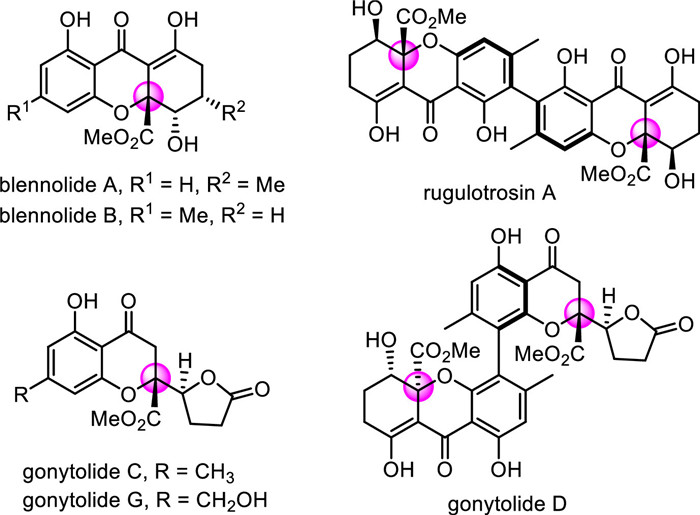
Scheme 1.
Representative examples of chromanone lactones and tetrahydroxanthones.
Asymmetric conjugated addition of aryl Grignard reagents for the construction of chromanones bearing quaternary stereogenic centers in batch and flow
Ya-Ling Li , Jia-Wei Ke , Yue Liu , Dong-Mei Yao , Jing-Dong Zhang , You-Cai Xiao , Fen-Er Chen

The chromanone lactones and tetrahydroxanthones are a class of fungal secondary metabolites with high structural and stereochemical complexity [1-5], such as blennolide A and C, rugulotrosin A, gonytolide C, D and G (Scheme 1). Many of them show attractive biological properties, including antitumor, antibacterial, antimalarial and anti-HIV activities [6-8]. In view of their notable structural features and broad-spectrum biological activities, the total synthesis of these natural products has drawn great interest [9-13]. The asymmetric construction of the quaternary stereocenter at the C2 position of chromanone scaffold is one of the major challenges of total synthesis. Shibasaki [14, 15] and Feng's [16] group independently reported the copper-catalyzed asymmetric vinylogous addition of siloxyfurans or butenolides to 2-ester-substituted chromones, affording the corresponding chromanone lactones in excellent diastereo- and enantioselectivities (Scheme 2a). Mattson's group also disclosed an enantioselective alkynylation of 2-ester chromones to build the tertiary ether stereocenters (Scheme 2b) [17, 18]. Nevertheless, direct methods to install the chiral quaternary stereocenter at the C2 position of chromanone are still rare. Thus, it is highly desirable to develop new protocols for the construction of chromanone derivatives comprising oxygen-containing tetra stereocenters.
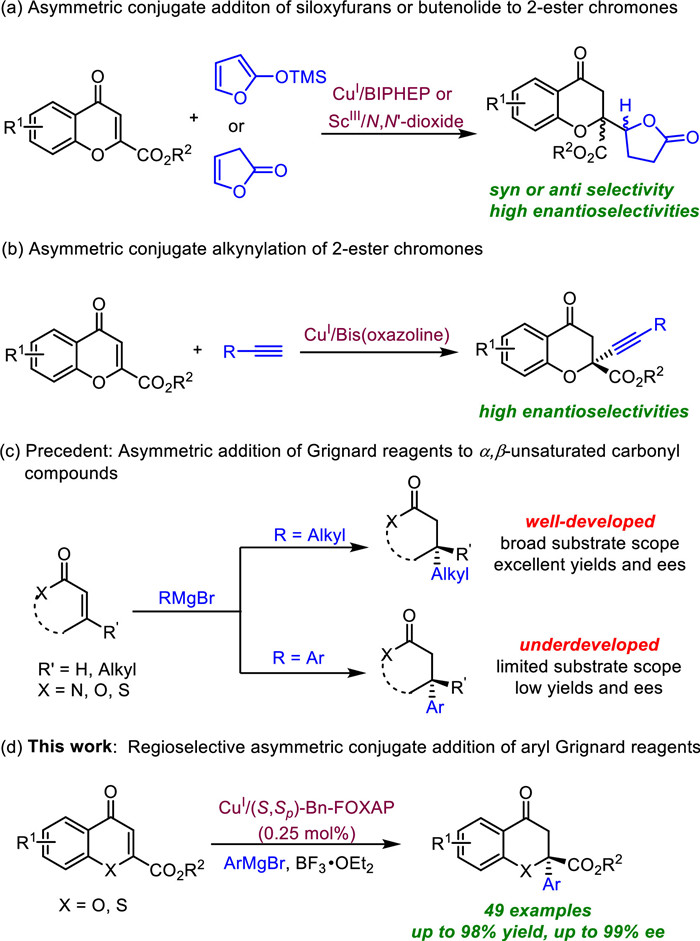
In the past decades, the asymmetric conjugate addition (ACA) of organometallic nucleophiles to trisubstituted enones have emerged as a robust and efficient strategy for the synthesis of all-carbon quaternary centers [19, 20]. Zinc [21-24], magnesium (Grignard) [25-28], aluminium [29-31] and boron species [32-35] have been successfully applied to the ACA reactions to form quaternary centers catalyzed by rhodium, copper or palladium complex. Among them, Grignard reagents have undoubtedly attracted the most attention for large-scale applications based on their availability, efficiency, economy and toxicity [36]. Although great progress has been made in the asymmetric conjugation of Grignard reagents to various cyclic or acyclic α, β-unsaturated compounds by Feringa, Alexakis, Harutyunyan's group [37-46], the substrate scope was mainly limited to alkyl Grignard reagents (Scheme 2c). The use of aryl Grignard reagents in asymmetric conjugation reaction remain underdeveloped, probably due to the non-catalyzed pathway outcompeting the slow catalytic reaction (Scheme 2c) [47]. Up to date, only several single examples were reported to apply aryl Grignard reagent as the nucleophile in asymmetric conjugation reaction, and the 1, 4-addition products were obtained in either low yields or enantioselectivities [48-51]. Based on our previous work on the asymmetric transformation of Grignard reagents [[52], [53]], we reported the regioselective asymmetric addition of aryl Grignard reagents to 2-ester (thio)chromones by chiral P, N ligand, delivering the tetra-substituted chromanones in excellent yields and ees at low catalyst loading (Scheme 2d).
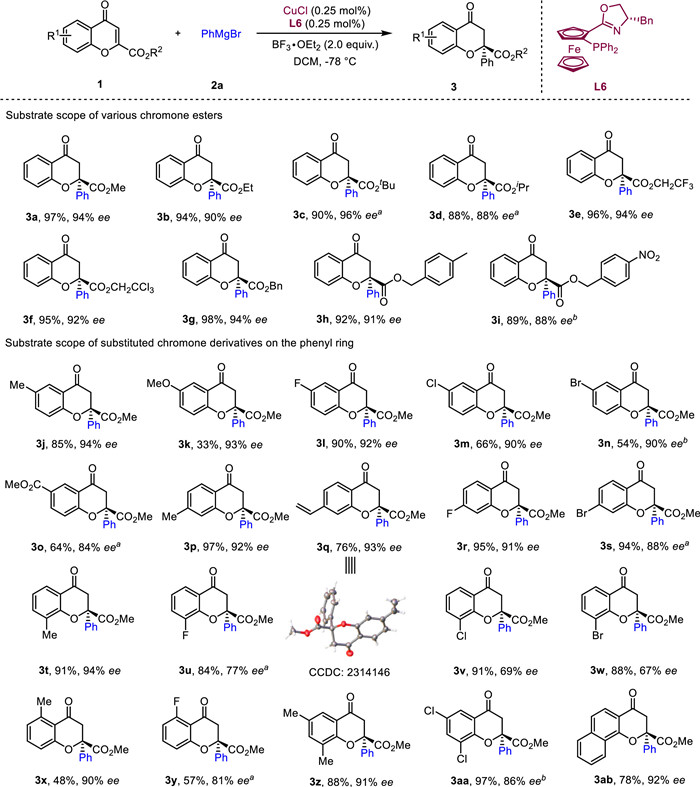
We initiated our investigation by assessing the copper-catalyzed enantioselective addition of ethylmagnesium bromide to 2-ester chromone 1a. Unfortunately, no stereochemical control was observed under various reaction conditions, albeit with moderate to high yields in some cases (see details in Supporting information). Then, we turned our attention to aryl magnesium reagent. As shown in Table 1, the model substrate 1a was treated with phenylmagnesium bromide 2a in the presence of copper metals and BF3·OEt2 using various chiral phosphine ligands (L1−L8) in DCM at −78 ℃. When ligand (R)-SegPhos (L1) was applied to this reaction, the conjugation product 3a could be obtained in 90% yield with excellent regioselectivity, albeit with 0% ee (entry 1). Chiral phosphine ligand (R, R)-pH-BPE (L2), phosphoramidite-type ligand L3 and rev-Josiphos (L4) could also catalyze this reaction but only very low enantioselectivities were obtained (entries 2–4). To our delight, the use of chiral P, N ligands shown promising results (L5−L7). The best performing ligand in terms of both yield and enantioselectivity was (S, Sp)-Bn-FOXAP (L6), which could deliver the conjugation product in almost quantitative yield and 95% ee within 5 min (entries 5–7). Furthermore, chiral P, N ligand (S)-tBu-Phox (L8) was also investigated, but no improved result was detected (entry 8). When changing the copper catalyst to CuCl or Cu(acac)2, both the reaction yields and enantioselectivities could be maintained (entries 9 and 10). Next, we investigated the reaction temperature and it was found that raising the temperature proved to be not beneficial for the stereoselective control (entries 11 and 12). Further investigation of the solvent effects indicated that DCM was the best solvent (entries 13 and 14). In the end, we examined the effects of catalyst loading. We found that reducing the amount of CuCl and L6 to 1.0 mol% does not influence the reaction result (entry 15). Slightly lower yields and enantioselectivities were obtained when further reducing the catalyst loading to 0.5 mol% or 0.25 mol% (entries 16 and 17).
Having identified the optimal reaction conditions, we first explored the scope of the enantioselective addition of Grignard reagents 2a with various chromone esters (Scheme 3). The 2-ethyl and tert‑butyl ester chromone could afford the corresponding products 3b and 3c in similar results, while isopropyl ester gave the tetra-substituted chromanones 3d in relatively lower yield and ee even using 1.0 mol% catalyst. Two more active substrate bearing trifluoroethyl and trichloroethyl groups were also amenable to this reaction, generating the products 3e and 3f in excellent yields (95%−96%) and enantioselectivities (92%−94%). Furthermore, the benzyl ester and its analogues 4-methylbenzyl, 4-nitrobenzyl ester derived chromones also proceeded smoothly to produce the desired products with high to excellent yields and ee (3g–3i, 89%−98% yields, 88%−94% ee). Next, a wide range of 2-methyl ester chromones containing diverse substituents at different positions on the phenyl ring underwent the conjugate additions in the same reaction condition. In general, electron-donating groups give better yields and ee compared with electron-withdrawing groups at the same position. 2-Methyl ester chromones bearing electron-donating groups (−Me and OMe) and electron-withdrawing groups (−F, Cl and Br) at C6-position on the benzene ring were suitable substrates for this reaction, affording the desired products 3j−3n in moderate to high yields (33%−90%) and excellent ee (90%−94%). Functional ester group at the same position was also accommodated, although a relatively lower ee was obtained (3o, 64% yield, 84% ee). Next, substituents (−Me, vinyl, F and Cl) at C7-position of 2-methyl ester chromones were also investigated, and similar results were obtained compared with the C6-substituted substrates (3p−3s, 76%−97% yields, 88%−93% ee). Although the ee value could be maintained using C8 methyl substituted chromones (3t, 91% yield, 94% ee), only moderate ee were obtained when electron-withdrawing groups (−F, Cl and Br) were introduced at the same position of chromones (3u−3w, 84%−91% yields, 67%−77% ee). Lastly, the more challenging C5-substituted chromones were explored in this reaction. Although both fluorine and methyl substituents decrease the reaction yields, the enantioselectivities remained high to excellent (3x−3y, 48%−57% yields, 81%−90% ee). It is noteworthy that both di-substituted and phenyl-fused chromones were also applicable to this reaction, providing the corresponding products 3z−3ab in high to excellent yields (78%−97%) and ee (86%−92%).
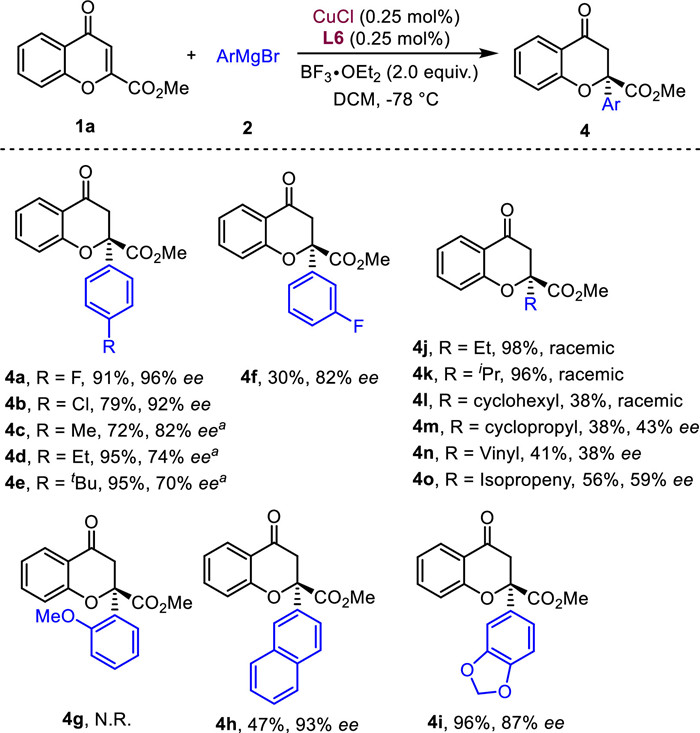
Next, we extended the substrate scope to aryl magnesium reagent with the optimal conditions (Scheme 4). Grignard reagents bearing electron-withdrawing groups (such as –F and Cl) at the para-position of phenyl ring were compatible to give the products 4a–4b in excellent ee (92%–96%), while electron-donating groups (such as –Me, Et, tBu) at the same position exhibited lower ee's, even increasing the catalyst loading to 1.0 mol% (4c–4e, 72%–95% yields, 70%–82% ee). A dramatically decreased yield was obtained when incorporating fluorine at the meta-position of phenyl ring (4f, 30% yield, 82% ee). Meanwhile, no obvious tetra-substituted chromanones 4g was detected when adding a methoxy group at the ortho-position of phenyl ring due to the steric hindrance. Gratifyingly, 2-naphthylmagnesium bromide and 3, 4-(methylene-dioxy)phenylmagnesium bromide exhibited excellent stereocontrol, delivering the functionalized products 4h and 4i in 93% and 87% ee respectively. In addition, the asymmetric conjugate additions using various alkyl magnesium reagents (−Et, iPr, cyclohexyl and cyclopropyl) were conducted under the identical conditions. No stereocontrol was observed (4j−4l, 68%−98% yields, racemic) except for cyclopropylmagnesium bromide, which could deliver the product 4m in 38% yield and 43% ee. Finally, the alkenyl magnesium bromide can also be applied to the reaction with moderate yields, albeit with low ee (4n–4o, 41%–56% yields, 38%–59% ee).
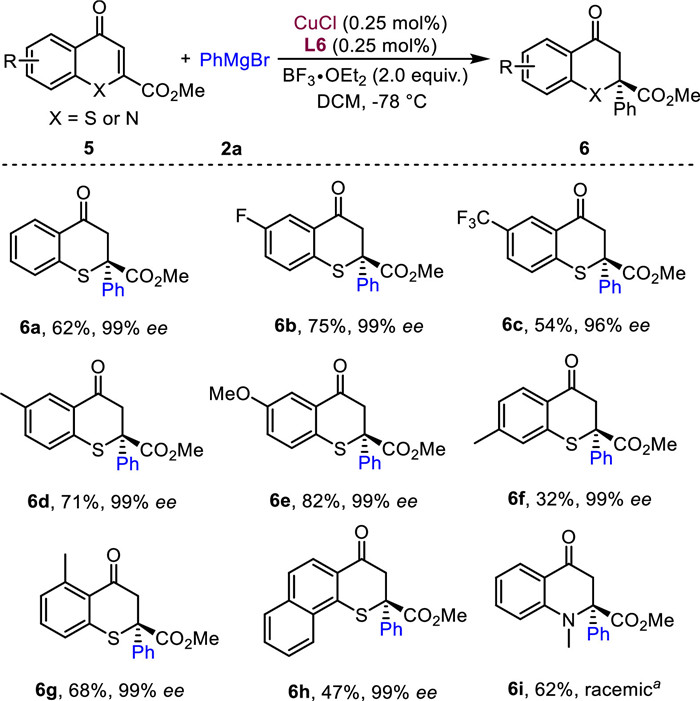
To further demonstrate the synthetic utility of this methodology, various 2-ester thiochromones were tested (Scheme 5). To our surprise, these substrates displayed elevated enantioselectivities compared with the 2-ester chromones. The desired products were all obtained in > 96% ee regardless of the substituent positions on the phenyl rings, albeit with moderate yields (6a−6g, 32%−82% yields, 96%−99% ee). Furthermore, phenyl-fused thiochromone was a suitable substrate in this reaction, affording the product 6h in 47% yield and 99% ee. Finally, N-methyl substituted 2-ester quinolone was applied to this reaction, only racemic product 6i was obtained in 62% yield. Changing the methyl group to other protecting groups such as Ac, Boc and Cbz failed to give the conjugation products. The limited successful examples of Grignard reagents in asymmetric conjugate addition for large-scale applications intrigue us to further explore the catalyst loading of this reaction (Section 2.6 in Supporting information).
Considering the high efficiency of this reaction (reaction was fully completed within 5–10 min in most cases). We wondered whether this reaction was applicable to continuous flow conditions (Table 2), which have demonstrated many advantages, including high safety, accurate control, easy amplification and high sustainability [54-56]. The flow system was set up as shown in Table 3. The substrate 1a and copper/L6 complex were first mixed together. The Lewis acid was further added under flow conditions (Flow rate: 0.1 mL/min, −78 ℃). Then, Grignard reagent 2b was mixed with the above reagents under the same conditions. To our delight, the desired product could be obtained in 57% yield and 90% ee (entry 1). Increasing the reaction length from 150 to 250 cm led to significant increase of the reaction yield (entry 2, 75% yield, 90% ee). Furthermore, increasing the reaction concentration up to 0.3 mol/L showed enhanced reaction yields and ee, which was not applicable in batch conditions (entries 3–5). Unfortunately, either increasing the flow rate or reaction temperature shows no benefit to the yield and stereocontrol (entries 6 and 7). In summary, the flow synthesis of this asymmetric conjugation was amenable with the following optimized conditions: 0.3 mol/L of 1a, 0.1 mL/min flow rate with 250 cm reaction length at −78 ℃.
In order to gain insight into the reaction mechanism, the effect of using different Lewis acids on the asymmetric conjugation was investigated (Table 3). In the absence of both Lewis acid and copper/L6 complex, no 1, 4-conjugate addition product was observed. In contrast, nucleophile addition products 3a' and 3a'' were obtained in 62% and 8% yield respectively (Table 4, entry 1). Meanwhile, 3a' became the main product under the optimal conditions without Lewis acid (entry 2). The 1, 4-conjugate addition product 3a could be obtained in high yields using various Lewis acids (BF3·OEt2, TMSOTf, TBSOTf) without the copper complex (entries 3–5). The above results indicated that Lewis acid has vital contribution to the regioselectivity of this reaction. However, only racemic products could be gained using Lewis acid TMSOTf instead of BF3·OEt2 under the standard conditions (entry 6). The augmentation of steric bulk at the silicon atom does not yield any discernible benefits for this reaction (entry 7). This phenomenon might be explained by the stronger Lewis acidity of TMSOTf and TBSOTf, which could promote the background reaction before the asymmetric conjugation occurred. In summary, the Lewis acid is the key regulation factor to control both the regioselectivity and enantioselectivity of the model reaction.
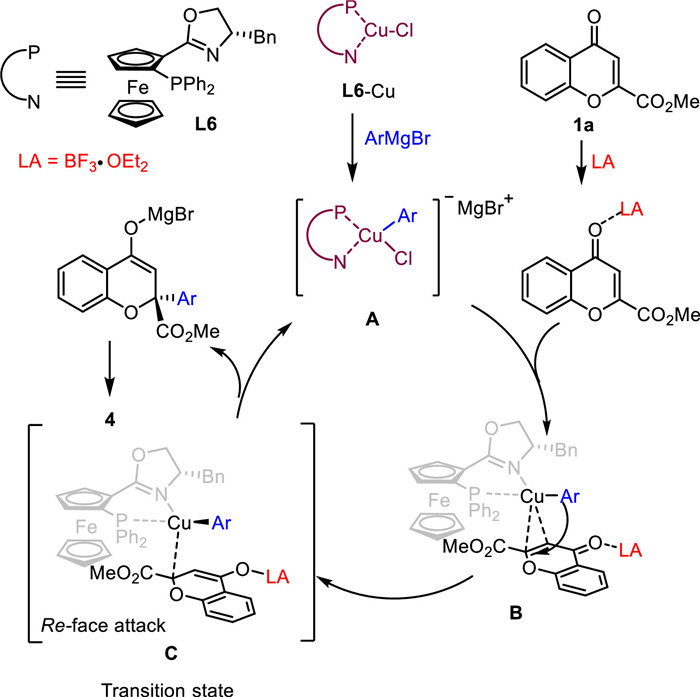
Based on the above results and previous reports [57-60], we proposed the mechanism in Scheme 6. The first step in the catalytic cycle is initiated by the formation of intermediate A, which is formed by Cu-L6 with Grignard reagent. Then intermediate B is formed via π-complexation between Lewis acid activated 2-ester chromone 1a and copper complex A. This is then followed by the formation of the key σ-complex C, which was quickly converted to the final product 4 by reductive elimination and reforming the intermediate A for the next catalytic cycle. The corresponding NMR spectroscopy based mechanistic studies have been deposited in Section 2.9 in Supporting information.
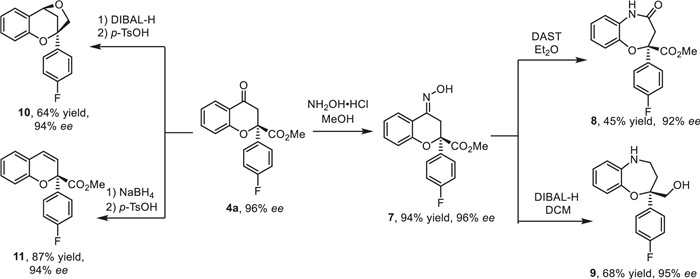
To further evaluate the practical utility of the synthetic methodology, several product transformations were performed (Scheme 7). For instance, 4a (96% ee) could be readily transformed to oxime 7 in 94% yield without any loss of enantioselectivity. Under Lewis acid condition, chiral 7 could undergo an efficient Beckmann rearrangement to product lactam 8 in 45% yield and 92% ee. Interestingly, when changing the condition to DIBAL-H, a cascade Beckmann and reduction reaction occurred, delivering the product 9 in 68% yield and 95% ee. In addition, compound 4a could undergo a two step reduction/intramolecular cyclization to product bridged product 10 in moderate yields and excellent ee. Finally, product 11 could be obtained in reasonable results via reduction /elimination reaction.
In conclusion, we have disclosed an efficient regio- and enantioselective conjugate additions of aryl Grignard reagent to tri-substituted enones in both batch and flow. The chiral biological relevant chromanone derivatives could be obtained in moderate to high yields, excellent enantioselectivities and high turnover number. The employment of the resulting chiral products is presented by various stereospecific functionalizations to provide valuable chromanone derivatives. Further application of the chiral P, N ligand system to other asymmetric transformations of aryl Grignard reagents is underway in our laboratory.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Ya-Ling Li: Writing – original draft. Jia-Wei Ke: Data curation. Yue Liu: Data curation. Dong-Mei Yao: Data curation, Conceptualization. Jing-Dong Zhang: Data curation. You-Cai Xiao: Writing – original draft. Fen-Er Chen: Writing – original draft.
The authors thank the financial support from the Sichuan Science and Technology Program (No. 2022NSFSC0619).
Supplementary material associated with this article can be found, in the online version, at doi:
S. Bräse, A. Encinas, J. Keck, C.F. Nising, Chem. Rev. 109 (2009) 3903–3990. doi: 10.1021/cr050001f
K.S. Masters, S. Bräse, Chem. Rev. 112 (2012) 3717–3776. doi: 10.1021/cr100446h
M. Isaka, A. Jaturapat, K. Rukseree, et al., J. Nat. Prod. 64 (2001) 1015–1018. doi: 10.1021/np010006h
W. Zhang, K. Krohn, Zia-Ullah, U. Flörke, et al., Chem. Eur. J. 14 (2008) 4913–4923. doi: 10.1002/chem.200800035
D. Rönsberg, A. Debbab, A. Mándi, et al., J. Org. Chem. 78 (2013) 12409–12425. doi: 10.1021/jo402066b
I.N. Siddiqui, A. Zahoor, H. Hussain, et al., J. Nat. Prod. 74 (2011) 365–373. doi: 10.1021/np100730b
H. Kikuchi, M. Isobe, M. Sekiya, et al., Org. Lett. 13 (2011) 4624–4627. doi: 10.1021/ol2018449
C. Wang, L. Engelke, D. Bickel, et al., Bioorg. Med. Chem. 27 (2019) 115044–115056. doi: 10.1016/j.bmc.2019.115044
X. Wu, T. Iwata, A. Scharf, et al., J. Am. Chem. Soc. 140 (2018) 5969–5975. doi: 10.1021/jacs.8b02535
T. Qin, T. Iwata, T.T. Ransom, J.A. Beutler, J.A. Porco Jr, J. Am. Chem. Soc. 137 (2015) 15225–15233. doi: 10.1021/jacs.5b09825
T. Qin, S.L. Skraba-Joiner, Z.G. Khalil, R.P. Johnson, R.J. Capon, J.A. Porco Jr, Nat. Chem. 7 (2015) 234–240. doi: 10.1038/nchem.2173
T. Qin, J.A. Porco Jr, Angew. Chem. Int. Ed. 53 (2014) 3107–3110. doi: 10.1002/anie.201311260
T. Qin, R.P. Johnson, J.A. Porco Jr, J. Am. Chem. Soc. 133 (2011) 1714–1717. doi: 10.1021/ja110698n
J. Cui, R. Oriez, H. Noda, T. Watanabe, M. Shibasaki, Angew. Chem. Int. Ed. 134 (2022) e202203128. doi: 10.1002/ange.202203128
J. Cui, R. Oriez, S. Samanta, H. Noda, M. Shibasaki, Org. Lett. 25 (2023) 8367–8371. doi: 10.1021/acs.orglett.3c03503
Y. Li, S. Xin, R. Weng, X. Liu, X. Feng, Chem. Sci. 13 (2022) 8871–8875. doi: 10.1039/d2sc02541h
Y. Guan, T.A. Buivydas, R.F. Lalisse, et al., ACS Catal. 11 (2021) 6325–6333. doi: 10.1021/acscatal.1c01095
Y. Guan, J.W. Attard, A.E. Mattson, Chem. Eur. J. 26 (2020) 1742–1747. doi: 10.1002/chem.201904822
Z. Wang, Org. Chem. Front. 7 (2020) 3815–3841. doi: 10.1039/d0qo00763c
C. Hawner, A. Alexakis, Chem. Commun. 46 (2010) 7295–7306. doi: 10.1039/c0cc02309d
J. Wu, D.M. Mampreian, A.H. Hoveyda, J. Am. Chem. Soc. 127 (2005) 4584–4585. doi: 10.1021/ja050800f
K.S. Lee, M.K. Brown, A.W. Hird, A.H. Hoveyda, J. Am. Chem. Soc. 128 (2006) 7182–7184. doi: 10.1021/ja062061o
M.K. Brown, T.L. May, C.A. Baxter, A.H. Hoveyda, Angew. Chem. Int. Ed. 46 (2007) 1097–1100. doi: 10.1002/anie.200604511
E. Fillion, A. Wilsily, J. Am. Chem. Soc. 128 (2006) 2774–2775. doi: 10.1021/ja056692e
D. Martin, S. Kehrli, M. d'Augustin, et al., J. Am. Chem. Soc. 128 (2006) 8416–8417. doi: 10.1021/ja0629920
H. Hénon, M. Mauduit, A. Alexakis, Angew. Chem. Int. Ed. 47 (2008) 9122–9124. doi: 10.1002/anie.200803735
M. Tissot, D. Poggiali, H. Hénon, et al., Chem. Eur. J. 18 (2012) 8731–8747. doi: 10.1002/chem.201200502
S. Kehrli, D. Martin, D. Rix, M. Mauduit, A. Alexakis, Chem. Eur. J. 16 (2010) 9890–9904. doi: 10.1002/chem.201000471
M. d'Augustin, L. Palais, A. Alexakis, Angew. Chem. Int. Ed. 44 (2005) 1376–1378. doi: 10.1002/anie.200462137
T.L. May, M.K. Brown, A.H. Hoveyda, Angew. Chem. Int. Ed. 47 (2008) 7358–7362. doi: 10.1002/anie.200802910
C. Hawner, K. Li, V. Cirriez, A. Alexakis, Angew. Chem. Int. Ed. 47 (2008) 8211–8214. doi: 10.1002/anie.200803436
R. Shintani, W.L. Duan, T. Hayashi, J. Am. Chem. Soc. 128 (2006) 5628–5629. doi: 10.1021/ja061430d
R. Shintani, Y. Tsutsumi, M. Nagaosa, T. Nishimura, T. Hayashi, J. Am. Chem. Soc. 31 (2009) 13588–13589. doi: 10.1021/ja905432x
B.T. Hahn, F. Tewes, R. Fröhlich, F. Glorius, Angew. Chem. Int. Ed. 49 (2010) 1143–1146. doi: 10.1002/anie.200905712
K. Kikushima, J.C. Holder, M. Gatti, B.M. Stoltz, J. Am. Chem. Soc. 133 (2011) 6902–6905. doi: 10.1021/ja200664x
G.P. Howell, Org. Process Res. Dev. 16 (2012) 1258–1272. doi: 10.1021/op200381w
B.L. Feringa, R. Badorrey, D. Peña, A.J. Minnaard, Proc. Natl. Acad. Sci. U.S.A. 101 (2004) 5834–5838. doi: 10.1073/pnas.0308008101
F. López, S.R. Harutyunyan, A.J. Minnaard, B.L. Feringa, J. Am. Chem. Soc. 126 (2004) 12784–12785. doi: 10.1021/ja046632t
C. Vila, V. Hornillos, M. Fañanás-Mastral, B.L. Feringa, Chem. Commun. 49 (2013) 5933–5935. doi: 10.1039/c3cc43105c
R.P. Jumde, F. Lanza, M. Veenstra, S.R. Harutyunyan, Science. 352 (2016) 433–437. doi: 10.1126/science.aaf1983
X. Yan, S.R. Harutyunyan, Nat. Commun. 10 (2019) 3402. doi: 10.1038/s41467-019-11345-z
Y. Guo, S.R. Harutyunyan, Angew. Chem. Int. Ed. 58 (2019) 12950–12954. doi: 10.1002/anie.201906237
Y. Guo, J. Kootstra, S.R. Harutyunyan, Angew. Chem. Int. Ed. 57 (2018) 13547–13550. doi: 10.1002/anie.201808392
M. Rodríguez-Fernández, X. Yan, J.F. Collados, P.B. White, S.R. Harutyunyan, J. Am. Chem. Soc. 139 (2017) 14224–14231. doi: 10.1021/jacs.7b07344
D. Martin, S. Kehrli, M. d'Augustin, H. Clavier, M. Mauduit, A. Alexakis, J. Am. Chem. Soc. 128 (2006) 8416–8417. doi: 10.1021/ja0629920
N. Germain A. Alexakis, Chem. Eur. J. 21 (2015) 8597–8606. doi: 10.1002/chem.201500292
S.R. Harutyunyan, T. den Hartog, K. Geurts, A.J. Minnaard, B.L. Feringa, Chem. Rev. 108 (2008) 2824–2852. doi: 10.1021/cr068424k
F. López, S.R. Harutyunyan, A.J. Minnaard, and B.L. Feringa, J. Am. Chem. Soc. 126 (2004) 12784–12785. doi: 10.1021/ja046632t
B.L. Feringa, R. Badorrey, D. Peña, S.R. Harutyunyan, A.J. Minnaard, Proc. Natl. Acad. Sci. U.S.A. 101 (2004) 5834–5838. doi: 10.1073/pnas.0308008101
D. Martin, S. Kehrli, M. d'Augustin, et al., J. Am. Chem. Soc. 128 (2006) 8416–8417. doi: 10.1021/ja0629920
T. Robert, J. Velder, H.G. Schmalz, Angew. Chem. Int. Ed. 47 (2008) 7718–7721. doi: 10.1002/anie.200803247
J. Li, Y. Wang, J. Huang, H. You, F.E. Chen, Chem. Sci. 14 (2023) 4351–4356. doi: 10.1039/d3sc00127j
J. Wang, J. Li, Y. Yang, et al., ACS. Catal. 12 (2022) 9629–9637. doi: 10.1021/acscatal.2c02056
J. Liao, S. Zhang, Z. Wang, et al., Green Synth, Catal. 1 (2020) 121–133. doi: 10.1016/j.gresc.2020.08.001
Y. Xin, S. Peng, J.X. Chen, Z.J. Yang, J.Y. Zhang, Chin. Chem. Lett. 31 (2020) 1448–1461. doi: 10.1016/j.cclet.2019.09.054
P. Liu, F. Zhao, J. Zhang, et al., Chin. Chem. Lett. 35 (2024) 109020–1461.
M. Rodríguez-Fernández, X. Yan, J.F. Collados, P.B. White, S.R. Harutyunyan, J. Am. Chem. Soc. 139 (2017) 14224–14231. doi: 10.1021/jacs.7b07344
Y. Guo, M. Castiñeria Reis, J. Kootstra, S.R. Harutyunyan, ACS Catal. 11 (2021) 8476–8483. doi: 10.1021/acscatal.1c01544
S. Somprasong, M. Castiñeria Reis. S.R. Harutyunyan, Angew. Chem. Int. Ed. 62 (2023) e202217328. doi: 10.1002/anie.202217328
S.R. Harutyunyan, F. López, W.R. Browne, et al., J. Am. Chem. Soc. 128 (2006), 9103–9118. doi: 10.1021/ja0585634

Scheme 2 Asymmetric conjugate addition of various nucleophiles to trisubstituted cyclic enones.

Scheme 3 Substrate scope of 2-ester chromones. Reaction conditions: 1 (0.2 mmol), 2a (0.4 mmol), CuCl (0.25 mol%), L6 (0.25 mol%), BF3·OEt2 (2.0 equiv.) in DCM (4.0 mL) at −78 ℃ for 5–10 min. a 1.0 mol% of CuCl and L6 were used. b 0.5 mol% of CuCl and L6 were used.

Scheme 4 Substrate scope of Grignard reagents 2. a Reaction conditions: 1a (0.2 mmol), 2 (0.4 mmol), CuCl (0.25 mol%), L6 (0.25 mol%), BF3·OEt2 (2.0 equiv.) in DCM (4.0 mL) at −78 ℃ for 5 − 10 min. a 1.0 mol% of CuCl and L6 were used.

Scheme 5 Substrate scope of 2-ester thiochromones. Reaction conditions: 5 (0.2 mmol), 2a (0.4 mmol), CuCl (0.25 mol%), L6 (0.25 mol%), BF3·OEt2 (2.0 equiv.) in DCM (4.0 mL) at −78 ℃ for 5−10 min. a TMSOTf (2.0 equiv.) was used instead of BF3·OEt2.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:




 下载:
下载:

