) inhibits iron death, and the phenylalanine tyrosine (FFY) motif not only acts as a linker, but also has self-assembly power. Both in vitro and in vivo experiments confirmed that BA-NFs have good targeting properties and can inhibit cardiomyocyte apoptosis and improve DOX-induced myocardial injury via the iron death pathway [128]. Recent advancements in SMDCs research are outlined in Table S5 (Supporting information).
Fibroblast activating protein (FAP), highly expressed in most malignant solid tumors, is a promising target for drug development. Bocci et al. introduced OncoFAP-SMDCs using MMAE and exatecan as payloads to create OncoFAP-GlyPro-MMAE and OncoFAP-GlyPro-Exatecan. The glycine-proline dipeptide moiety acts as a FAP-sensitive linker that is rapidly and specifically cleaved in the TME. In vivo and in vitro experiments have shown these SMDCs achieve more effective tumor cell killing and high targeted delivery performance at optimal release doses [129].
VCit dipeptide linkers are widely used in drug conjugates; however, the instability of VCit linkers leads to the premature release of cytotoxic loads into the systemic circulation. Stability can be optimized by considering the linker and targeting ligand. To improve stability, Amin et al. developed trifunctional molecules (TFMs) by adding an extra arm containing the hydrophilic thyroid transthyroid hormone (TTR)-binding ligand AG10 to a typical bifunctional SMDC, replacing the VCit dipeptide linker with the glutamic acid-valine-citrulline (EVCit) tripeptide linker [130,131]. The results showed that EVCit-TFM (TFM with the EVCit linker) had a higher plasma stability and was more sensitive to cathepsin B than VCit as a linker, leading to improved efficacy due to its extended half-life. The introduction of hydrophilic glutamic acid into the linker enhanced serum stability without affecting the linker's cleavage by cathepsin B. Zheng et al. designed an SMDC-Fc scaffold using MMAF as the payload. They added a single amino acid X-linker to the C-terminal carboxyl group of MMAF as an enzyme-cleavable linker for folate-targeting ligands. In vivo and in vitro experimental results demonstrated that using X-linkers to attach couplings led to significantly prolonged blood circulation and antitumor efficacy in xenografted mice with small or large advanced KB tumors [132]. Continued research is advancing new methods in ApDCs development (Table S6 in Supporting information).
Photodynamic therapy (PDT) has gained prominence for its minimal trauma, rapid recovery, and low side effects in cancer treatment. PDT relies on the interaction of light, photosensitizers, and oxygen to induce apoptosis through the production of singlet and active oxygen. However, the utility of singlet oxygen is limited owing to its short lifetime and diffusion range. Therefore, Yang et al. designed a circular aptamer-polyethylene glycol (PEG) structure capable of responding to the acidic TME, designed to selectively recognize cancer cells. This structure was coupled with pyrochlorophyll A (PA), a potent photosensitizer, to form PA-Apt ApDCs [133,134]. These conjugates penetrate solid tumors and enable selective bioimaging and PDT in vivo [135,136]. The PA-Apt-ApDCs, with two terminal amino groups, were coupled with aldehyde groups at both terminals via Schiff base formation to form PA-Apt-CHO-PEG. At physiological pH 7.4, PA-Apt-CHO-PEG maintains its colloidal stability, prevents normal cell absorption, and prolongs blood circulation. However, upon entry into the TME at pH 6.5, the Schiff base linkage between the aptamer and PEG is rapidly cleaved, allowing the conjugate to penetrate the dense extracellular matrix and target cancer cells effectively [137,138]. In vitro and in vivo experiments showed that PA-Apt-CHO-PEG nanostructures are efficient PDT agents with dramatic tumor inhibitory efficacy. Compared to traditional ApDCs, PA-Apt-CHO-PEG offers prolonged blood circulation and stealth-like behavior due to the conjugated PEG, which helps avoid non-specific recognition [139].
Traditional ApDCs are single-responsive, meaning that they respond only to a single signal molecule, such as pH or GSH. Therefore, the stability and targeting ability may be improved by introducing a multilevel-responsive targeting ligand. For example, free radicals are substances produced by the human metabolism that have a high degree of chemical activity, serving as an effective defense system in the body. However, excessive free radicals can cause irreversible oxidative damage and cell death. By carefully balancing free radical concentrations, it is possible to achieve therapeutic effects without causing cellular harm. Xuan et al. introduced biorthogonal chemistry and prodrug design to create novel ApDC micelles capable of dual TME targeting. The engineered ApDC micelles consist of three components: an aptamer as the tumor-recognizing moiety, Fe2+-activatable tetraoxane ("T") as the free-radical prodrug, and hemin as the TME-responsive Fe2+ source for in situ activation of "T". A single prodrug base can act as a hydrophobic tail to trigger the self-assembly of aptamers into a rigid multicomponent supramolecular structure, thereby improving the drug-loading capacity and minimizing unwanted drug leakage [140,141]. ApDC micelles are inactive under physiological conditions but trigger cascading orthogonal responses upon receptor-mediated uptake by cancer cells. The loaded Fe3+ is reduced to Fe2+ via intracellular GSH, triggering "T" activation that directly releases toxic C-based free radicals. This process does not rely on tumor acidity or H2O2 levels. "T" is attached to the 5′ end of a DNA aptamer (AS1411), with six G bases serving as a linker to form ApDCs (Ap-6G-"T"). The incorporation of intermolecular G-quadruplexes helps minimize nonspecific membrane insertion caused by serum albumin disruption [142-144]. The results showed that ApDC micelles could specifically recognize cancer cells via receptor-mediated targeting, with enhanced cellular uptake compared to non-targeted micelles. After endocytosis, ApDC micelles can produce toxic radicals more efficiently than non-targeted micelles, contributing to significant oxidative damage in cancer cells and enhanced antiproliferative effects.
The synthesis of these ApDCs using phosphoramidite chemistry allows for precise control over conjugation site and prodrug base density, facilitating scalable production and potential clinical translation. With these advantages, we believe that combined bioorthogonal chemistry and prodrug design can be extended to other aptamers to create an arsenal of advanced ApDCs and provide new insights into free-radical-related molecular mechanisms [145].
Triptolide (TP), the active ingredient of triptolide, not only exerts anti-tumor activity in immune diseases but also produces a synergistic effect with chemotherapeutic drugs to enhance its therapeutic effect. However, the clinical application of TP is severely limited owing to its high toxicity, poor solubility, and low bioavailability [146]. Miao et al. reported an AS1411–triptolide conjugate (ATC) that greatly reduced the toxicity of TP. Experiments on serum stability, targeting, and in vivo anti-tumor activity revealed that ATC specifically targets the MDA-MB-231 cell line, both in vitro and in vivo. Introduction of the aptamer significantly enhanced the in vivo anti-tumor activity of triptolide in the treatment of TNBC. In contrast, free triptolide shows minimal tumor inhibition at the same dosage [147]. This advancement marks a significant development in RDC research (Table S7 in Supporting information).
The treatment of mCRPC is challenging due to the disease's heterogeneity and the presence of multiple anti-apoptotic pathways. Radiopharmaceuticals offer promising new avenues for cancer diagnosis and treatment. Therefore, the introduction of radioactive elements may be the most promising avenue for treating mCRPC. Bidka et al. designed and synthesized 225Ac-DOTA-YS5 (Fig. 6A), which targets CD46 and maintains a high level of expression in prostate cancer across differentiation patterns. A biodistribution study showed that 225Ac-DOTA-YS5 delivered higher levels of radiation to tumor tissues than to healthy organs (Fig. 6B) [148,149]. Moreover, 225Ac-DOTA-YS5 suppressed tumor size and prolonged survival in cell- and patient-derived xenograft models (Figs. 6C–F). Overall, this preclinical study confirmed that 225Ac-DOTA-YS5 is a highly effective treatment, supporting the clinical development of CD46-targeted radioligand therapy for prostate cancer [150].

The high molecular weight of mAbs makes passive transport across the BBB difficult, limiting the use of ADCs in the brain. Intracranial uptake can be enhanced using receptor-mediated endocytosis (RMT) by attaching a "molecular trojan horse". Compared to other radionuclides, 89Zr is more widely used in clinical practice, has better physical properties for positron emission tomography (PET) imaging, and is more suitable for coupling. Wuensche et al. reported that imaging of the specific brain uptake of the bispecific aducanumab brain shuttle antibody Adu-8D3 in APP/PS1 TG mice with 89Zr was possible when using the chelator desferrioxamine analogues (DFO*). The brain uptake and targeting efficacy of radiolabeled Adu-8D3 in APP/PS1 TG mice in vivo and in vitro using the bispecific anti-HIV-monoclonal antibody mAbB12-scFab and wild-type animals as controls were evaluated using PET imaging [151]. The findings suggest that 89Zr-based immuno-PET holds promise for future neurological applications due to its availability, cost-effectiveness, and superior imaging quality [152,153].
Most RDCs often serve a single purpose: Either diagnostic imaging or therapeutic intervention. However, combining radionuclides with a payload that has both diagnostic and therapeutic capabilities can enhance efficacy and provide a synergistic effect. For instance, 89Zr-DFO-α CD11b has demonstrated high tumor uptake and can be used to display tumor-associated myeloid cells (TAMCs) in situ genetic mouse gliomas via preclinical immune-PET imaging [154,155]. However, these drugs lack the cytotoxicity required for radiopharmaceutical therapy (TRT). To address this, Foster et al. developed Lumi804, a macrocyclic bifunctional chelator that binds to both 89Zr and 177Lu radionuclides, and conjugated it with anti-CD11B antibodies to form LUMI804-ACD11B. The study found that 89Zr/177Lu-labeled Lumi804-aCD11b effectively enables non-invasive imaging of TAMCs in mouse gliomas, reduces TAMC numbers in the spleen and tumor, and improves the efficacy of checkpoint immunotherapy. Therefore, it holds promise as a therapeutic combination for monitoring and reducing TAMCs in gliomas, potentially improving immunotherapy outcomes [154,156].
The development of advanced drug delivery systems for cancer treatment has led to the emergence of drug conjugates as a promising strategy for precise cancer treatment, effectively reducing potential toxicity and enhancing therapeutic efficacy. This review focuses on five distinct classes of targeted molecules and payloads, evaluates the advantages and disadvantages of the two linker types, and provides a concise overview of recent advancements in coupled drug therapy over the past 5 years. Despite a limited number of approved drug conjugates, notable improvements in design have been achieved.
Currently, ADCs remain the most popular avenue for drug conjugation, with the largest number of drug conjugates in the market entering clinical trials. Various targeting ligands such as peptides, small molecules, and nucleic acid aptamers have been developed to address the challenges and limitations associated with ADCs in clinical applications. However, some common unresolved problems remain, including drug resistance and off-target toxicity. To enhance targeting ligands, modifications are made to improve specificity and affinity. These include introducing small side-chain structures, utilizing dual-targeting molecules for precise localization, and shielding the Fab domain with peptide masks that are easily cleaved by tumor-overexpressing proteases. Ongoing efforts also focus on discovering new targets that are either highly expressed in tumors or absent in normal tissues. From the linker perspective, new linkers are continually being developed, while existing ones are adjusted in terms of chemical length, composition, and rotational freedom to reduce off-target effects and cytotoxicity from paracellular effects. The emphasis is on achieving controlled payload release independent of endogenous enzyme-mediated cleavage and optimizing coupling techniques. Addressing drug resistance involves the discovery of novel payloads, employing low-efflux or bi-isotopic payloads, and using radionuclides for targeted killing. Additionally, future research is focusing on developing molecules that can serve as both targeting agents and payloads.
Overall, the multifunctionality of targeted molecules, along with the exploration of new ligands and cytotoxic payloads, positions drug conjugates at the frontier of next-generation treatments for various diseases. A systematic evaluation of each component of the coupled drugs and an in-depth exploration of their mechanisms of action and resistance will provide important insights into the mechanistic basis of drug conjugate design and provide an opportunity to better understand the impact of changes in drug conjugate characteristics on therapeutic activity and safety. Drug conjugates show great promise for controlling or eliminating tumors and may offer new therapeutic options for clinical management in the future. It is anticipated that new efficient drug conjugates will continue to emerge in the future to play a more important role in combating tumors and other diseases.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Jia Deng: Writing – review & editing, Writing – original draft. Jianbin Shi: Writing – review & editing, Writing – original draft. Dan Li: Writing – review & editing, Writing – original draft. Xue Jiao: Writing – original draft. Jinrui Liu: Conceptualization. Haowen Tian: Conceptualization. Na Liu: Conceptualization. Cong Luo: Project administration. Ken-ichiro Kamei: Project administration. Chutong Tian: Project administration, Funding acquisition, Conceptualization.
This work was generously provided by the Project of China-Japan Joint International Laboratory of Advanced Drug Delivery System Research and Translation of Liaoning Province (No. 2024JH2/102100007), the open fund of National Key Laboratory of Advanced Drug Formulations for Overcoming Delivery Barriers (No. 2024-KFB-003), the National Natural Science Foundation of China (No. 82104109) and Scientific Research Project of Liaoning Department of Education (No. LJ212410163045).
Supplementary material associated with this article can be found, in the online version, at doi:
H. Sung, J. Ferlay, R.L. Siegel, et al., CA Cancer J. Clin. 71 (2021) 209–249.
F. Peyraud, C. Allenet, M. Gross-Goupil, et al., Cancer Treat. Rev. 90 (2020) 102087. doi: 10.1016/j.ctrv.2020.102087
S. Kumari, S. Mukherjee, D. Sinha, et al., Int. J. Mol. Sci. 21 (2020) 8151. doi: 10.3390/ijms21218151
C. Li, X. You, X. Xu, et al., Adv. Sci. 9 (2022) e2104134. doi: 10.1002/advs.202104134
S.Y. Yi, M.Z. Wei, L. Zhao, Crit. Rev. Oncol. Hematol. 196 (2024) 104313. doi: 10.1016/j.critrevonc.2024.104313
X. Zhu, J. Wu, W. Shan, et al., Angew. Chem. Int. Ed. 55 (2016) 3309–3312. doi: 10.1002/anie.201509183
N. Bertrand, J. Wu, X. Xu, et al., Adv. Drug Deliv. Rev. 66 (2014) 2–25. doi: 10.1016/j.addr.2013.11.009
Z. Zhang, R. Sun, C. Bian, et al., Chin. Chem. Lett. 36 (2025) 109784. doi: 10.1016/j.cclet.2024.109784
B. Hu, L. Zhong, Y. Weng, et al., Signal Transduct. Target. Ther. 5 (2020) 101. doi: 10.1038/s41392-020-0207-x
L. Zhou, Y. Lu, W. Liu, et al., Exp. Hematol. Oncol. 13 (2024) 26. doi: 10.1186/s40164-024-00493-8
K. Strebhardt, A. Ullrich, Nat. Rev. Cancer 8 (2008) 473–480. doi: 10.1038/nrc2394
Z. Fu, S. Li, S. Han, et al., Signal Transduct Target. Ther. 7 (2022) 93. doi: 10.1038/s41392-022-00947-7
K. Tsuchikama, Y. Anami, S.Y.Y. Ha, C.M. Yamazaki, Nat. Rev. Clin. Oncol. 21 (2024) 203–223. doi: 10.1038/s41571-023-00850-2
L. Gong, H. Zhao, Y. Liu, et al., Acta Pharm. Sin. B 13 (2023) 3659–3677. doi: 10.1016/j.apsb.2023.02.013
C. Fu, L. Yu, Y. Miao, et al., Acta Pharm. Sin. B 13 (2023) 498–516. doi: 10.1016/j.apsb.2022.07.020
A. Juzeniene, V.Y. Stenberg, Ø. S. Bruland, R.H. Larsen, Cancers 13 (2021) 779. doi: 10.3390/cancers13040779
R. Eychenne, C. Bouvry, M. Bourgeois, et al., Molecules 25 (2020) 4012. doi: 10.3390/molecules25174012
D.Y. Ruan, H.X. Wu, Q. Meng, R.H. Xu, Cancer Commun. 44 (2024) 3–22. doi: 10.1002/cac2.12517
J.G. Salfeld, Nat. Biotechnol. 25 (2007) 1369–1372. doi: 10.1038/nbt1207-1369
M. De Cecco, D.N. Galbraith, L.L. McDermott, Expert Opin. Biol. Ther. 21 (2021) 841–847. doi: 10.1080/14712598.2021.1880562
L. Qian, X. Lin, X. Gao, et al., Chem. Rev. 123 (2023) 7782–7853. doi: 10.1021/acs.chemrev.2c00915
M.S. Dennis, H. Jin, D. Dugger, et al., Cancer Res. 67 (2007) 254–261. doi: 10.1158/0008-5472.CAN-06-2531
Y.S. Zhu, K. Tang, J. Lv, Trends Pharmacol. Sci. 42 (2021) 857–869. doi: 10.1016/j.tips.2021.07.001
J. Xie, Y. Bi, H. Zhang, et al., Front. Pharmacol. 11 (2020) 697. doi: 10.3389/fphar.2020.00697
M.B. Hock, K.E. Thudium, M. Carrasco-Triguero, N.F. Schwabe, AAPS J. 17 (2015) 35–43. doi: 10.1208/s12248-014-9684-6
M.A. Firer, G. Gellerman, J. Hematol. Oncol. 5 (2012) 70. doi: 10.1186/1756-8722-5-70
N. Vale, D. Duarte, S. Silva, et al., Pharmacol. Res. 162 (2020) 105231. doi: 10.1016/j.phrs.2020.105231
S. Dissanayake, W.A. Denny, S. Gamage, V. Sarojini, J. Control. Release 250 (2017) 62–76. doi: 10.1016/j.jconrel.2017.02.006
L. Ge, X. You, K. Huang, et al., Biomater. Sci. 6 (2018) 125–135. doi: 10.1039/C7BM00776K
X. Sun, H. Li, Z. Chen, et al., Nucleic Acids Res. 53 (2025) D1476–D1485. doi: 10.1093/nar/gkae859
J. Zhou, N. Meng, L. Lu, et al., J. Control. Release 369 (2024) 722–733. doi: 10.1016/j.jconrel.2024.04.011
J. Pan, Y. Lai, S. Zhang, et al., Adv. Mater. 35 (2023) e2305798. doi: 10.1002/adma.202305798
Y. Cao, X. Ge, Y. Wei, et al., Chin. Chem. Lett. 35 (2024) 108672. doi: 10.1016/j.cclet.2023.108672
V. Akbari, C.P. Chou, D. Abedi, Biochim. Biophys. Acta. Rev. Cancer 1874 (2020) 188448. doi: 10.1016/j.bbcan.2020.188448
T. Todorovski, D. Kalafatovic, D. Andreu, Pharmaceutics 15 (2023) 357. doi: 10.3390/pharmaceutics15020357
D.A. Mendonça, M. Bakker, C. Cruz-Oliveira, et al., Bioconjug. Chem. 32 (2021) 1067–1077. doi: 10.1021/acs.bioconjchem.1c00123
T. Todorovski, D.A. Mendonça, L.O. Fernandes-Siqueira, et al., Pharmaceutics 14 (2022) 738. doi: 10.3390/pharmaceutics14040738
N. Kaur, P. Popli, N. Tiwary, R. Swami, J. Control. Release 355 (2023) 417–433. doi: 10.1016/j.jconrel.2023.01.032
M. Srinivasarao, C.V. Galliford, P.S. Low, Nat. Rev. Drug Discov. 14 (2015) 203–219. doi: 10.1038/nrd4519
C. Zhuang, X. Guan, H. Ma, et al., Eur. J. Med. Chem. 163 (2019) 883–895. doi: 10.1016/j.ejmech.2018.12.035
Q. Chen, Z. Wu, H. Zhu, et al., J. Med. Chem. 67 (2024) 19586–19611. doi: 10.1021/acs.jmedchem.4c01910
C.F. Lo, T.Y. Chiu, Y.T. Liu, et al., J. Med. Chem. 65 (2022) 12802–12824. doi: 10.1021/acs.jmedchem.2c00631
X. Wang, J. Zhang, K. Zheng, et al., J. Pharm. Anal. 13 (2023) 776–787. doi: 10.1016/j.jpha.2023.02.010
J. He, Q. Duan, C. Ran, et al., Acta Pharm. Sin. B 13 (2023) 1358–1370. doi: 10.1016/j.apsb.2023.01.017
S. He, Y. Du, H. Tao, H. Duan, Int. J. Biol. Macromol. 238 (2023) 124173. doi: 10.1016/j.ijbiomac.2023.124173
R. Thevendran, S. Sarah, T.H. Tang, M. Citartan, J. Control. Release 323 (2020) 530–548. doi: 10.1016/j.jconrel.2020.04.051
C. Yang, Y. Jiang, S.H. Hao, et al., J. Mater. Chem. B 10 (2021) 20–33.
Y. Liu, Y. Hu, Y. Tan, et al., Chin. Chem. Lett. 36 (2025) 110289. doi: 10.1016/j.cclet.2024.110289
V. Bagalkot, O.C. Farokhzad, R. Langer, S. Jon, Angew. Chem. Int. Ed. 45 (2006) 8149–8152. doi: 10.1002/anie.200602251
Y. Hu, J. Duan, Q. Zhan, et al., PLoS One 7 (2012) e31970. doi: 10.1371/journal.pone.0031970
Z. Liu, J.H. Duan, Y.M. Song, et al., J. Transl. Med. 10 (2012) 148. doi: 10.1186/1479-5876-10-148
Z. Su, D. Xiao, F. Xie, et al., Acta Pharm. Sin. B 11 (2021) 3889–3907. doi: 10.1016/j.apsb.2021.03.042
S. Kumari, S. Raj, M.A. Babu, et al., Arch. Pharm. Res. 47 (2024) 40–65. doi: 10.1007/s12272-023-01479-6
M. Alas, A. Saghaeidehkordi, K. Kaur, J. Med. Chem. 64 (2021) 216–232. doi: 10.1021/acs.jmedchem.0c01530
H. Wang, L. Wang, Y. Gao, Y. Ding, Chin. Chem. Lett. 32 (2021) 1041–1045. doi: 10.1016/j.cclet.2020.08.044
N. Dan, S. Setua, V.K. Kashyap, et al., Pharmaceuticals 11 (2018) 32. doi: 10.3390/ph11020032
F. Bryden, C. Martin, S. Letast, et al., Org. Biomol. Chem. 16 (2018) 1882–1889. doi: 10.1039/C7OB02780J
Q. Song, X. Chuan, B. Chen, et al., Drug Deliv. 23 (2016) 1734–1746.
S. Jaracz, J. Chen, L.V. Kuznetsova, I. Ojima, Bioorg. Med. Chem. 13 (2005) 5043–5054. doi: 10.1016/j.bmc.2005.04.084
X. Lin, L. Wang, L. Zhao, et al., Food Funct. 11 (2020) 4146–4159. doi: 10.1039/D0FO00260G
G. Wu, Y.Z. Fang, S. Yang, et al., J. Nutr. 134 (2004) 489–492. doi: 10.1093/jn/134.3.489
J.P.M. António, J.I. Carvalho, A.S. André, et al., Angew. Chem. Int. Ed. 60 (2021) 25914–25921. doi: 10.1002/anie.202109835
L. Meng, K. Sefah, M.B. O'Donoghue, et al., PLoS One 5 (2010) e14018. doi: 10.1371/journal.pone.0014018
M. Walles, A. Connor, D. Hainzl, Curr. Top. Med. Chem. 17 (2017) 3463–3475.
Z.X. Phuna, P.A. Kumar, E. Haroun, et al., Life Sci. 347 (2024) 122676. doi: 10.1016/j.lfs.2024.122676
P. Khongorzul, C.J. Ling, F.U. Khan, et al., Mol. Cancer Res. 18 (2020) 3–19. doi: 10.1158/1541-7786.MCR-19-0582
L. Conilh, L. Sadilkova, W. Viricel, C. Dumontet, J. Hematol. Oncol. 16 (2023) 3. doi: 10.1186/s13045-022-01397-y
Y.T. Tai, P.A. Mayes, C. Acharya, et al., Blood 123 (2014) 3128–3138. doi: 10.1182/blood-2013-10-535088
A. Gazzah, P.L. Bedard, C. Hierro, et al., Ann. Oncol. 33 (2022) 416–425. doi: 10.1016/j.annonc.2021.12.012
H. Suzuki, S. Nagase, C. Saito, et al., Mol. Cancer Ther. 23 (2024) 257–271.
X. Zhang, D. Li, J. Huang, et al., J. Mater. Chem. B 7 (2019) 251–264. doi: 10.1039/C8TB02474J
K.C. Nicolaou, S. Rigol, Acc. Chem. Res. 52 (2019) 127–139. doi: 10.1021/acs.accounts.8b00537
M.J. McPherson, A.D. Hobson, A. Hernandez Jr., et al., Sci. Transl. Med. 16 (2024) eadd8936. doi: 10.1126/scitranslmed.add8936
T. Wieland, H. Faulstich, Experientia 47 (1991) 1186–1193. doi: 10.1007/BF01918382
P. Pal, P. Zhang, S.K. Poddar, G. Zheng, Expert Opin. Ther. Pat. 32 (2022) 1003–1026. doi: 10.1080/13543776.2022.2116311
K. Holen, L.B. Saltz, E. Hollywood, et al., Invest. New Drugs 26 (2008) 45–51. doi: 10.1007/s10637-007-9083-2
S. Gervasoni, I. Öztürk, C. Guccione, et al., J. Chem. Inf. Model. 63 (2023) 4924–4933. doi: 10.1021/acs.jcim.3c00712
R. Lengacher, A. Marlin, D. Śmiłowicz, E. Boros, Chem. Soc. Rev. 51 (2022) 7715–7731. doi: 10.1039/D2CS00407K
T.J. Wadas, E.H. Wong, G.R. Weisman, C.J. Anderson, Chem. Rev. 110 (2010) 2858–2902. doi: 10.1021/cr900325h
S. Sheikhbahaei, M.S. Sadaghiani, S.P. Rowe, L.B. Solnes, AJR Am. J. Roentgenol. 217 (2021) 495–506. doi: 10.2214/AJR.20.23349
N. Vahidfar, S. Farzanehfar, M. Abbasi, et al., Cancers 14 (2022) 1914. doi: 10.3390/cancers14081914
H. Maecker, V. Jonnalagadda, S. Bhakta, et al., mAbs 15 (2023) 2229101. doi: 10.1080/19420862.2023.2229101
J.K. McGavin, C.M. Spencer, Drugs 61 (2001) 1317–1322. doi: 10.2165/00003495-200161090-00007
S.H. Petersdorf, K.J. Kopecky, M. Slovak, et al., Blood 121 (2013) 4854–4860. doi: 10.1182/blood-2013-01-466706
J. Lambert, C. Pautas, C. Terré, et al., Haematologica 104 (2019) 113–119. doi: 10.3324/haematol.2018.188888
H.M. Kantarjian, D.J. DeAngelo, M. Stelljes, et al., N. Engl. J. Med. 375 (2016) 740–753. doi: 10.1056/NEJMoa1509277
A.D. Ricart, Clin. Cancer Res. 17 (2011) 6417–6427. doi: 10.1158/1078-0432.CCR-11-0486
N. Zein, A.M. Sinha, W.J. McGahren, G.A. Ellestad, Science 240 (1988) 1198–1201. doi: 10.1126/science.3240341
S. Girish, M. Gupta, B. Wang, et al., Cancer Chemother. Pharmacol. 69 (2012) 1229–1240. doi: 10.1007/s00280-011-1817-3
S. Verma, D. Miles, L. Gianni, et al., N. Engl. J. Med. 367 (2012) 1783–1791. doi: 10.1056/NEJMoa1209124
N.W. van de Donk, E. Dhimolea, mAbs 4 (2012) 458–465. doi: 10.4161/mabs.20230
J.A. Francisco, C.G. Cerveny, D.L. Meyer, et al., Blood 102 (2003) 1458–1465. doi: 10.1182/blood-2003-01-0039
A. Younes, A.K. Gopal, S.E. Smith, et al., J. Clin. Oncol. 30 (2012) 2183–2189. doi: 10.1200/JCO.2011.38.0410
B. Pro, R. Advani, P. Brice, et al., J. Clin. Oncol. 30 (2012) 2190–2196. doi: 10.1200/JCO.2011.38.0402
D. Dornan, F. Bennett, Y. Chen, et al., Blood 114 (2009) 2721–2729.
L.H. Sehn, A.F. Herrera, C.R. Flowers, et al., J. Clin. Oncol. 38 (2020) 155–165.
E.Y. Yu, D.P. Petrylak, P.H. O'Donnell, et al., Lancet Oncol. 22 (2021) 872–882. doi: 10.1016/S1470-2045(21)00094-2
Y. Ogitani, K. Hagihara, M. Oitate, et al., Cancer Sci. 107 (2016) 1039–1046. doi: 10.1111/cas.12966
S. Modi, C. Saura, T. Yamashita, et al., N. Engl. J. Med. 382 (2020) 610–621. doi: 10.1056/NEJMoa1914510
K. Shitara, Y.J. Bang, S. Iwasa, et al., N. Engl. J. Med. 382 (2020) 2419–2430. doi: 10.1056/NEJMoa2004413
U. Hennrich, K. Kopka, Pharmaceuticals 12 (2019) 114. doi: 10.3390/ph12030114
S. Dhillon, Drugs 81 (2021) 963–969. doi: 10.1007/s40265-021-01522-0
U. Hennrich, M. Eder, Pharmaceuticals 15 (2022) 1292. doi: 10.3390/ph15101292
I. Vergote, C.P. Leamon, Ther. Adv. Med. Oncol. 7 (2015) 206–218. doi: 10.1177/1758834015584763
J.Y. Park, Y.L. Cho, J.R. Chae, et al., Mol. Ther. Nucleic Acids 12 (2018) 543–553. doi: 10.1016/j.omtn.2018.06.003
S. Cavdarli, S. Groux-Degroote, P. Delannoy, Biomolecules 9 (2019) 311. doi: 10.3390/biom9080311
D.V. Kalinovsky, I.V. Kholodenko, A.V. Kibardin, et al., Int. J. Mol. Sci. 24 (2023) 1239. doi: 10.3390/ijms24021239
I. Nessler, E. Khera, S. Vance, et al., Cancer Res. 80 (2020) 1268–1278. doi: 10.1158/0008-5472.CAN-19-2295
L. Jolivet, I. Ait Mohamed Amar, C. Horiot, et al., Pharmaceutics 14 (2022) 114063.
A.S. André, J.N.R. Dias, S. Aguiar, et al., Sci. Rep. 13 (2023) 4837. doi: 10.1038/s41598-023-31568-x
L. He, L. Wang, Z. Wang, et al., J. Med. Chem. 64 (2021) 15716–15726. doi: 10.1021/acs.jmedchem.1c00961
S.Y.Y. Ha, Y. Anami, C.M. Yamazaki, et al., Mol. Cancer Ther. 21 (2022) 1449–1461. doi: 10.1158/1535-7163.MCT-22-0362
D.A. Rose, J.W. Treacy, Z.J. Yang, et al., J. Am. Chem. Soc. 144 (2022) 6050–6058. doi: 10.1021/jacs.2c01136
D. Wei, Y. Mao, H. Wang, et al., Chin. Chem. Lett. 34 (2023) 108091. doi: 10.1016/j.cclet.2022.108091
Z. Ma, H. He, F. Sun, et al., J. Cancer Res. Clin. Oncol. 143 (2017) 1929–1940. doi: 10.1007/s00432-017-2436-0
F. Sun, Y. Wang, X. Luo, et al., Cancer Res. 79 (2019) 3395–3405.
K. Johnson, J.C. Delaney, T. Guillard, et al., PLoS Pathog. 19 (2023) e1011612. doi: 10.1371/journal.ppat.1011612
C.R. Raetz, C. Whitfield, Annu. Rev. Biochem. 71 (2002) 635–700. doi: 10.1146/annurev.biochem.71.110601.135414
A. Han, O. Olsen, C. D'Souza, et al., J. Med. Chem. 64 (2021) 11958–11971. doi: 10.1021/acs.jmedchem.1c00541
D.N. Louis, A. Perry, G. Reifenberger, et al., Acta Neuropathol. 131 (2016) 803–820. doi: 10.1007/s00401-016-1545-1
E.I. Deryugina, M.A. Bourdon, R.A. Reisfeld, A. Strongin, Cancer Res. 58 (1998) 3743–3750.
D.V. Hingorani, C.N. Lippert, J.L. Crisp, et al., PLoS One 13 (2018) e0198464. doi: 10.1371/journal.pone.0198464
D. Hua, L. Tang, W. Wang, et al., Adv. Sci. 8 (2021) 2001960. doi: 10.1002/advs.202001960
A. Montero, F. Fossella, G. Hortobagyi, V. Valero, Lancet Oncol. 6 (2005) 229–239. doi: 10.1016/S1470-2045(05)70094-2
G.S. Karagiannis, J.M. Pastoriza, Y. Wang, et al., Sci. Transl. Med. 9 (2017) eaan0026. doi: 10.1126/scitranslmed.aan0026
C. Li, J. Lang, Y. Wang, et al., Acta Pharm. Sin. B 13 (2023) 3849–3861. doi: 10.1016/j.apsb.2023.03.024
M.J. Hinner, R.S.B. Aiba, T.J. Jaquin, et al., Clin. Cancer Res. 25 (2019) 5878–5889. doi: 10.1158/1078-0432.CCR-18-3654
Y. Zeng, X. Liao, Y. Guo, et al., J. Control. Release 366 (2024) 838–848. doi: 10.1016/j.jconrel.2023.12.034
M. Bocci, A. Zana, L. Principi, et al., J. Control. Release 367 (2024) 779–790. doi: 10.1016/j.jconrel.2024.02.014
T.U. Amin, R. Emara, A. Pal, et al., J. Med. Chem. 65 (2022) 15473–15486. doi: 10.1021/acs.jmedchem.2c01423
A. Pal, W. Albusairi, F. Liu, et al., Mol. Pharm. 16 (2019) 3237–3252. doi: 10.1021/acs.molpharmaceut.9b00432
Y. Zheng, R. Xu, H. Cheng, W. Tai, Mol. Ther. 32 (2024) 1048–1060. doi: 10.1016/j.ymthe.2024.02.020
T.J. Dougherty, C.J. Gomer, B.W. Henderson, et al., J. Natl. Cancer Inst. 90 (1998) 889–905. doi: 10.1093/jnci/90.12.889
K. Lu, C. He, W. Lin, J. Am. Chem. Soc. 136 (2014) 16712–16715. doi: 10.1021/ja508679h
N.M. Idris, M.K. Gnanasammandhan, J. Zhang, et al., Nat. Med. 18 (2012) 1580–1585. doi: 10.1038/nm.2933
Y. Yang, J. Liu, X. Sun, et al., Nano Res. 9 (2016) 139–148. doi: 10.1007/s12274-015-0898-4
S. Wilhelm, A.J. Tavares, Q. Dai, et al., Nat. Rev. Mater. 1 (2016) 16014. doi: 10.1038/natrevmats.2016.14
D. Ling, M.J. Hackett, T. Hyeon, Nat. Mater. 13 (2014) 122–124. doi: 10.1038/nmat3860
Y. Yang, W. Zhu, L. Cheng, et al., Biomaterials 246 (2020) 119971. doi: 10.1016/j.biomaterials.2020.119971
T.P. Szatrowski, C.F. Nathan, Cancer Res. 51 (1991) 794–798.
P.T. Schumacker, Cancer Cell 10 (2006) 175–176. doi: 10.1016/j.ccr.2006.08.015
H. Ranji-Burachaloo, P.A. Gurr, D.E. Dunstan, G.G. Qiao, ACS Nano 12 (2018) 11819–11837. doi: 10.1021/acsnano.8b07635
Z. Zhou, J. Song, R. Tian, et al., Angew. Chem. Int. Ed. 56 (2017) 6492–6496. doi: 10.1002/anie.201701181
A. Rösler, G.W. Vandermeulen, H.A. Klok, Adv. Drug Deliv. Rev. 53 (2001) 95–108. doi: 10.1016/S0169-409X(01)00222-8
W. Xuan, Y. Xia, T. Li, et al., J. Am. Chem. Soc. 142 (2020) 937–944. doi: 10.1021/jacs.9b10755
A. Lee, M.B.A. Djamgoz, Cancer Treat. Rev. 62 (2018) 110–122. doi: 10.1016/j.ctrv.2017.11.003
J. He, T. Peng, Y. Peng, et al., J. Am. Chem. Soc. 142 (2020) 2699–2703. doi: 10.1021/jacs.9b10510
N.K. Tafreshi, M.L. Doligalski, C.J. Tichacek, et al., Molecules 24 (2019) 4341. doi: 10.3390/molecules24234341
T. Langbein, G. Chaussé, R.P. Baum, J. Nucl. Med. 59 (2018) 1172–1173. doi: 10.2967/jnumed.118.214379
A.P. Bidkar, S. Wang, K.N. Bobba, et al., Clin. Cancer Res. 29 (2023) 1916–1928. doi: 10.1158/1078-0432.CCR-22-3291
D. Sehlin, X.T. Fang, L. Cato, et al., Nat. Commun. 7 (2016) 10759. doi: 10.1038/ncomms10759
G. van Dongen, W. Beaino, A.D. Windhorst, et al., J. Nucl. Med. 62 (2021) 438–445. doi: 10.2967/jnumed.119.239558
T.E. Wuensche, N. Stergiou, I. Mes, et al., Theranostics 12 (2022) 7067–7079. doi: 10.7150/thno.73509
N. Kamran, P. Kadiyala, M. Saxena, et al., Mol. Ther. 25 (2017) 232–248. doi: 10.1016/j.ymthe.2016.10.003
S. Nigam, L. McCarl, R. Kumar, et al., Mol. Imaging Biol. 22 (2020) 685–694. doi: 10.1007/s11307-019-01427-1
A. Foster, S. Nigam, D.S. Tatum, et al., EBioMedicine 71 (2021) 103571. doi: 10.1016/j.ebiom.2021.103571

Figure 2 Structures of FDA-approved drug conjugates: AdcetrisⓇ (A), EnhertuⓇ (B), LutatheraⓇ (C), and PluvictoⓇ (D).

Figure 3 (A) Reaction scheme of the generation of scFv and minibody conjugates with monomethyl auristatin E (MMAE) or F (MMAF). (B) Polyacrylamide gel electrophoresis of the FDCs in reducing conditions. 1, molecular weight protein markers; 2, scFv; 3, scFv-MMAE; 4, scFv-MMAF; 5, minibody; 6, minibody-MMAE; 7, minibody-MMAF. (C) Direct enzyme-linked immunosorbent assay (ELISA) of minibodies/scFv fragments and minibody-based FDCs/scFv-based FDCs. (D) Viability of cell lines analyzed by MTT assay following 72 h incubation with scFv-MMAE and minibody-MMAE, scFv-MMAF and minibody-MMAF. Copied with permission [110]. Copyright 2023, Springer Nature.

Figure 5 (A) Illustration of the chemical structure and mechanism of SynB 3-PVGLIG-PTX. (B) The effect of SynB3‐PVGLIG‐PTX on cell proliferation in different GBM cells using the MTS assay. (C) In vivo bioluminescence images of the intracranial glioma xenograft nude mice. (D) Bioluminescent images of different organs. (E) MMP‐2 expression was efficiently down‐regulated by transfecting MMP‐2 siRNA in U87MG. (F) The weights of mice. (G) The bioluminescence quantification results on days 7, 14, 21, and 28 after implantation. (H) The concentration‐time profiles of PTX in different brain tissues. Copied with permission [123]. Copyright 2020, Wiley-VCH GmbH.

Figure 6 (A) Schematic diagram of 225Ac-DOTA-YS5 structure. (B) The distribution of 225Ac-DOTA-YS5 in organs collected from day 1 to day 17. (C) Cell-associated molecules of 225Ac-DOTA-YS5 in 22Rv1 and DU145 cells after the treatment for different timepoints. (D) MTT assay showing dose-dependent reduction of cell viability. (E) Liver and kidney function tests results showing increase in creatinine, blood urea nitrogen, and alkaline phosphatase. (F) iQID camera digital autoradiographic imaging for distribution of the225Ac-DOTA-YS5 in 22Rv1 xenograft tumor and healthy tissues studied from day 1 to day 7 after injections. Copied with permission [150]. Copyright 2023, American Association for Cancer Research.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:
