
Scheme 1.
Photorelease reaction of Guo from BP-Guo.
Photochemical reaction mechanism of benzophenone protected guanosine at N7 position
Yan Guo , Hongtao Bian , Le Yu , Jiani Ma , Yu Fang

Establishing the precise conditional control of biological function comparable to that of the natural system is an important tool for elucidating the mechanisms of cellular processes [1,2]. One of the most excellent strategies is photoregulation by introducing photolabile protecting group (PPG) to small molecules, proteins, and nucleic acids [3-9]. After light irradiation at a suitable wavelength, the protecting group is irreversibly cleaved whereby the masked native functionality and biological activity of the protected compound can be restored. Among the photoprotected biomolecules, nucleic acids with PPGs that can modify at nucleobases, phosphate moieties, or sugar, open a particularly active field and are extensively applied to light-regulation of gene expression, photomodulation of protein function, and synthesis of DNA or RNA by polymerases [10-14].
Various PPGs toward protecting nucleic acids have been exploited that were mostly pioneered on the class of o-nitrobenzyl and coumarin. For example, two-photon PPG nitrodibenzofuran (NDBF) was discovered, via a Norrish type II photoprotecteion mechanism, enabling deeper tissue penetration and efficient deprotection because of their higher extinction coefficient at deprotecting wavelength. Deiters applied NDBF in successful protecting of thymidine phosphoramidite at N3 [15]. And Heckel developed NDBF-caged deoxycytidine and deoxyadenosine that can be incorporated into an oligonucleotide [16]. Hocek reported nitropiperonyl-caged 5-(hydroxymethyl)uracil derivative that is a good substrate for DNA polymerases and is efficiently deprotected under blue light [17]. In addition, diethylaminocoumarin (DEACM) derivatives as PPGs exhibit superior photochemical properties with high deprotecting efficiency at wavelengths over 400 nm. Utilization of DEACM to protect deoxyguanosine in oligonucleotides [18], deoxythymidine in DNA duplex [19], and 5′ untranslated region of an mRNA [20,21] have been reported. And to prevent occurrence of undesired recombination reactions, an optimized strategy to the design of DEACM-caged thymidine was proposed using a self-immolative spacer to separate the DEACM and thymidine [22]. Furthermore, the other distinct PPG on protecting specific nucleic acids [23], is a thiochromone S, S-dioxide group installed on antisense oligonucleotides at O4 of thymidine.
Unlike the typical protecting strategies of nucleobases, in which PPG replaces a hydrogen atom with modifications and does not change the charge distribution of the molecule, Rentmeister developed a novel strategy to protect nucleic acids by introducing a positive charge on nucleobase [24]. Specifically, taking aryl ketone derivatives as PPGs attached to guanosine N7 position or adenosine N1 position. And this strategy is extended to more complex biomolecules dinucleotide and RNA, achieving to spatiotemporally control biologically relevant functions. Benzophenone protected guanosine at N7 position (denoted as BP-Guo) was selected as the representative system to study the photodeprotect mechanism (Scheme 1). Irradiation of BP-Guo in a buffer solution containing EDTA and glycerol (GI) releases guanosine and 4-methylbenzophenone. And a possible photorelease reaction mechanism on benzophenone protected guanosine at N7 position was proposed that the photorelease was triggered by the hydrogen atom transfer between triplet state of BP chromophore and the solvent (Scheme S1 in Supporting information) [24].
However, regarding the photorelease mechanism of BP-Guo, certain pivotal issues are still left to be addressed. The information of key transient intermediates generated during photorelease of BP-Guo, especially the spectroscopic and kinetic characterizations for intermediates on the ultrafast time scale are absent. As well, as a classical photosensitizer BP can cause damage to nucleic acids or DNA through a variety of processes including triplet energy transfer (TTET), hydrogen atom transfer (HAT), or electron transfer [25,26]. It is noted that TTET plays an essential role in photophysical photochemical reaction [27-31]. Will BP-Guo occur these reactions between the two chromophores and further compete with HAT reaction between BP-Guo and solvent molecules to trigger photorelease? Furthermore, no theoretical investigation is done on study of photoreaction paths of BP-Guo by now. Here BP-Guo was selected as an example for aryl ketone protected nucleic acids, and femtosecond transient absorption (fs-TA), nanosecond transient absorption (ns-TA), and nanosecond transient resonance Raman (ns-TR2) spectroscopy coupling with density functional theory (DFT) were employed to thoroughly investigate the photorelease mechanism.
The photorelease of BP-Guo in Gl-acetonitrile (ACN)-H2O was monitored by UV–vis spectra under 266 nm irradiation (Fig. S1a in Supporting information). As irradiation time increases, the absorption bands of BP-Guo decreased and subsequently shifted to 252 and 270 nm, where the later agrees with the characteristic bands of Guo (Fig. S1b in Supporting information). The TD-DFT calculation on BP-Guo as shown in Table S1 (Supporting information). The S0 → S1 transition is predominantly contributed by the HOMO−5 → LUMO nπ* excitation. It is noted that the most of the charge is localized on the BP moiety with a locally excited character. And the C=O bond of the carbonyl group exhibits an elongation from 1.22 Å to 1.29 Å upon excitation of ground state of BP-Guo to its S1 state. BP is known to have a high ISC quantum yield after photoexciting to its S1 (nπ*) state [32,33]. fs-TA experiments were performed for BP-Guo in Gl-ACN—H2O (3:4:16, v: v: v). Upon irradiation, the fs-TA spectra displayed absorption features that essentially coincided with the typical signals of BP chromophore (Figs. 1a-c) [34], where the singlet state of BP moiety at 340 and 580 nm converted via an intersystem crossing (ISC) to the triple state (labeled as 3[BP]-Guo) at 330 and 535 nm. Thereafter, the 330 nm band decreased and red-shifted to 335 nm, as well as the band at 535 nm attenuated, a signal with a shoulder band at 375 and 450 nm appeared.
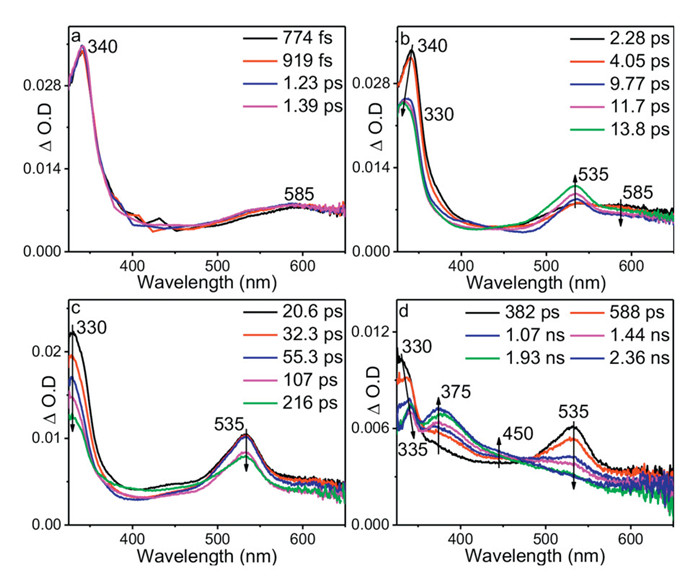
Fig. 2 shows the ns-TA spectra of BP-Guo in Gl-ACN—H2O (3:4:16, v: v: v). The ns-TA spectra recorded under the open air condition are resemble with the fs-TA spectra recorded in later delay time, where predominate bands at 375 and 450 nm, and a sharp band at 335 nm and a broad signal at 545 nm were detected. The relative absorbance of 335 and 375 nm changes with the time delay increase, indicating that the signals were contributed by the overlapping signals from different intermediates.
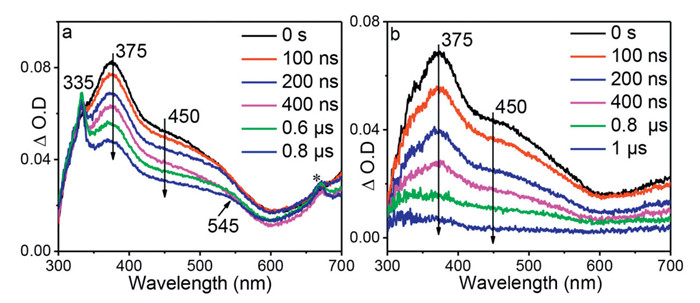
By comparison with the reported transient absorption data on BP [35], the intermediate at 335 and 545 nm was assigned to ketyl radical that formed from HAT between BP moiety and C—H bond of glycerol (denoted as ketyl-Guo). In the late delay time, the signals of ketyl-Guo are more intense than the species at 375 and 450 nm. Therefore, the characteristic spectrum of ketyl-Guo could be deduced by subtracting the 0 ns spectrum from the 0.6 µs spectrum using a proper scale factor with the band at 375 nm as the reference. The obtained spectrum reasonably agrees with the calculated UV–vis spectrum of ketyl-Guo (Fig. S2 in Supporting information). In an oxygen-purged solution, the signal of ketyl-Guo was reduced evidently (Fig. 2b), which is consistent with previous reports that organic radicals can be quenched by oxygen via addition reactions [36-38]. This result further supports the above analysis on the attribution of ketyl-Guo.
The decay time constants at 375 and 450 nm were 2.1 and 2.3 µs, respectively, and decrease to 468 and 434 ns in the presence of oxygen (Fig. 2b and Fig. S3 in Supporting information). Both absorption bands have close decay time constants and could be quenched by oxygen, suggesting that they probably arise from one triplet species. According to previous studies, photosensitized TTET occurs from BP to nucleobases [25]. Herein, the intramolecular TTET between triple BP and Guo chromophore is tentatively proposed, and thus the intermediate at 375 and 450 nm was ascribed to the triplet state of Guo (noted BP-3[Guo] hereafter). This assignment is consistent with a related work, where triplet acetone occurs TTET to give rise the triplet of guanosine monophosphates with a similar maximum absorption feature at 380 nm [39]. Furthermore, DFT and TD-DFT calculations (UM062X/6–311G**) are conducted. The simulated UV–vis spectrum of 3[BP]-Guo and BP-3[Guo] resembled well with the experiment spectra at 32.3 ps and 0 ns, respectively (Fig. 3), further validating the assignments for TA data. Calculation results clearly demonstrate that the spin density localized at benzophenone for 3[BP]-Guo, while localized on guanosine for BP-3[Guo] (Fig. 3). The energy for 3[BP]-Guo is 3.0 kcal/mol higher than BP-3[Guo], showing that the TTET from 3[BP]-Guo to BP-3[Guo] is energetically feasible.
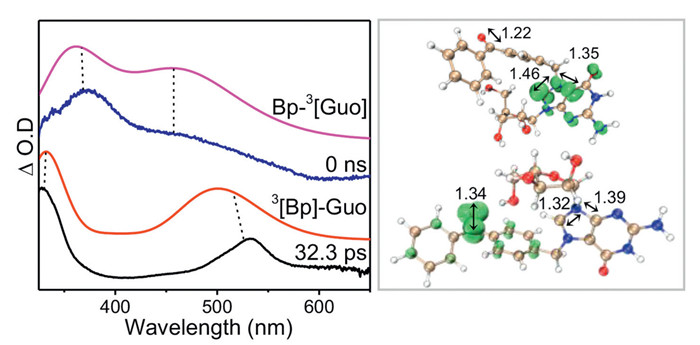
Both experimental spectral data and energy calculations indicate that the BP-Guo undergoes an intramolecular TTET process from the BP moiety to the Guo moiety. This conclusion comes as a bit of a surprise to us, as it is somewhat inconsistent with the results reported in the literature. TTET between triplet state of BP and guanine derivatives is a disfavored process because triplet state of BP is not energetic enough to populate guanine [25,40,41]. In our work, it is still observed, which could be explained by the fact that Guo moiety of BP-Guo exists as a cationic form with a lower triple excited state energy (2.85 eV) than that of the normal form of Guo (3.26 eV) [42]. This is consistent with the trend of calculation data as shown in Table S2 (Supporting information), where the triplet energy value decreases from 3.48 eV to 3.10 eV on going from Guo to its cationic form.
After a clear attribution of the signals from the time-resolved spectra, we conclude that by irradiating the BP-Guo with formation of 3[BP]-Guo in Gl-ACN—H2O, two pathways occurred. One is the TTET process to give rise BP-3[Guo], and the other is the HAT to form the ketyl-Guo.
The effect of the excitation wavelength and solvents on the photoreaction pathways was fully characterized. On one hand, the ns-TA experiment for BP-Guo was performed upon excitation by 355 nm (Fig. S4 in Supporting information), where only the BP chromophore could be excited. No significant difference in ns-TA spectra was observed compared to the results excited by 266 nm. These results indicated that the signal of BP-3[Guo] is generated by the intramolecular TTET from 3[BP]-Guo, excluded the occurrence of the ISC process form the excited singlet state of Guo. On the other hand, the ns-TA experiments were performed for BP-Guo in neat hydrogen donor and non-hydrogen donor solvents of MeOH and DMSO, respectively (Fig. S5 in Supporting information). Similar ns-TA result was obtained for BP-Guo in MeOH with that in Gl-ACN—H2O, by detecting the signals of BP-3[Guo] and ketyl-Guo, suggesting that BP-Guo also occurs both intermolecular HAT and intramolecular TTET processes. In DMSO only the TTET process is probed with the signal of BP-3[Guo], while no HAT is found.
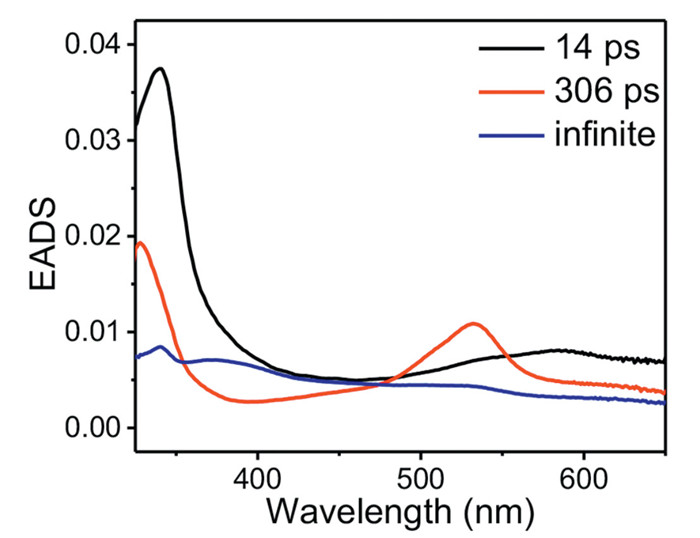
To gain a more detailed insight, the fs-TA data of BP-Guo in GI-ACN—H2O was analyzed by a global fitting in terms of a sequential model that results in an evolution associated difference spectra (EADS) as presented in Fig. 4. Three time constants, 14 ps, 306 ps and infinite, are required for global analysis. The first EADS with absorption bands at 340 and 585 nm is the singlet state of BP-Guo. It evolves to 3[BP]-Guo in 14 ps with signal around 330 and 535 nm, which represents the process of ISC. Then, the evolution from 3[BP]-Guo to the third EADS in 306 ps contains bands 335, 375 and 450 nm as the overlapping spectral features between BP-3[Guo] and ketyl-Guo. The third EADS with time constant of infinite (>3 ns) represents decay of the residual absorption exceeds our instrument detection window. The kinetic trace of BP-3[Guo] absorption feature was fit at 380 nm yielding a rise time of 523 ps assigned to BP-3[Guo] formation time constant (Fig. S6 in Supporting information), which is slower than the decay of 3[BP]-Guo (306 ps). This suggests that 3[BP]-Guo is at least partially deactivated though other pathways than TTET, in line with an HAT process occurring from 3[BP]-Guo. The observed decay rate constant of 3[BP]-Guo (k = 1/τ) is the sum of the rates of TTET and HAT processes (k = kTTET + kHAT). Using τ = 306 ps and τTTET = 523 ps, a HAT time constant τHAT of 746 ps was obtained. And the deactivation percentage of 3[BP]-Guo is determined to be 59% by TTET and 41% by HAT.
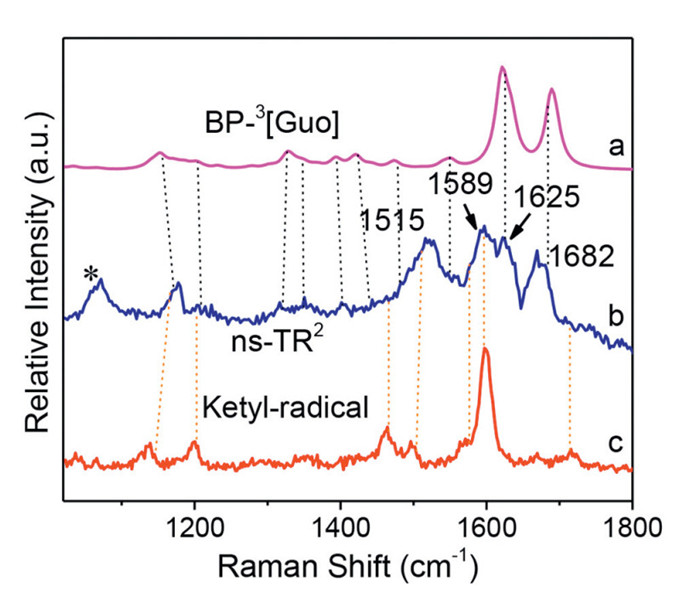
The ns-TR2 experiment was performed for BP-Guo in MeOH, where transient species that appear during the duration of the laser pulse (10 ns) could readily be detected. Major Raman bands were detected around 1515, 1589, 1625 and 1682 cm−1 (Fig. 5). Given the same detection time scale for ns-TR2 spectrum with that of the ns-TA spectra, it is proposed that BP-3[Guo] and ketyl-Guo were the intermediates probed here. The ns-TR2 peaks at 1589 and 1515 cm−1 closely resemble the Raman bands for ketyl radical of BP. And the Raman signals at 1625 and 1682 cm−1 (indicated with black dash lines) agree well with the predicted DFT normal Raman spectrum of BP-3[Guo]. Therefore, the structure information obtained from ns-TR2 here solidly demonstrated that the species observed by ns-TA in MeOH could be ascribed to BP-3[Guo] and ketyl-Guo respectively.
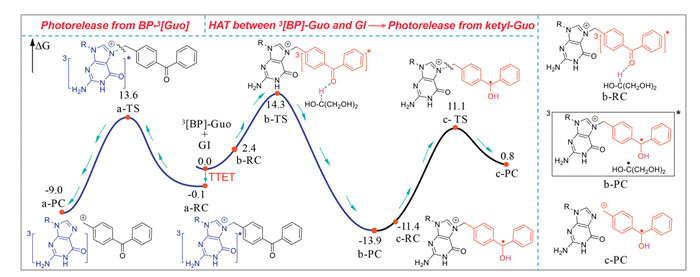
Herein, the free energy profiles for the reaction process starting from 3[BP]-Guo were constructed at the M062X/6–311G** (H2O) level (Fig. 6). The sum of the free energies of BP-3[Guo] and GI is used as the zero point to obtain relative free energies of all transition states (TS) and minima. In order to keep conservation of electron and atom numbers in the system, the free energy of GI is added to the free energies of a-TS, a-RC and a-PC, respectively. In addition, the free energy for ketyl radical of GI is added to the free energies of c-TS, c-RC and c-PC, respectively. On one hand, 3[BP]-Guo underwent TTET to give rise BP-3[Guo] (a-RC), which would conduct C—N bond cleavage through a-TS with a barrier height of 13.7 kcal/mol to release Guo and produce the cation species of BP. And the free energy of the product (a-PC) lies 8.9 kcal/mol lower with respect to the reactant (a-RC). The computed result revealed that the photodeprotection from BP-3[Guo] is both kinetically and thermodynamically feasible. On the other hand, 3[BP]-Guo could proceed HAT with GI by overcoming a 14.3 kcal/mol barrier (b-TS) and yielded to radical pair b-PC. After ketyl radical releasing from b-PC, the generated ketyl-Guo (c-RC) might go through c-TS with a rather high activation barrier of 25.0 kcal/mol to release Guo. Additionally, the strongly endothermic nature of this c-PC generation process (14.7 kcal/mol) implying the reaction equilibrium would favor the c-RC side. The relative lower barrier height for C—N bond breaking from BP-3[Guo] suggesting which would be the primary photodeprotection pathway.
The photodeprotection mechanism of BP-Guo is depicted (Scheme 2). After generation of 3[BP]-Guo, two competing reaction routes occurred to trigger the photodeprotection. One route is the TTET to yield BP-3[Guo], which breaks C—N bond to release the Guo along with BP cation that finally trapped by solvent to give the final product. The photolysis products of BP-Guo were analyzed by GC–MS. The data revealed a product with a mass of m/z 210, which was also detected during the irradiation of 4-(hydroxymethyl)BP and would correspond to oxidation product 4-benzoylbenzaldehyde of the 4-(hydroxymethyl)BP. Therefore, it is proposed that the BP cation was trapped by solvent to give the 4-(hydroxymethyl)BP, which was further oxidizes to 4-benzoylbenzaldehyde under irradiation of light. The other is the HAT with formation of ketyl-Guo, followed by releasing Guo and the BP radical cation. The later undergoes electron transfer and further tautomerizes to 4-methylBP. The present results provide an in-depth insight into the inactivation pathway for BP protected nucleic acids after light irradiation. In particular, the photosensitized TTET pathway from BP leads to the generation of nucleic acid in their triplet states, which, in addition to potentially initiating photodeprotection, may also trigger well-known cyclization reactions that brings about DNA mutagenic adducts, commonly referred to as triple photodamage [43,44]. The latter needs further investigation and should be avoided in developing improved strategies for photoprotecting nucleic acids.

The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Yan Guo: Writing – review & editing, Writing – original draft, Investigation, Funding acquisition, Data curation. Hongtao Bian: Writing – review & editing, Resources. Le Yu: Writing – review & editing. Jiani Ma: Writing – review & editing, Supervision, Resources, Project administration, Funding acquisition. Yu Fang: Resources.
The research was sponsored by grants from the National Natural Science Foundation of China (Nos. 22322301, 22303046), the Shaanxi Science Fund for Distinguished Young Scholars (No. 2021JC-38), and the Fundamental Research Funds for the Central Universities (Nos. GK202207001, GK202304008) and the Shaanxi Province Postdoctoral Science Foundation (No. 2023BSHEDZZ187).
Supplementary material associated with this article can be found, in the online version, at doi:
G. Mayer, A. Heckel, Angew. Chem. Int. Ed. 45 (2006) 4900–4921. doi: 10.1002/anie.200600387
S. Jia, E.M. Sletten, ACS Chem. Biol. 17 (2022) 3255–3269. doi: 10.1021/acschembio.1c00518
S.K. Jia, S.X. Yang, H.M. Ji, et al., Chin. Chem. Lett. 31 (2020) 1104–1108. doi: 10.1016/j.cclet.2019.10.021
Y.J. Li, M.L. Wang, F. Wang, S. Lu, X.Q. Chen, Smart Mol. 1 (2023) e20220003. doi: 10.1002/smo.20220003
V.M. Lechner, M. Nappi, P.J. Deneny, et al., Chem. Rev. 122 (2022) 1752–1829. doi: 10.1021/acs.chemrev.1c00357
N. Ankenbruck, T. Courtney, Y. Naro, A. Deiters, Angew. Chem. Int. Ed. 57 (2018) 2768–2798. doi: 10.1002/anie.201700171
P. Klán, T. Šolomek, C.G. Bochet, et al., Chem. Rev. 113 (2013) 119–191. doi: 10.1021/cr300177k
R. Weinstain, T. Slanina, D. Kand, P. Klán, Chem. Rev. 120 (2020) 13135–13272. doi: 10.1021/acs.chemrev.0c00663
Z.C. Situ, W.B. Chen, S.R. Yang, et al., J. Phys. Chem. B 126 (2022) 3338–3346. doi: 10.1021/acs.jpcb.2c01440
J.M. Govan, R. Uprety, J. Hemphill, M.O. Lively, A. Deiters, ACS Chem. Biol. 7 (2012) 1247–1256. doi: 10.1021/cb300161r
K. Ohno, D. Sugiyama, L. Takeshita, et al., Bioorg. Med. Chem. 25 (2017) 6007–6015. doi: 10.1016/j.bmc.2017.09.032
S. Boháčová, Z. Vaníková, L.P. Slavětínská, M. Hocek, Org. Biomol. Chem. 16 (2018) 5427–5432. doi: 10.1039/c8ob01106k
F. Debart, C. Dupouy, J.J. Vasseur, J. Org. Chem. 14 (2018) 436–469. doi: 10.3762/bjoc.14.32
V. Dhamodharan, Y. Nomura, M. Dwidar, Y. Yokobayashi, Chem. Commun. 54 (2018) 6181–6183. doi: 10.1039/c8cc02290a
H. Lusic, R. Uprety, A. Deiters, Org. Lett. 12 (2010) 916–919. doi: 10.1021/ol902807q
F. Schäfer, K.B. Joshi, M.A.H. Fichte, et al., Org. Lett. 13 (2011) 1450–1453. doi: 10.1021/ol200141v
S. Boháčová, L. Ludvíková, L. Poštová Slavětínská, et al., Org. Biomol. Chem. 16 (2018) 1527–1535. doi: 10.1039/c8ob00160j
C. Menge, A. Heckel, Org. Lett. 13 (2011) 4620–4623. doi: 10.1021/ol201842x
P. Seyfried, M. Heinz, G. Pintér, et al., Chem. Eur. J. 24 (2018) 17568–17576. doi: 10.1002/chem.201804040
D.Y. Zhang, C.Y. Zhou, K.N. Busby, S.C. Alexander, N.K. Devaraj, Angew. Chem. Int. Ed. 57 (2018) 2822–2826. doi: 10.1002/anie.201710917
D.Y. Zhang, S.J. Jin, X.J. Piao, N.K. Devaraj, ACS Chem. Biol. 15 (2020) 1773–1779. doi: 10.1021/acschembio.0c00205
S.Z. Tang, J. Cannon, K. Yang, et al., J. Org. Chem. 85 (2020) 2945–2955. doi: 10.1021/acs.joc.9b02617
S. Hikage, Y. Sasaki, T. Hisai, et al., J. Photochem. Photobiol. A 331 (2016) 175–183. doi: 10.1016/j.jphotochem.2016.01.007
L. Anhäuser, N. Klöcker, F. Muttach, et al., Angew. Chem. Int. Ed. 59 (2020) 3161–3165. doi: 10.1002/anie.201914573
M.C. Cuquerella, V. Lhiaubet-Vallet, J. Cadet, M.A. Miranda, Acc. Chem. Res. 45 (2012) 1558–1570. doi: 10.1021/ar300054e
W. Adam, M.A. Arnold, W.M. Nau, U. Pischel, C.R. Saha-Möller, J. Am. Chem. Soc. 124 (2002) 3893–3904. doi: 10.1021/ja017600y
R. Gao, D.P. Yan, Chem. Sci. 8 (2017) 590–599. doi: 10.1039/C6SC03515A
Y.S. Yang, K.Z. Wang, D.P. Yan, Chem. Commun. 53 (2017) 7752–7755. doi: 10.1039/C7CC04356B
B. Zhou, D.P. Yan, Adv. Funct. Mater. 29 (2019) 1807599. doi: 10.1002/adfm.201807599
H.Y. Lin, X.P. Chang, D.P. Yan, W.H. Fang, G.L. Cui, Chem. Sci. 8 (2017) 2086–2090. doi: 10.1039/C6SC04354B
B. Zhou, Q. Zhao, L.C. Tang, D.P. Yan, Chem. Commun. 56 (2020) 7698–7701. doi: 10.1039/d0cc02730h
P.F. McGarry, C.E. Doubleday, C.H. Wu, H.A. Staab, N.J. Turro, J. Photochem. Photobiol. A 77 (1994) 109–117. doi: 10.1016/1010-6030(94)80033-2
Y.A. Wang, C. Wang, J.R. Zhang, et al., Chin. Chem. Lett. 34 (2023) 108062. doi: 10.1016/j.cclet.2022.108062
J.N. Ma, X.T. Zhang, D.L. Phillips, Acc. Chem. Res. 52 (2019) 726–737. doi: 10.1021/acs.accounts.8b00619
V. Lhiaubet-Vallet, N. Belmadoui, M.J. Climent, M.A. Miranda, J. Phys. Chem. B 111 (2007) 8277–8282. doi: 10.1021/jp071524p
I. Andreu, F. Palumbo, F. Tilocca, et al., Org. Lett. 13 (2011) 4096–4099. doi: 10.1021/ol2016059
S. Jockusch, N.J. Turro, J. Am. Chem. Soc. 121 (1999) 3921–3925. doi: 10.1021/ja9837194
D. Hristova-Neeley, D. Neshchadin, G. Gescheidt, J. Phys. Chem. B 119 (2015) 13883–13887. doi: 10.1021/acs.jpcb.5b04263
I.G. Gut, P.D. Wood, R.W. Redmond, J. Am. Chem. Soc. 118 (1996) 2366–2373. doi: 10.1021/ja9519344
A.A. Lamola, M. Gueron, T. Yamane, J. Eisinger, R.G. Shulman, J. Chem. Phys. 47 (1967) 2210–2217. doi: 10.1063/1.1703293
M.C. Cuquerella, V. Lhiaubet-Vallet, F. Bosca, M.A. Miranda, Chem. Sci. 2 (2011) 1219–1232. doi: 10.1039/c1sc00088h
J.W. Longworth, R.O. Rahn, R.G. Shulman, J. Chem. Phys. 45 (1966) 2930–2939. doi: 10.1063/1.1728048
W.M. Kwok, C.S. Ma, D.L. Phillips, J. Am. Chem. Soc. 130 (2008) 5131–5139. doi: 10.1021/ja077831q
T. Climent, I. González-Ramírez, R. González-Luque, M. Merchán, L. Serrano-Andrés, J. Phys. Chem. Lett. 1 (2010) 2072–2076. doi: 10.1021/jz100601p

Figure 1 (a-d) fs-TA spectra of BP-Guo at various delay times after photoexcitation at 266 nm in Gl-ACN—H2O (3:4:16, v: v: v).

Figure 2 ns-TA spectra of BP-Guo at various delay times after photoexcitation at 266 nm in Gl-ACN—H2O (3:4:16, v: v: v) under (a) air and (b) oxygen-saturated conditions. The peak marked with asterisk (*) represents frequency-doubled signal.

Figure 3 (Left) Comparison of ns-TA spectra at 32.3 ps and 0 ns with computed UV−vis spectra of 3[BP]-Guo and BP-3[Guo], respectively. The calculations were performed on level of TD-M062X/6–311G** (H2O) with a scale factor of 1.12 and a half-width of 2000 cm−1. (Right) The spin densities (isovalue: 0.02) calculated for the triplet states localized on the BP group (3[BP]-Guo) and Guo chromophore (BP-3[Guo]).

Figure 5 Comparison of (b) ns-TR2 spectrum of BP-Guo in MeOH with (a) calculated normal Raman spectrum of BP-3[Guo] (with a scale factor of 0.974 and a half-width of 10 cm−1) and (c) ns-TR2 for ketyl radical of BP.

Figure 6 The free energy profiles of BP-Guo were mapped employing M062X/6–311G**/SMD (H2O) calculations. Paths a, b, and c represent photorelease of Guo from BP-3[Guo], HAT between 3[BP]-Guo and GI, and photorelease of Guo from ketyl-Guo, respectively (free energy in kcal/mol).

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:

