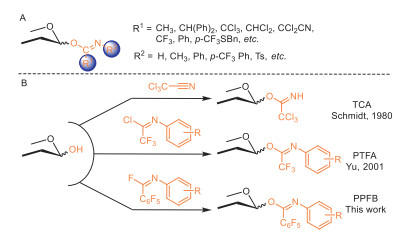
Figure 1.
Overview of glycosyl imidates. (A) Diversified substituents on the carbon and nitrogen atoms of different imidates. (B) Two representative glycosyl imidates and new design in this report.
Glycosyl N-phenyl pentafluorobenzimidates as a new generation of imidate donors for catalytic glycosylation
Xin Zhou , Guangyao Liu , Meifang Yang , Mengyu Li , Xiaodi Yang , Weiliang Gu , Yitian Zhao , Houchao Tao

The synthesis of carbohydrate has become pressingly demanded [1], given the expanding recognition of glycans' significant physiological roles [2] and the importance of glycosyl modifications in various biomolecules [3]. Additionally, carbohydrate-based drugs [4] hold remarkable pharmacological and therapeutic potential. However, the biosynthetic production of carbohydrates with precisely defined structures remains challenging because of their natural microheterogeneity [5]. Despite substantial progress in both chemical [6] and enzymatic synthesis [7] over the past decades, the synthesis of carbohydrates still lags behind that of oligopeptides and oligonucleotides, which can be produced with reliability and affordability through well-established methods. A primary obstacle in carbohydrate synthesis is the adequate and effective construction of glycosyl linkage.
Innovations in glycosylations are continually emerging [8], with glycosyl imidates standing out as pioneering donors for catalytic glycosylation [9]. Since their initial conception [10], the variety and utility of imidates have been greatly expanded (Fig. 1). Spectacularly, glycosyl trichloroacetimidate (TCA) has been instrumental in synthesizing a wide range of natural products [11]. TCA donors can be conveniently prepared in a single step by reacting hemiacetals with commercially available trichloroacetonitrile. However, despite improvements over the early acetimidates, TCA donors generally lack long-term stability and require careful handling, particularly with "armed" substrates. Another important class of imidates, glycosyl N-phenyl trifluoroacetimidate (PTFA), features a trifluoromethyl surrogate and a phenyl group attached to the nitrogen. These donors are produced from corresponding imidoyl chloride [12]. PTFA donors typically exhibit higher stability and effectively prevent back attack from the leaving moiety, which can interrupt the desired glycosylation process [13,14]. Interestingly, this design allows the preparation of the otherwise inaccessible ketosyl and ulosonyl imidates [15–17]. Additional imidates have been developed with various substituents, such as dichloromethyl [18], dichloro-cyanomethyl [19], and para-trifluoromethylthiopheny [20] groups on the carbon atom, and para-trifluoromethylphenyl, tosyl [21] on the nitrogen atom [22–30]. The ongoing evolution of imidates continuously introduces new merits, such as improved accessibility, stability, and tunability, through these innovations often require balancing structural modifications to meet specific synthetic needs.
Overview of known imidates highlights the critical role of electron-withdrawing groups (EWGs) on the carbon and/or nitrogen atoms. Recognizing the effectiveness of substituents like trichloromethyl and trifluoromethyl as EWGs, we envisioned that pentafluorophenyl could serve as a comparable EWG. Herein, a new type of imidate donor, glycosyl N-phenyl pentafluorobenzimidate (PPFB) was accordingly designed. PPFB donors were prepared from N-phenyl pentafluorobenzimidoyl fluoride (Fig. 1), produced via a clean and environmentally friendly photo-irradiated reaction between perfluorobenzene and phenyl isonitriles [31]. This method is noteworthy for its tolerance of various electronically substituents, including acid-labile groups, thus potentiating the reactivity tunability of the resulting donors. In comparison, traditional imidoyl chlorides are typically synthesized from corresponding amides under highly acidic conditions using phosphorous reagents [32] which are environmentally unfavored. It should be noted that the synthesis of N-phenyl pentafluorobenzimidoyl fluoride requires the use of excess hexafluorobenzene in benzene to introduce the pentafluorophenyl group. Fortunately, these volatile solvents can be recycled following the completion of the reaction. Moreover, imidoyl fluorides offer superior intrinsic stability compared to imidoyl chlorides. They are stable in air and moisture, and are odorless solids, facilitating easier storage and handling. Despite their stability, imidoyl fluorides retain sufficient reactivity to couple with glycosyl 1-OH (1) under condition similar to that used for preparing PTFA (Scheme 1). This reactivity, together with the stability, marks a significant advancement in the field of glycosylation chemistry.
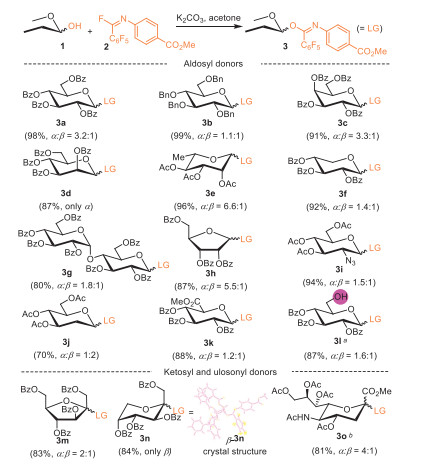
In this report, we utilized an ester-functionalized imidoyl fluoride (2) due to its nearly quantitative preparation [31]. A wide array of PPFB donors (3a-3o) were accordingly prepared, all in excellent yields (Scheme 1). Remarkably, beyond the common aldosyl donors (3a-3k), we successfully synthesized ketosyl and ulosonyl PPFB donors (3m-3o), including both furano- and pyrano-fructose, as well as sialic acid derivative, which are otherwise inaccessible as TCA donors [33]. It should be noted that the amount of imidoyl fluoride (3 equiv.) was less required than that of N-phenyl trifluoromethyl imidoyl chloride (10 equiv.) in the preparation of ketosyl donors [15]. It was also found that a Cs2CO3-DMSO system could accelerate this reaction and increase the yield. Additionally, we were able to synthesize a bifunctional PPFB donor (3l), or namely an imidate acceptor with a free 6‑hydroxy group, through a selective substitution reaction with the hemiacetal hydroxy group. This bifunctional donor, which is again unapproachable for TCA donor [34], is particularly useful in one-pot [35,36] glycosylation schemes. It can initially serve as an acceptor in the first glycosylation and subsequently as a donor. Such a unique feature of PPFB donors made it be possible to further distinguish them from TCA donors. This selectivity is likely due to the generally reduced reactivity of imidoyl fluoride in its interaction with alcohols, compared to its chloride counterpart and trichloroacetonitrile.
Unlike PTFA donor, which exhibits a miscellaneous configuration of the carbon-nitrogen double bond at ambient temperature, PPFB donors displayed clean 1H NMR spectra (Supporting information). The crystal structure of the frucotopyranosyl donor (β−3n, CCDC 2376950, Scheme 1) further confirms that the N-phenyl group is oriented syn to the pentafluophenyl group while anti to the bulky sugar moiety. Interestingly, the N-phenyl group lies within the plane of imidate (O–C=N), distinguishing it from that of PTFA donor [37]. Instead, the pentafluophenyl group is positioned perpendicular to this plane, likely due to favorable π-π stacking interaction with N-phenyl group. This structural elucidation highlights the structural clarity of the PPFB donors.
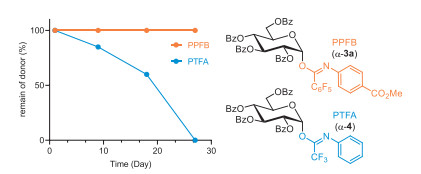
These PPFB donors demonstrated greater stability than their PTFA counterparts, as confirmed by 19F NMR evaluation over an extended period on the two perbenzoylated glucopyronosyl donors (Fig. 2). Over a test period of approximately four weeks, PPFB donor (3a) remained virtually intact after being stored at room temperature without special protection from air and moisture. In contrast, PTFA donor (4) showed significant decomposition, with a half-life of about 20 days.
With these PPFB donors prepared, we explored their glycosylation potential (Scheme 2). Using a typical promotor for imidates (TMSOTf, 0.05 equiv.), the glycosylation reactions proceeded efficiently with the isolation and characterization of the byproduct (7) through X-ray diffraction analysis (CCDC: 2376953). First, the glucopyronosyl donor (3a) was tested with a range of representative acceptors (5), resulting in O-glycosylation products (6a-6h) in good to excellent yields. This included secondary alcohols such as menthol and cholesterol, tertiary alcohols like 1-adamantanol, and even less nucleophilic phenols. Glycosylation of 3a appeared to be influenced by the electronic properties of the acceptor, with higher yields for electron-donating groups (99% for 6d with p-OMe phenol), compared to EWGs (77% for 6e with p-CO2Me phenol). It is important to note that neither of these reactions resulted in the formation of any C-glycoside products. Glycosylation with glycosyl acceptors also showed slight sensitivity to steric hindrance, achieving 95% yields for 6f and 6g with 6-OH acceptors, but a lower yield of 88% for 6 h with 2-OH acceptor. Furthermore, N-glycosylation (6i-6j) was investigated for conjugation with two typical nucleobases. Cytosine, after being pre-treated with N,O-bis(trimethylsilyl)trifluoroacetamide, was glycosylated quantitively (6j). A Boc-protected purine derivative was also glycosylated, though in a lower yield (6i, 40%). It should be noted that N-glycosylation of purines is rarely achieved with imidate donors like TCA and PTFA, due to the generally poor nucleophilicity and solubility of purines [38]. This successful glycosylation represents a significant advancement in the synthesis of glycosylated nucleosides by PPFB donors.
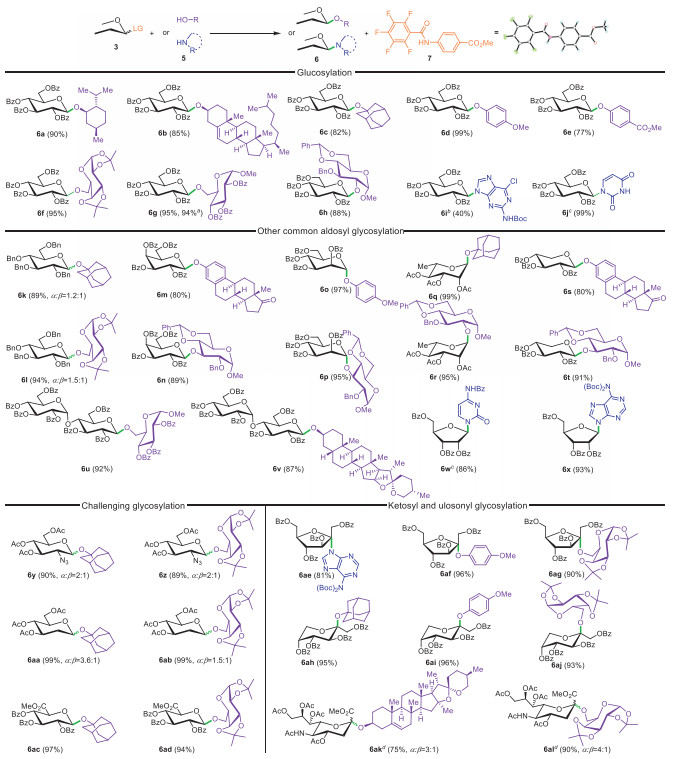
We further examined the glycosylation capabilities of many other aldosyl donors, including a per-benzylated glucosyl donor (3b), and donors derived from D-galactose (3c), D-mannose (3d), L-rhamnose (3e), L-xylose (3f), maltose (3g) and D-ribose (3h). These donors, consistently yielded O-glycosylation products in excellent yields when reacted with phenols and alcohol acceptors, either glycosyl or nonglycosyl. In addition, the N-glycosylation of the D-ribofuranosyl donor (3h) with both pyrimidine and purine derivatives also resulted in high yields. Besides these ordinary glycosyl bonds, we successfully constructed more challenging glycosyl bonds using glucopyranosyl donors with varied electronic effects, like 2-deoxy 2-N3 (3i), 2-deoxy (3j), and glucuronate (3k). These donors, whether "armed"or "disarmed" [39], when reacted with the galactosyl acceptor and 1-adamantanol, provided the corresponding products in high yields, albeit mostly as anomeric mixtures for the former two donors due to the absence of neighbouring participating groups. Furthermore, it should be highly noted that the coupling of ketosyl donors (3m-3o) [15–17] with various acceptors again produced the desired glycosides in satisfactory yields. The broad substrate scope, encompassing both donors and acceptors, underscores the versatility and general applicability of PPFB donors in the synthesis of complex molecules such as saponins [40], saccharides [41], nucleosides [42] and vaccines [43].
With the established methodology in place, we pursued the opportunity for PPFB donors in one-pot synthesis of glycans. Imidates, known for their high reactivity, are typically schemed as the initial donors in many orthogonally promoted one-pot synthesis [35]. For the same reason, achieving sufficient reactivity differences between imidates for chemoselective activation has been challenging. To date, only two studies have demonstrated sequential activation of two "armed" TCA and PTFA donors using Yb(OTf)3 [44] or Kass' thiourea catalyst [45]. In contrast, no precedent exists for the one-pot pairing of "disarmed" TCA and PTFA donors, likely due to the poor activity of "disarmed" donors and the reduced activity difference under harsher promoters. To address this gap, we focused on "disarmed" imidate donors and investigated the reactivity gradient of PPFB donors in comparison to their TCA and PTFA counterparts.
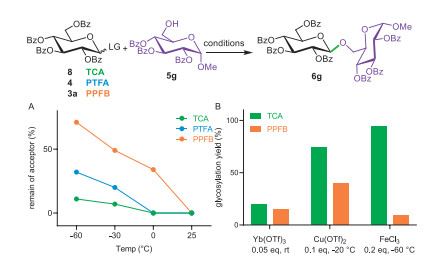
We compared the activities of a set of perbenzoylated glucosyl imidates—TCA (8), PTFA (4) and PPFB (3a)—under identical promotion conditions using TMSOTf at varying temperatues (Fig. 3A). The presence of acceptor (5g) could trap the generated oxonium ion, leading either to the glycosyl product (6g) or an orthoester. The activation of imidate was approximately correlated with the dissapearance of acceptor, as determined from the integration of the characteristic anomeric methoxy group in 1H NMR spectra. All imidates were fully activated at ambient temperature. However, as the temperture decreased, a reactivity gradient in reactivity among these imidates became evident. The TCA donor remained highly reactive even at −60 ℃, followed by the PTFA donor, while the PPFB donor showed the lowest reactivity. This descending order of imidate activity (TCA > PTFA > PPFB) suggested that PPFB donors, with their reduced reactivity and therefore greater differential from TCA donors, could serve as better candidates for new plan of one-pot glycosylation synthesis. Nevertheless, the reactivity difference under TMSOTf promotion was insufficient (the maximal difference was observed at −60 ℃, with 89% for TCA donor vs. 29% for PPFB donor). Additionally, glycosylation with the "disarmed" TCA donor at −60 ℃ predominantly resulted in orthoester formation (> 62%). To address this, we screened other promoters for TCA and PPFB donors (Fig. 3B). While Yb(OTf)3 efficiently activated "armed" TCA donors [44], it failed to drive complete glycosylation for both "disarmed" TCA and PPFB donors. In comparison, Cu(OTf)2 [46] facilitated glycosylation in good yield even at −20 ℃, but it also increased the yield for PPFB donor, thereby diminishing the reactivity difference. Ultimately, FeCl3 [47,48] demonstrated improved selectivity for glycosylation between these two imidates at −60 ℃, achieving a 94% yield for the TCA donor compared to just 9% for the PPFB donor.
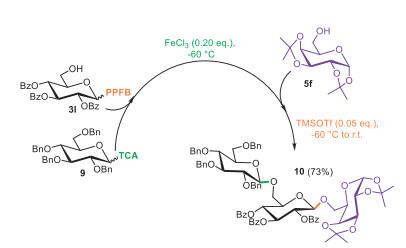
With the optimized selective promotion condition and the already available bifunctional building block (3l), we investigated the fully catalytic one-pot synthesis by combining imidates TCA and PPFB (Scheme 3). The first glycosylation reaction between TCA donor (9) and bifunctional intermediate (3l) proceeded smoothly, yielding β-disaccharide under the promotion of FeCl3 (0.2 equiv.) at −60 ℃ [47]. After approximately 0.5 h, once the donor (9) was fully consumed, acceptor (4f) and TMSOTf (0.05 equiv.) were added to the reaction mixture. The second glycosylation went complete within an additional hour during which the reaction was allowed to warm to ambient temperature. Finally, trisaccharide (10) was isolated with an overall yield of 73%. Mass spectrum analysis suggested that the lower yield was ascribed to the partial activation of PPFB donor during the first glycosylation. This synthesis demonstrates the practicality of PPFB donors in a fully catalytic one-pot synthesis. Future work will focus on further optimizing the promotion conditions, including organocatalysis [49] or transition metal catalysis [50,51] that have proven successful for imidate donors. Additionally, new chemical entities will be further introduced and refined to enhance the reactivity differential, thereby improving overall efficiency and yield.
In summary, we have developed a new generation of imidate donors, glycosyl N-phenyl pentafluorobenzimidates (PPFBs), for the efficient synthesis of glycans and glycal conjugates. PPFBs demonstrate broad substrate scope, accommodating comprehensively including all aldosyl, ketosyl and ulosonyl donors, as well as both O- and N-glycosylation acceptors. These donors are readily accessible through nuclephilic substitution on corresponding imidoyl fluorides, which are user-friendly, odorless and storable solid. Compared to imidoyl chlorides used for PTFA synthesis, imidoyl fluorides offer reduced reactivity, entailing selective reactions with the anomeric hydroxyl group and facilitating the preparation of bifunctional intermediate essential for one-pot synthesis strategies. Remarkably, PPFBs demonstrate even lower reactivity than PTFA, creating a greater reactivity difference from TCA donors, which is crucial for chemoselective one-pot synthesis. These benficial properties promise expanded applications of PPFBs in constructing glycosyl bonds in both natural and synthetic products, a potential we are continuing to explore in our laboratory.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Xin Zhou: Writing – original draft, Validation, Methodology, Formal analysis, Data curation. Guangyao Liu: Visualization, Validation, Investigation, Formal analysis, Data curation. Meifang Yang: Visualization, Validation, Methodology, Formal analysis, Data curation. Mengyu Li: Visualization, Validation, Methodology, Formal analysis, Data curation. Xiaodi Yang: Visualization, Validation, Software, Investigation. Weiliang Gu: Writing – original draft, Supervision, Resources, Investigation, Funding acquisition. Yitian Zhao: Visualization, Validation, Software, Project administration, Methodology, Investigation, Formal analysis, Data curation. Houchao Tao: Writing – review & editing, Supervision, Project administration, Funding acquisition, Conceptualization.
The authors are thankful to the National Natural Science Foundation of China (No. 21672147) and Shanghai University of Traditional Chinese Medicine for financial support. The authors gratefully acknowledge Prof. B. Yu for insightful instructions.
Supplementary material associated with this article can be found, in the online version, at doi:
I. Shin, K.S. Kim, Chem. Soc. Rev. 42 (2013) 4267–4269. doi: 10.1039/c3cs90030d
L. Su, Y. Feng, K. Wei, et al., Chem. Rev. 121 (2021) 10950–11029. doi: 10.1021/acs.chemrev.0c01338
C.R. Bertozzi, L.L. Kiessling, Science 291 (2001) 2357–2364.
X. Cao, X. Du, H. Jiao, et al., Acta Pharm. Sin. B 12 (2022) 3783–3821.
C.T. Campbell, K.J. Yarema, Genome Biol. 6 (2005) 236. doi: 10.1186/gb-2005-6-11-236
A. Malik, P.H. Seeberger, D. Varón Silva, Adv. Biochem. Eng. Biotechnol. 175 (2021) 201–230.
A.S. Rowan, C.J. Hamilton, Nat. Prod. Rep. 23 (2006) 412–443. doi: 10.1039/b409898f
X. Zhu, R.R. Schmidt, Angew. Chem. Int. Ed. 48 (2009) 1900–1934. doi: 10.1002/anie.200802036
M.M. Nielsen, C.M. Pedersen, Chem. Rev. 118 (2018) 8285–8358. doi: 10.1021/acs.chemrev.8b00144
J.R. Pougny, J.C. Jacquinet, M. Nassr, et al., J. Am. Chem. Soc. 99 (1977) 6762–6763. doi: 10.1021/ja00462a052
R.R. Schmidt, J. Michel, Angew. Chem. Int. Ed. 19 (1980) 731–732.
B. Yu, H. Tao, Tetrahedron Lett. 42 (2001) 2405–2407.
B. Yu, J. Sun, Chem. Commun. 46 (2010) 4668–4679. doi: 10.1039/c0cc00563k
U. Huchel, Dissertation, Universität Konstanz, 1998.
S. Cai, B. Yu, Org. Lett. 5 (2003) 3827–3830.
G. Lian, Q. Gao, F. Lin, Carbohydr. Res. 343 (2008) 2992–2996.
F. Lin, G. Lian, Q. Xu, et al., Tetrahedron 69 (2013) 1128–1132.
R.R. Schmidt, J. Michel, M. Roos, Ann. Liebigs Chem. 1984 (1984) 1343–1357. doi: 10.1002/jlac.198419840710
U. Schmelzer, Z. Zhang, R.R. Schmidt, J. Carbohydr. Chem. 26 (2007) 223–238. doi: 10.1080/07328300701410650
H. Chiba, S. Funasaka, T. Mukaiyama, Bull. Chem. Soc. Jpn. 76 (2003) 1629–1644.
S. Liu, P. Yannan, P. Wang, et al., Synlett 23 (2012) 1501–1504.
M. Palme, A. Vasella, Bioorg. Med. Chem. 2 (1994) 1169–1177.
S. Hanessian, J.J. Condé, B. Lou, Tetrahedron Lett. 36 (1995) 5865–5868.
U. Huchel, C. Schmidt, R.R. Schmidt, Eur. J. Org. Chem. 1998 (1998) 1353–1360.
A.V. Demchenko, N.N. Malysheva, C. De Meo, Org. Lett. 5 (2003) 455–458.
U. Huchel, P. Tiwari, R.R. Schmidt, J. Carbohydr. Chem. 29 (2010) 61–75. doi: 10.1080/07328301003597673
S.S. Nigudkar, A.R. Parameswar, P. Pornsuriyasak, et al., Org. Biomol. Chem. 11 (2013) 4068–4076. doi: 10.1039/c3ob40667a
S.S. Nigudkar, K.J. Stine, A.V. Demchenko, J. Am. Chem. Soc. 136 (2014) 921–923. doi: 10.1021/ja411746a
T. Tanaka, N. Kikuta, Y. Kimura, et al., Chem. Lett. 44 (2015) 846–848. doi: 10.1246/cl.150201
G. St-Pierre, S. Hanessian, Org. Lett. 18 (2016) 3106–3109. doi: 10.1021/acs.orglett.6b01263
A. Dewanji, C. Mück-Lichtenfeld, K. Bergander, et al., Chem. Eur. J. 21 (2015) 12295–12298. doi: 10.1002/chem.201502298
K. Tamura, H. Mizukami, K. Maeda, et al., J. Org. Chem. 58 (1993) 32–35. doi: 10.1021/jo00053a011
T. Müller, R. Schneider, R.R. Schmidt, Tetrahedron Lett. 35 (1994) 4763–4766.
C. Uriel, A.M. Gomez, J. Cristóbal López, et al., J. Carbohydr. Chem. 24 (2005) 665–675. doi: 10.1080/07328300500176528
S.S. Kulkarni, C.C. Wang, N.M. Sabbavarapu, et al., Chem. Rev. 118 (2018) 8025–8104. doi: 10.1021/acs.chemrev.8b00036
X. Zhong, X. Zhao, J. Ao, et al., Org. Chem. Front. 9 (2022) 4151–4157. doi: 10.1039/d2qo00727d
B. Yu, H. Tao, J. Org. Chem. 67 (2002) 9099–9102.
Q. Zhang, J. Sun, Y. Zhu, et al., Angew. Chem. Int. Ed. 50 (2011) 4933–4936. doi: 10.1002/anie.201100514
B. Fraser-Reid, J.C. López, Armed–disarmed effects in carbohydrate chemistry: history, synthetic and mechanistic studies, in: B. Fraser-Reid, J. Cristóbal López (Eds.), Reactivity Tuning in Oligosaccharide Assembly, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 1–29.
Z. Zhang, R. Wu, S. Cao, et al., Sci. Adv. 10 (2024) eadn1305.
D. Cai, F. He, S. Wu, et al., Carbohydr. Polym. 327 (2024) 121668.
X. Tang, Y. Zhou, Y. Wang, et al., J. Am. Chem. Soc. 146 (2024) 8768–8779. doi: 10.1021/jacs.4c01780
C. Jordan, K. Siebold, P. Priegue, et al., J. Am. Chem. Soc. 146 (2024) 15366–15375. doi: 10.1021/jacs.4c03179
M. Adinolfi, A. Iadonisi, A. Ravidà, Synlett 2006 (2006) 0583–0586.
K. Xiao, Y. Hu, Y. Wan, et al., Chem. Sci. 13 (2022) 1600–1607. doi: 10.1039/d1sc05772c
H. Park, H. min hee, F. Chang, et al., Bull. Kor. Chem. Soc. 34 (2013) 1339–1344. doi: 10.5012/bkcs.2013.34.5.1339
M.M. Mukherjee, N. Basu, R. Ghosh, RSC Adv. 6 (2016) 105589–105606.
F. Yang, W. Hou, D. Zhu, et al., Chem. Eur. J. 28 (2022) e202104002.
M.M. Nielsen, T. Holmstrøm, C.M. Pedersen, Angew. Chem. Int. Ed. 61 (2022) e202115394.
P. Peng, R.R. Schmidt, J. Am. Chem. Soc. 137 (2015) 12653–12659. doi: 10.1021/jacs.5b07895
J. Li, H.M. Nguyen, Acc. Chem. Res. 55 (2022) 3738–3751. doi: 10.1021/acs.accounts.2c00636

Figure 1 Overview of glycosyl imidates. (A) Diversified substituents on the carbon and nitrogen atoms of different imidates. (B) Two representative glycosyl imidates and new design in this report.

Scheme 1 Synthesis of PPFB donors. Standard conditions unless otherwise stated: 1 (1.0 mmol), 2 (1.2 mmol), K2CO3 (3.0 mmol), acetone (5 mL), r.t., 3 h. Isolated yield, and α:β ratio was determined by 1H NMR. a 1 (0.6 mmol), 2 (0.72 mmol), K2CO3 (3.0 mmol), acetone (3 mL), r.t., 3 h. b 1 (0.1 mmol), 2 (0.3 mmol), Cs2CO3 (0.4 mmol), DMSO (1 mL), r.t., 10 min.

Figure 2 Stability comparison between PPFB and PTFA donors. Remaining amount of each donor was determined by 19F NMR spectra after being stored without protection from air and moisture under ambient conditions (20 ℃, 60% RH) at indicated time.

Scheme 2 Scope of the glycosylation reaction with PPFB donors. Reagents and conditions: Standard conditions unless otherwise stated: 3 (0.12 mmol), 5 (0.10 mmol), TMSOTf (0.005 mmol), dry CH2Cl2 (2 mL), 4 Å MS (300 mg), r.t., 2 h. a Reaction was carried out in 1 mmol scale. b TMSOTf (0.3 equiv.) was used. c BSTFA (0.4 mmol), 5 (0.2 mmol), dry CH3CN (1 mL), 3 (0.1 mmol), TMSOTf (0.05 mmol), 4 Å MS (300 mg), r.t., 2 h. d 3 (0.075 mmol), 5 (0.05 mmol), TMSOTf (0.01 mmol), dry CH2Cl2/CH3CN (0.5 mL: 0.5 mL), 4 Å MS (150 mg), −65 ℃ to r.t., 2 h.

Figure 3 Reactivity comparison of imidates. (A) Activation profiles of imidates (8, 4, and 3a) under TMSOTf (0.05 equiv.) promotion at various temperatures. Imidate activity was inversely correlated with the remaining amount of acceptor (5g), as quantified from integration of 1H NMR spectra. (B) Glycosylation efficiency of imidates (8 and 3a) with acceptor (5g) under different promotion conditions. Yields were determined from 1H NMR spectra.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
