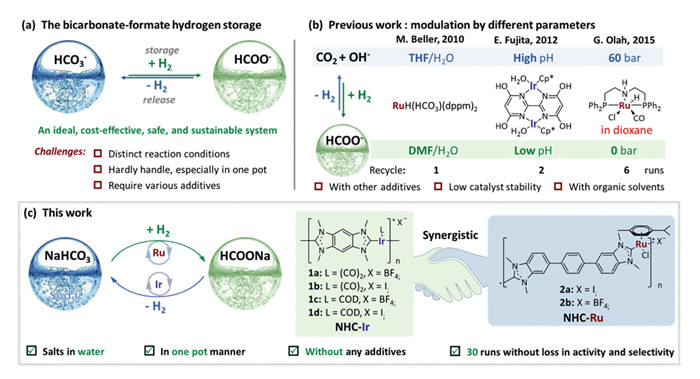
Figure 1.
Catalytic transformation approaches for the bicarbonate-formate hydrogen storage.
A rechargeable and portable hydrogen storage system grounded on soda water
Lingyun Shen , Shenxiang Yin , Qingshu Zheng , Zheming Sun , Wei Wang , Tao Tu

The US Department of Energy released three Energy Earthshots Initiatives in 2021, Hydrogen Shot, Long Duration Storage Shot and Carbon Negative Shot [1]. These Earthshots have been challenging the scientific community to develop practically useful technologies for green energy production, storage and application. Among the technologies developed to date, the bicarbonate-formate (HCO3− – HCO2−) interconversion stands out for the ability to make impacts on all three of these Earthshots since the chemistry can produce, store and release hydrogen (H2) [2-11]. Furthermore, aqueous solution of formate ions (HCO2−) in the bicarbonate–formate cycle constitutes an inexpensive, safe and easily handled chemical hydrogen carriers [2,3,7-11], in contrast to common liquid organic carriers [12-24], and thus provides substantial potential as a clean energy technology towards carbon neutrality [25-29].
Given these unrivaled features, and the high stability of the earth abundant salts of bicarbonate and formate, which can also be produced by the capture and utilization of CO2 [30,31], it becomes critical to develop cost-effective, environmentally friendly and robust strategies to produce H2 from the cycle, the core tactic in the technology. The establishment of such a viable reversible hydrogen storage system (Fig. 1a), however, presents daunting challenges [32]. In the core technology, the dehydrogenation of formates and hydrogenation of bicarbonates are generally carried out under the distinct reaction conditions [11,33-35], thus making operation complicated. Beller and colleagues demonstrated for the first time that the hydrogenation of CO2 and dehydrogenation of NaHCO3 could be conducted in the presence of [{RuCl2(benzene)}2] and 1,2-bis(diphenylphosphino)methane (dppm) by charging/discharging H2 in different organic solvents (DMF/H2O for hydrogenation and THF/H2O for dehydrogenation, respectively) under basic conditions (Fig. 1b) [35]. By modulating the pH value, Himeda et al. achieved the reversible hydrogen storage of formates/bicarbonates but with limited cycles (Fig. 1b) [36]. Prakash, Olah and coworkers then realized the reversible transformation with commercially available Ru pincer complexes in dioxane aqueous solution by varying the pressure swing of H2 (Fig. 1b) [37]. Despite these impressive advances, from a practical concern, a rechargeable hydrogen storage and liberation system based on formates/bicarbonates cycle under simple operation in aqueous phase without the help of organic solvent has not been established. A practical rechargeable hydrogen storage system should fulfill these criteria: (ⅰ) reversibility and simplicity: the H2 storage and release should be reversible in one pot/container and can be easily regulated by an operationally simple pressure swing of H2; (ⅱ) stability and reusability: the catalytic systems should be robust in a considerable time frame without loss of catalytic activities and selectivity, especially, recyclable and reusable for dozens of runs; (ⅲ) safe and environmental-friendliness: the system should ideally use only water without additional toxic organic solvents or additives.
Addressing the demanding challenges requires understanding the specific mechanisms of dehydrogenation of formates and hydrogenation of bicarbonates, especially, developing compatible catalytic systems that can tolerate the two distinct processes independently but in a cooperative manner and reduce catalyst deactivation in aqueous media. Recently, we reported the reductive amination of levulinic acid (LA) to unprotected lactam by using N-heterocyclic carbene metal (NHC-M, M = Ir, Ru) coordination assemblies 1 and 2 as catalysts [38], in which the ammonium formate was found to be partially decomposed along with hydrogen release, even without LA. Inspired by the unexpected discovery and our recent studies on valorization of biomass via hydrogenation and dehydrogenation with NHC-M catalysts [39-49], we envisioned that the NHC-M coordination assemblies might serve as suitable catalysts towards formates/bicarbonates equilibrium (Fig. 1c). Herein, we uncover a highly efficient rechargeable and portable hydrogen storage system with soda water by simply mixing two different NHC-Ir and NHC-Ru assemblies as the solid molecular catalysts in one pot. We observe that the aqueous sodium formate can readily release H2 in the presence of solid NHC-Ir catalyst, while, the resulting soda water (bicarbonate) is effectively hydrogenated by the solid NHC-Ru catalyst to complete the carbon neutral charge-discharge cycle of H2. This one-pot process is simply conducted by charging or releasing H2, no additional operation or treatment is required. The charge-discharge cycle of H2 could be repeated for >30 runs without obvious decrease of yield and selectivity. The efficiency, simplicity and robustness of our newly developed system highlight a great potential for the practical application of the rechargeable hydrogen storage system.
Based on our previous synthetic procedures for the fabrication of NHC-M (M = Ir, Ru) coordination assemblies [43-46], bis-benzimidazolium salts or p-phenylene-bridged bis-benzimidazolium salts were employed to react with iridium or ruthenium precursors under alkaline conditions, producing corresponding coordination assemblies 1a-d and 2a-b (Fig. 1), respectively. The obtained solids were insoluble in all tested organic solvents (DCM, MeOH, MeCN, THF, DMF, DMSO, etc.) and water. X-ray photoelectron spectroscopy (XPS) of catalysts 1a and 2a revealed the metal centers were IrⅠ and RuⅡ (Fig. S28 vs. Fig. S30 in Supporting information), respectively. Scanning electron microscopy (SEM), transmission electron microscopy (TEM), energy dispersive spectroscopy (EDS), and powder X-ray diffractions (XRD) indicated that the obtained catalysts were amorphous irregular solids with uniformly distributed single-site Ir or Ru atoms in the assemblies matrix (Figs. S3–S8, S13-S18, S23, S25, S32-S37 in Supporting information). The porosity of solid coordination assemblies was further characterized by N2 adsorption/desorption studies, which revealed that solid 2a (28 m2/g) had a higher Brunauer-Emmett-Teller (BET) specific surface area than 1a (8 m2/g), indicating their nonporous nature [38,44,50].
With these solid catalysts in hand, a number of commercially available formates with different cations (NH4+, Li+, K+ and Na+, 2 mol/L) were heated at 80 ℃ in the presence of 0.03 mol% of NHC-Ir coordination assemblies 1a for hydrogen liberation. Sodium formate was found to be the best hydrogen source, although its hydrogen content is not the highest, likely due to Na⁺'s ability to promote dehydrogenation (Fig. 2a) [51]. Notably, 20 mmol of sodium formate could be converted into sodium bicarbonate quantitatively, along with the liberation of ca. 500 mL of hydrogen gas within 6 h (measured with a gas burette). Further gas chromatography (GC) revealed that the liberated gas was free from CO or CO2 (< 10 ppm, below the detection limit, Fig. S2 in Supporting information), which was a prerequisite for use in present-day hydrogen fuel cells [11]. Thus, the generated hydrogen gas can be used directly for further energy applications.
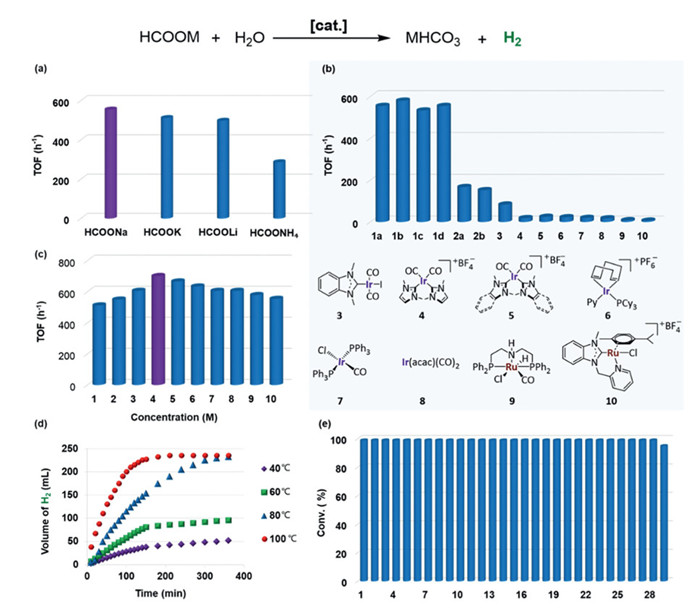
The catalytic efficiencies of different NHC-M coordination assemblies were then compared under the otherwise identical reaction conditions (Fig. 2b). Both the coordination assemblies NHC-Ir 1a-d and NHC-Ru 2a-b could achieve quantitative conversion of formate (Fig. S1 in Supporting information), but significantly higher TOF values were attained by the former Ir catalysts than the latter Ru catalysts (Fig. 2b). In consideration of that different auxiliary ligands and anions for the coordination assemblies NHC-Ir 1a-1d did not significantly affect their catalytic efficiency (TOF: 533–580 h−1) in this reaction, and that the solid catalyst 1a was readily prepared, it was chosen for further studies. Other viable homogenous catalysts 3–10 including mono- or bis-NHC-Ir complexes and the metal precursor Ir(acac)(CO)2 were also involved, which all led to inferior results (TOF: 6–83 h−1, Fig. 2b), highlighting the superiority of our solid molecular catalysts. These results were also consistent with our previous reports [49], confirming that the "self-supporting" strategy could effectively suppress the formation of inactive iridium dimers/clusters commonly occurred in homogeneous catalysis process [52].
We continued to optimize the concentration of sodium formate with 1a and found that the best concentration is 4 mol/L (TOF: 701 h−1, Fig. 2c). It was found that the conventional additive (DMSO or i-PrOH) showed a negative effect towards the dehydrogenation, all resulting in much lower TOF values (89 h−1 and 249 h−1, entries 1 and 2 in Table S1 in Supporting information). Decreasing the catalyst loading to 0.01 mol% also led to an inferior result (TOF: 156 h−1, entry 3, Table S1). Reaction profile studies revealed that elevating reaction temperature could effectively accelerate the H2 evolution efficiency (Fig. 2d). The H2 liberation amount reached the maximum within 5 h at 80 ℃, while lower temperature would dramatically slow down the reaction rate. The H2 evolution could be completed within 3 h at 100 ℃.
Arrhenius analysis on the initial rate of gas evolution as a function of temperature gave an activation energy of Ea = 33 ± 2 kJ/mol (calculation details see Supporting information), which was lower than the recorded value (41 ± 2 kJ/mol) [9,46], further confirmed that the high catalytic efficiency of the coordination assemblies NHC-Ir 1a. For the dehydrogenation of HCOONa, the TOF increased from 943 h−1 to 1020 h−1 when NaOH was added, whereas acidic conditions hampered the activity of 1a (TOF: 500 h−1 vs. 943 h−1), suggesting that basic conditions may favor formate dehydrogenation (Table S7 in Supporting information).
Deuterium labeling studies were carried out to get a further insight of the isotopic effects on the reaction rate of dehydrogenation (Table 1 and Fig. S49 in Supporting information). When sodium formate and/or water was replaced by their deuterated counterparts, the reaction rate decreased dramatically. The deuterium kinetic isotopic effect (KIE) value [36] obtained from DCOONa was higher than that with D2O (1.4 vs. 2.4, Table 1, entries 2 vs. 3), which suggests that the generation of an iridium-hydride intermediate (NHC-Ir-H) from the formate was the rate-limiting step for the dehydrogenation [53].
In light of the insolubility of the solid molecular catalysts in all test organic solvents and water, the catalysts were readily recovered by centrifugation after the reaction completion. The recyclability of the solid coordination assemblies 1a was investigated under the same optimum reaction conditions (Fig. 2e). Solid 1a could be reused for >25 times without any activation process. Even in the 29th run, 95% conversion of the substance was still observed within 24 h, and a full conversion could be achieved by extending reaction time to 30 h. The TOF values were measured for each run and the highest TOF value was 833 h−1 (Table S2 in Supporting information, and the cumulative TON for 29 runs can reach up to 96, 667). It worth to mention that the reaction mixture containing the solid coordination assemblies was stable at room temperature without any hydrogen gas evolution, indicating the H2 release could be simply controlled by temperature and thus making it possible for safe and practical transportation of H2.
The filtrates after each hydrogen liberation were analyzed by inductively coupled plasma atomic emission spectroscopy (ICP-AES, Table S3 in Supporting information) for potential iridium leaching. Trace amount of iridium leaching was observed in the initial 4 runs, which could be attributed to possible low molecular weight NHC-Ir oligomers entrapped in the solid matrix [40]. No further iridium leaching was detected and the high efficiency of the reaction was well sustained in the subsequent recycles, demonstrating the robustness of the catalysts, which was further confirmed by the similar morphology and particle size of the recovered catalysts with the freshly prepared catalysts (Fig. S3 vs. S9, Fig. S13 vs. S19, Fig. S32 vs. S38 in Supporting information). Additionally, XPS spectra showed the valence state of iridium was consistent with the newly prepared catalyst 1a (64.7 eV vs. 64.7 eV, 61.6 eV vs. 61.6 eV, Fig. S28 vs. S29 in Supporting information).
As one type of bi-functional catalysts with dehydrogenation and hydrogenation ability [38,43,44,47-49,52], the solid NHC-M coordination assemblies were then applied to the hydrogenation of soda water (Fig. 3) after being evaluated in the dehydrogenation of sodium formate. Initially, screening of coordination assemblies NHC-M revealed that the solid NHC-Ru 2a was the best catalyst, which showed quantitative conversion and 71% yield with 0.03 mol% catalyst loading in 2 mol/L soda water under 50 atm of H2 at 140 ℃ for 24 h (Fig. 3a). Inferior results were obtained with other viable homogenous catalysts 5–10 (3%−58% yield of formate, Fig. 3). Other (bi)carbonates with different cations were then evaluated under the otherwise identical conditions, but delivered inferior yields (3%−60%, Fig. 3b). Sodium bicarbonate (soda water) was therefore selected for the hydrogenation study, which was also beneficial for constructing a potential hydrogen storage system in one-pot.
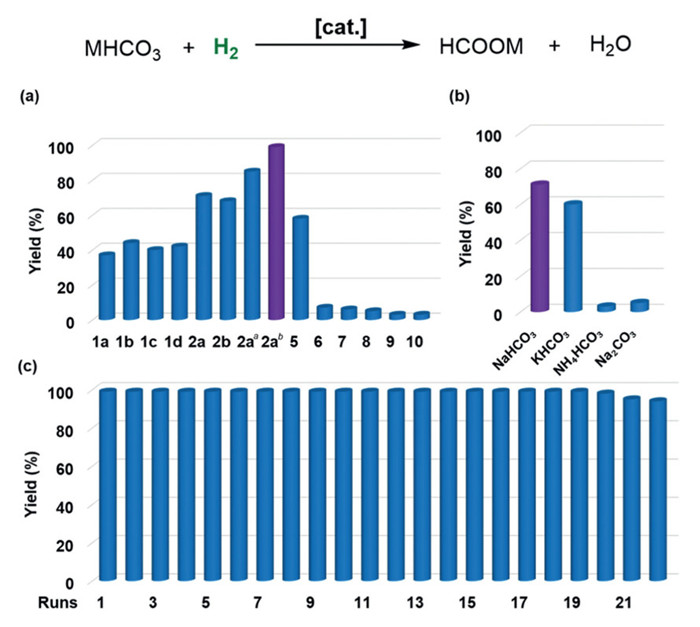
The concentration strongly effected the transformation, increasing or decreasing the concentration of sodium bicarbonate all led to inferior results (entries 1 and 2, Table S4 in Supporting information). When the reaction time was extended to 48 h and 72 h, the yields of sodium formate could reach 85% and >99%, respectively (Fig. 3a). Upon reducing the catalyst loading to 0.01 mol%, a satisfactory yield could also be achieved by extending reaction time to 120 h (82%, entry 4, Table S4). When reducing the pressure to 40 atm of H2, the yield dropped to 81% (Table S4, entry 5). Increasing or decreasing reaction temperature both led to lower yields (41%−92% yield of formate, entries 6–8 in Table S4). No product was formed without solid catalyst addition (entry 9, Table S4). The pH value also affected hydrogenation. Both acidic and basic conditions inhibited the transformation of bicarbonate (Table S8 in Supporting information). When 1 equiv. NaOH was added, the yield decreased from 99% to 78% (Table S8, entry 1), and when 1 equiv. CH₃COOH was added, a 90% yield of HCOONa was obtained (Table S8, entry 7).
After centrifugation and filtration, the solid molecular catalyst 2a was quantitatively recovered, which could be directly re-used in the second run of hydrogenation, simply by further charging with sodium bicarbonate and water, without any additional activation steps. The solid catalyst 2a could be reused for around 20 runs (Fig. 3c), and no significant loss in yield and selectivity of the sodium formate. The filtrates after each run were also analyzed by ICP-AES for potential ruthenium leaching. Similar to the aforementioned analytic results in dehydrogenation with solid catalyst 1a, except for the first 3 times, no further metal leaching was detected in the subsequent recycling runs (Table S5 in Supporting information).
In light of that different catalysts were privileged in the dehydrogenation and hydrogenation reactions, a solid mixture of coordination assemblies 1a and 2a in 1:1 mole ratio was directly applied as the catalyst in the hydrogen charging-discharging process in one-pot. As the dehydrogenation and hydrogenation reactions were both carried out under near neutral conditions without any additive, the charging-discharging process could be directly recycled without further pH modulation, additive addition or any further manipulation.
An autoclave reactor was charged with soda water (1 mol/L, 10 mL), a mixed catalyst of solids 1a (0.03 mol%), and 2a (0.03 mol%) under 50 bar H2, which was then heated at 140 ℃ for the hydrogenation process. After complete conversion, the autoclave was cooled to room temperature and then the remaining pure hydrogen gas was released. The autoclave was then attached to a gas burette and heated at 100 ℃. After the evolution of gas was stopped, the autoclave was cooled to room temperature, and around 248 mL of hydrogen gas was collected, which was close to the theoretical maximum hydrogen storage capacity of the sodium formate at room temperature (250.7 mL).
It is noteworthy that the same hydrogen storage and liberation cycle was also performed only with the single catalyst 1a or 2a under otherwise identical reaction conditions for comparison. However, though 1a achieved quantitative conversion in the dehydrogenation step, only 22% yield was obtained in the hydrogenation step (entry 11, Table S4). A contrary trend was observed for the catalyst 2a, in which 49% and 98% yields were achieved for the dehydrogenation and hydrogenation step, respectively. These results were significantly lower than those achieved with the mixture of solid catalysts 1a and 2a, suggesting that the two catalysts worked in a cooperative manner and thus endowing the catalyst mixture in one pot with superior catalytic performance than the single catalyst.
Notably, the released gas generated by the rechargeable system is pure H2, in which CO2 content is below the detection limitation (Fig. S2 in Supporting information), and could potentially be directly applied in a fuel cell without further purification [37,54]. Moreover, the fugitive hydrogen loss during dehydrogenation was only 1.2%, demonstrating the excellent gas-tightness of this hydrogen production system [55]. The mixture in the same autoclave could then be charged with H2 again and used in the second hydrogen storage and liberation cycle without any further treatment. We were able to repeat this charging/discharging recycling >30 times without significant decrease in the volume of hydrogen evolution (Fig. 4a). The deficiency after the 36th cycle was attributed to the accumulated loss of soda water caused by the inevitable liberation of trace CO2 in successive runs, rather than the solid catalyst deficiency. Indeed, upon recharging the recycled mixture of the two solid molecular catalysts to fresh soda water (the 37th run), the rechargeable hydrogen storage system re-exhibited full catalytic efficiency, and quantitative hydrogen evolution was still observed, confirming the robustness of the solid molecular catalysts.

SEM and TEM images demonstrated that the recovered catalysts were still irregular solid (Figs. 4b, c, e and f). EDX mapping (insets in Fig. 4f) indicated that the metal centers were uniformly well dispersed in the solid matrix without formation of nanoparticles, and the two catalysts were well mixed with each other. This is further supported by XRD spectra (Figs. S40 and S41 in Supporting information), in which no obvious diffraction peaks corresponding to metal nanoparticles were observed. XPS study revealed that the IrⅠ and RuⅡ barely changed their valence states after multiple runs of recycle (Figs. 4d and g), confirming the robustness of the solid molecular catalysts. In addition, no loss in storage efficiency was observed upon leaving the hydrogen storage system to stand for months. These results highlighted the great potential of our reversible hydrogen storage system for practical application.
The present work outlines a practical approach for achieving carbon-neutral hydrogen storage and release. By employing a combination of NHC-Ir and NHC-Ru coordination assemblies as solid molecular catalysts, we have successfully developed a straightforward and reversible chemical hydrogen storage and release system within soda water. This innovation not only validates the viability of utilizing soda water as a novel, safe, and cost-effective hydrogen storage medium but also streamlines the process by allowing for a seamless manipulation of the reversible charging and discharging cycle within a single system, without the need for adjustments to other parameters or the use of organic solvents. Notably, this storage system demonstrates remarkable stability and reusability, retaining its catalytic activity even after >30 cycles of reuse, and remaining stable for extended periods at room temperature. This robust, cost-effective, environmentally friendly, and reversible hydrogen storage and release system holds immense practical potential for advancing hydrogen fuel cell technology.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Lingyun Shen: Writing – original draft, Investigation, Data curation. Shenxiang Yin: Data curation. Qingshu Zheng: Writing – original draft, Data curation. Zheming Sun: Investigation, Data curation. Wei Wang: Investigation. Tao Tu: Writing – original draft, Investigation.
Financial support from the National Natural Science Foundation of China (No. 22271060). The Department of Chemistry at Fudan University and the School of Chemistry and Chemical Engineering at Ningxia University are gratefully acknowledged.
Supplementary material associated with this article can be found, in the online version, at doi:
Energy Earthshots Initiative.
M. Calabrese, D. Russo, A. di Benedetto, et al., Renew. Sustain. Energy Rev. 173 (2023) 113102–113115. doi: 10.1016/j.rser.2022.113102
A. Kumar, P. Daw, D. Milstein, Chem. Rev. 122 (2022) 385–441. doi: 10.1021/acs.chemrev.1c00412
A. Bahuguna, Y. Sasson, ChemSusChem 14 (2021) 1258–1283. doi: 10.1002/cssc.202002433
S. Chatterjee, I. Dutta, Y. Lum, et al., Energy Environ. Sci. 14 (2021) 1194–1246. doi: 10.1039/d0ee03011b
K. Sordakis, C. Tang, L.K. Vogt, et al., Chem. Rev. 118 (2018) 372–433. doi: 10.1021/acs.chemrev.7b00182
K. Nakajima, M. Tominaga, M. Waseda, et al., ACS Sustain. Chem. Eng. 7 (2019) 6522–6530. doi: 10.1021/acssuschemeng.8b04698
C. Guan, Y. Pan, E.P.L. Ang, et al., Green Chem. 20 (2018) 4201–4205. doi: 10.1039/c8gc02186d
Q. Bi, J. -D. Lin, Y.M. Liu, et al., Angew. Chem. Int. Ed. 53 (2014) 13583–13587. doi: 10.1002/anie.201409500
G. Papp, J. Csorba, G. Laurenczy, et al., Angew. Chem. Int. Ed. 50 (2011) 10433–10435. doi: 10.1002/anie.201104951
A. Boddien, F. Gärtner, C. Federsel, et al., Angew. Chem. Int. Ed. 50 (2011) 6411–6414. doi: 10.1002/anie.201101995
R. Sang, Z. Wei, Y. Hu, et al., Nat. Catal. 6 (2023) 543–550. doi: 10.1038/s41929-023-00959-8
D. Wei, X. Shi, H. Junge, et al., Nat. Commun. 14 (2023) 3726–3735. doi: 10.1038/s41467-023-39309-4
R. Qu, K. Junge, M. Beller, Chem. Rev. 123 (2023) 1103–1165. doi: 10.1021/acs.chemrev.2c00550
R. Verron, Puig, E., P. Sutra, et al., ACS Catal. 13 (2023) 5787–5794. doi: 10.1021/acscatal.3c00476
D. Wei, R. Sang, P. Sponholz, et al., Nat. Energy 7 (2022) 438–447. doi: 10.1038/s41560-022-01019-4
W. Peng, S. Liu, X. Li, et al., Chin. Chem. Lett. 33 (2022) 1403–1406. doi: 10.1016/j.cclet.2021.08.033
A.J. McNeece, K.A. Jesse, A.S. Filatov, et al., Chem. Commun. 57 (2021) 3869–3872. doi: 10.1039/d0cc08236h
S. Wang, S. Hou, C. Wu, et al., Chin. Chem. Lett. 30 (2019) 398–402. doi: 10.1016/j.cclet.2018.06.021
Y. Xie, P. Hu, Y. Ben-David, et al., Angew. Chem. Int. Ed. 58 (2019) 5105–5109. doi: 10.1002/anie.201901695
A. Kumar, T. Janes, N.A. Espinosa-Jalapa, et al., J. Am. Chem. Soc. 140 (2018) 7453–7457. doi: 10.1021/jacs.8b04581
X. Wang, Q. Meng, L. Gao, et al., Int. J. Hydrog. Energy 43 (2018) 7055–7071. doi: 10.1016/j.ijhydene.2018.02.146
J. Eppinger, K.W. Huang, ACS Energy Lett. (2017) 188–195. doi: 10.1021/acsenergylett.6b00574
S. Chatterjee, I. Dutta, Y. Lum, et al., Energy Environ. Sci. 14 (2021) 1194–1246. doi: 10.1039/d0ee03011b
S. Chatterjee, K.W. Huang, Nat. Commun. 11 (2020) 3287. doi: 10.1038/s41467-020-17203-7
C. Wei, R.R. Rao, J. Peng, et al., Adv. Mater. 31 (2019) 1806296. doi: 10.1002/adma.201806296
Z.P. Cano, D. Banham, S. Ye, et al., Nat. Energy 3 (2018) 279–289. doi: 10.1038/s41560-018-0108-1
M. Czaun, J. Kothandaraman, A. Goeppert, et al., ACS Catal. 6 (2016) 7475–7484. doi: 10.1021/acscatal.6b01605
S. Chu, A. Majumdar, Nature 488 (2012) 294–303. doi: 10.1038/nature11475
L. Lin, X. He, X.G. Zhang, et al., Angew. Chem. Int. Ed. 62 (2023) e202214959. doi: 10.1002/anie.202214959
W. Ma, S. Xie, X.G. Zhang, et al., Nat. Commun. 10 (2019) 892. doi: 10.1038/s41467-019-08805-x
O.Y. Gutiérrez, K. Grubel, J. Kothandaraman, et al., Green Chem. 25 (2023) 4222–4233. doi: 10.1039/d3gc00219e
F. Bertini, I. Mellone, A. Ienco, et al., ACS Catal. 5 (2015) 1254–1265. doi: 10.1021/cs501998t
C. Ziebart, C. Federsel, P. Anbarasan, et al., J. Am. Chem. Soc. 134 (2012) 20701–20704. doi: 10.1021/ja307924a
C. Federsel, R. Jackstell, A. Boddien, et al., ChemSusChem 3 (2010) 1048–1050. doi: 10.1002/cssc.201000151
J.F. Hull, Y. Himeda, W.H. Wang, et al., Nat. Chem. 4 (2012) 383–388. doi: 10.1038/nchem.1295
J. Kothandaraman, M. Czaun, A. Goeppert, et al., ChemSusChem 8 (2015) 1442–1451. doi: 10.1002/cssc.201403458
Z. Sun, J. Chen, T. Tu, Green Chem. 19 (2017) 789–794. doi: 10.1039/C6GC02591A
Y. Shen, Q. Zheng, Z.N. Chen, et al., Angew. Chem. Int. Ed. 60 (2021) 4125–4132. doi: 10.1002/anie.202011260
J. Wu, L. Shen, S. Duan, et al., Angew Chem. Int. Ed. 59 (2020) 13871–13878. doi: 10.1002/anie.202004174
J. Wu, L. Shen, Z.N. Chen, et al., Angew. Chem. Int. Ed. 59 (2020) 10421–10425. doi: 10.1002/anie.202002403
Y. Shen, Q. Zheng, H. Zhu, et al., Adv. Mater. 32 (2020) 1905950. doi: 10.1002/adma.201905950
J. Wang, J. Wu, Z.N. Chen, et al., J. Catal. 389 (2020) 337–344. doi: 10.1016/j.jcat.2020.06.004
L. Shen, Z.N. Chen, Q. Zheng, et al., ACS Catal. 11 (2021) 12833–12839. doi: 10.1021/acscatal.1c04354
J. Chen, J. Wang, T. Tu, Chem. Asian J. 13 (2018) 2559–2565. doi: 10.1002/asia.201800759
J. Chen, H. Zhu, J. Chen, et al., Chem. Asian J. 12 (2017) 2809–2812. doi: 10.1002/asia.201701303
Z. Lu, Q. Zheng, G. Zeng, et al., Sci. China Chem. 64 (2021) 1361–1366. doi: 10.1007/s11426-021-1017-0
S. Yang, Z. Lu, J. Ji, et al., Sci. China Chem. 67 (2024) 914–921. doi: 10.1007/s11426-023-1843-8
Z. Sun, Y. Liu, J. Chen, et al., ACS Catal. 5 (2015) 6573–6578. doi: 10.1021/acscatal.5b01782
C. Qian, Q. Zheng, J. Ji, et al., Chem. Catal. 3 (2023) 100787. doi: 10.1016/j.checat.2023.100787
T. Wonglakhon, P. Surawatanawong, Dalton Trans. 47 (2018) 17020–17031. doi: 10.1039/c8dt03283a
L.S. Sharninghausen, J. Campos, M.G. Manas, et al., Nat. Commun. 5 (2014) 5084–5092. doi: 10.1038/ncomms6084
S. Kar, M. Rauch, G. Leitus, et al., Nat. Catal. 4 (2021) 193–201. doi: 10.1038/s41929-021-00575-4
X. Xu, Q. Zhou, D. Yu, Int. J. Hydrog. Energy 47 (2022) 33677–33698. doi: 10.1016/j.ijhydene.2022.07.261
I. Dutta, R. Parsapur, S. Chatterjee, et al., ACS Energy Lett. 8 (2023) 3251–3257. doi: 10.1021/acsenergylett.3c01098

Figure 1 Catalytic transformation approaches for the bicarbonate-formate hydrogen storage.

Figure 2 Screening of experimental variables and the reusability of 1a for dehydrogenation of formates. Optimization of (a) formates, (b) catalysts, (c) concentration of sodium formate, and (d) reaction temperature for the dehydrogenation of formate aqueous solution under the otherwise identical reaction conditions. Conversion was determined by 1H NMR data with NaOAc·3H2O as an internal standard. TOF was calculated based on hydrogen gas evolution quantified by gas burette and further confirmed by 1H NMR spectra of the reaction mixture. (e) The reusability of the NHC-Ir coordination assemblies 1a in dehydrogenation reactions of sodium formate: 20 mmol scale with 0.03 mol% 1a, 5 mL H2O at 100 ℃ for 24 h in each run.

Figure 3 The optimization of reaction conditions for hydrogenation of (bi)carbonates and the reusability of catalyst 2a. The yield of formate in the hydrogenation reaction of NaHCO3 with (a) different solid catalysts, and (b) different (bi)carbonates. Reactions were carried out with 20 mmol sodium bicarbonate, 0.03 mol% [cat.], 10 mL H2O in the presence of 50 bar H2 at 140 ℃ for 24 h. Yield was determined by 1H NMR spectroscopy with NaOAc·3H2O as an internal standard. a For 48 h. b For 72 h. (c) The reusability of the coordination assemblies NHC-Ru 2a in the hydrogenation reaction of soda water.

Figure 4 The rechargeable hydrogen storage system and characterizations of freshly prepared and recovered mixture of catalysts 1a and 2a. (a) The rechargeable hydrogen storage system involving 10 mL H2O, 0.03 mol% 1a and 2a. The hydrogenation reactions of soda water were carried out under 50 atm of H2 at 140 ℃ for 72 h. The dehydrogenation reactions of sodium formate were carried out in air at 80 ℃ for 24 h. Yield was determined by 1H NMR spectroscopy with NaOAc·3H2O as an internal standard. (b) SEM, (c) TEM (with EDX mapping as insets), and (d) XPS spectra of IrⅠ in the freshly prepared mixed catalysts and recovered mixed catalysts. (e) SEM, (f) TEM (with EDX mapping as insets), and (g) XPS spectra of RuⅡ in the freshly prepared mixed catalysts and recovered mixed catalysts.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载: