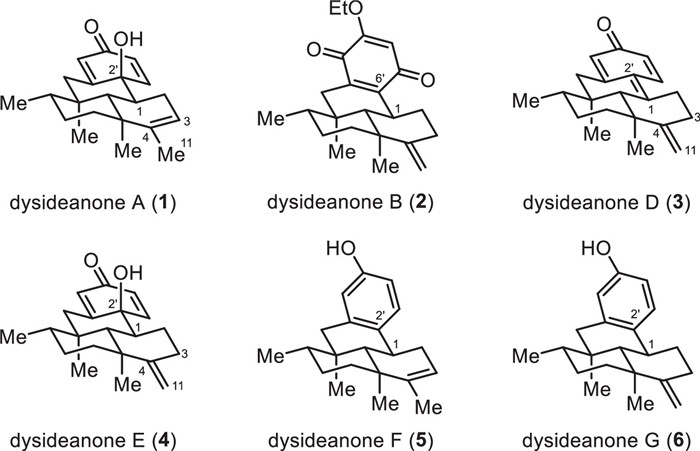
Figure 1.
Structures of dysideanones A, B, and D–G (1–6).
Divergent total synthesis of sesquiterpene (hydro)quinone meroterpenoids dysideanones A and E–G
Qunlong Zhang , Jingyi Kang , Jingwen Wang , Tiancheng Tan , Zhaoyong Lu

Sesquiterpene (hydro)quinones, biosynthetically consisting of two different origins and generally isolated from marine resources, are a group of structurally diverse and biologically important meroterpenoids [1-5]. Dysideanone A (1, Fig. 1), isolated by Lin and co-workers from the South China Sea sponge Dysidea avara in 2014, possesses an unprecedented 6/6/6/6-fused tetracyclic carbon skeleton and represents the first sesquiterpene quinone meroterpenoid having dysideanane ring system [6]. Dysideanone B (2, Fig. 1) was isolated from the same sponge and shared the same ring system with dysideanone A (1) but had different oxidation states and an extra ethoxy group on the quinone moiety [6]. The isolation of dysideanone D (3, Fig. 1) was reported by Zhou et al. in a patent in 2015 [7]. Except for the position of the double bond on the sesquiterpene moiety, dysideanone D (3) has a double bond between C1 and C2′ instead of a single bond and a tertiary alcohol within dysideanone A (1) [7]. In 2016, Lin and colleagues reported the isolation of dysideanone E (4, Fig. 1) from the sponge Dysidea avara [8]. The only difference between dysideanone E (4) and dysideanone A (1) is the position of the double bond in the decalin motif. Very recently, Lin and co-workers disclosed the isolation and structure elucidation of dysideanones F (5, Fig. 1) and G (6, Fig. 1) [9]. These two meroterpenoids share the same ring system with dysideanones A (1) and E (4) but are the phenol form of the latter.
Preliminary bioactivity evaluation showed that dysideanone B (2) exhibited respective IC50 values of 7.1 and 9.4 µmol/L cytotoxicity against HeLa and HepG2 [6], whereas dysideanone D (3) exhibited IC50 values of 6.77 µmol/L cytotoxic activity against HeLa and 29–41 µmol/L cytotoxicity against three other human cancer cell lines, HeLa, A549, and HCT15 [7]. The scarcity of material hampered the bioactivity evaluation of other dysideanone members [6-9]. With their varied oxidation states on the (hydro)quinone moiety and unwell-evaluated bioactivity due to sparse availability from natural resources, dysideanones attracted much attention from the synthetic community. Jana and co-workers constructed the 6/6/6/6-fused skeleton of dysideanone B (2) in 2017 [10] and evaluated the anti-cancer bioactivity of a series of synthetic analogs of this natural product in 2018 [11]. Li and co-workers also forged the 6/6/6/6-fused ring system of dysideanone B (2) and evaluated the antifungal bioactivity of some synthetic analogs in 2020 [12]. Our research group accomplished the first total synthesis of dysideanone B (2) and its congeners in 2021 [13]. Nevertheless, the total synthesis of the remaining members of dysideanones has not been accomplished. As a continuous endeavor towards the efficient synthesis of sesquiterpene (hydro)quinone meroterpenoids for further in-depth investigation of their biological functions, we here report an enantioselective and divergent total synthesis of dysideanones A and E–G (1 and 4–6).
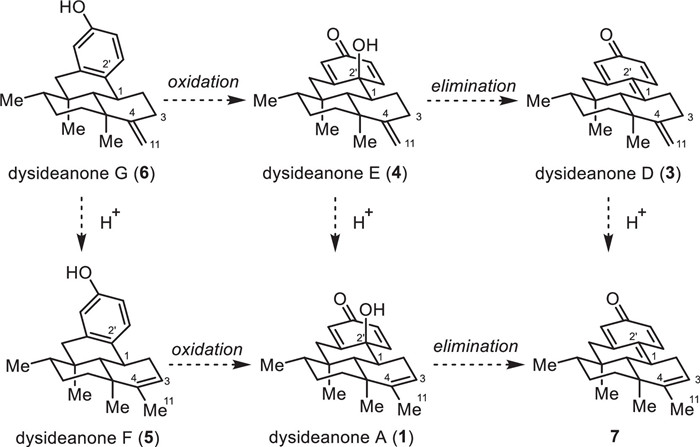
From a biosynthetic point of view, dysideanone G (6) might be a precursor compound for dysideanones A and D–F (1 and 3–5). As shown in Fig. 2, the double bond migration of dysideanone G (6) would give rise to dysideanone F (5). Oxidation of dysideanones G (6) and F (5) would render dysideanones E (4) and A (1), respectively. Further elimination of dysideanones E (4) and A (1) would afford dysideanone D (3) and its regioisomer 7.
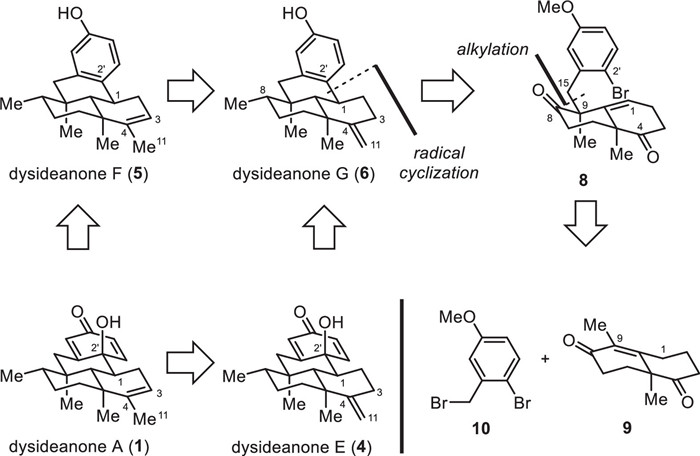
With the above biosynthetic transformations in mind, a retrosynthetic analysis of dysideanones A and E–G (1 and 4–6) was carried out, which was depicted in Fig. 3. Since dysideanone F (5) could be easily transformed from dysideanone G (6), and dysideanones A (1) and E (4) could be prepared through the oxidation of the corresponding phenol group of dysideanones F and G (6), respectively, our retrosynthetic analysis attention was focused on dysideanone G (6). As shown in Fig. 3, we envisioned that the methyl group at C8 and the terminal alkene at C4 of dysideanone G (6) could be converted from the corresponding carbonyl groups respectively. Disassembly of the C1–C2′ single bond of dysideanone G (6) would give rise to alkene bromide 8. Further disconnection of the C9–C15 single bond of 8 would trace back to Wieland–Miescher ketone derivative 9 and benzyl bromide 10.
Guided by the above retrosynthetic analysis, we started our adventure for the synthesis of dysideanones. Our synthesis commenced with the preparation of dysideanone G (6). As shown in Fig. 4, the more reactive C4 carbonyl group of Wieland–Miescher ketone derivative 9 was chemoselectively protected as a glycol acetal to give enone 11 in 94% yield [13]. The site-selective and diastereoselective alkylation reaction of 11 with benzyl bromide 10 proceeded smoothly under thermodynamical conditions (t-BuOK in THF at 40 ℃), giving the desired C9-alkylation product ketone 12 as a single diastereoisomer in satisfactory isolated yield (78%).
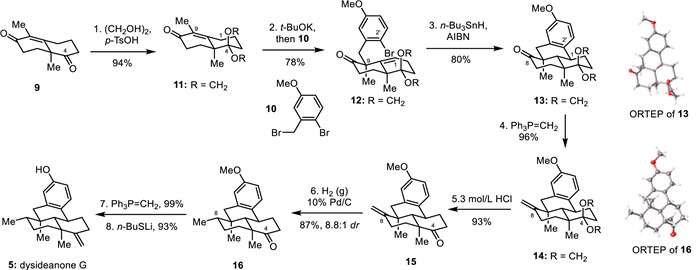
With alkene bromide 12 readily available, we switched to the key cyclization reaction to forge the core 6/6/6/6-fused tetracyclic ring system of dysideanones. Subjection of ketone 12 to radical reaction conditions [n-Bu3SnH and AIBN (2,2′-azobis(2-methylpropionitrile))] gave cyclized product 13 in 80% yield. The newly-formed single bond and the stereochemistry of the newly generated stereocenter were ambiguously confirmed by X-ray crystallographic analysis of cyclized compound 13 (see Supporting information for details).
With the tetracyclic ring system efficiently constructed, we moved forward to its transformation to dysideanone G (6). As depicted in Fig. 4, methylenation of tetracyclic ketone 13 using Wittig reagent (Ph3P=CH2) gave tetracyclic terminal olefin 14 in very high yield (96%). The glycol acetal protecting group was then removed under acidic conditions (3 mol/L HCl), rendering tetracyclic ketone 15 in 93% yield. Homogeneous hydrogenation of the terminal double bond of tetracyclic ketone 15 with RhCl(PPh3)3 under a hydrogen atmosphere gave high yield of reduction products (90%) but in a relatively low diastereoselectivity (1.2:1 dr), affording tetracyclic ketone 16 in 49% isolated yield, whose structure and absolute configuration were confirmed by X-ray crystallographic analysis (see Supporting information for details). The diastereoselectivity was significantly improved to 8.8:1 by using heterogeneous hydrogenation conditions with 10% Pd/C. The carbonyl group at C4 of ketone 16 was then converted to exo olefin with Wittig reagent (Ph3P=CH2) in an almost quantitively isolated yield (99%). Finally, the removal of the methyl protecting group of the phenol with n-BuSLi rendered dysideanone G (6) in 93% yield.
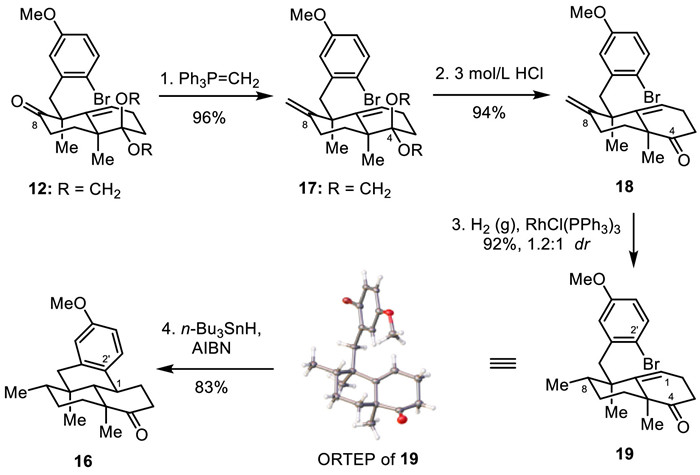
Alternatively, tetracyclic ketone 16 could be prepared from olefin bromide 12 by a late-stage cyclization sequence. As shown in Fig. 5, methylenation of the C8 carbonyl group of 12 with Ph3P=CH2 led to olefin 17 in 96% yield. Acidic-promoted deprotection of the glycol acetal of 17 with 3 mol/L HCl released the C4 carbonyl group, giving 18 in 94% yield. Homogeneous reduction of the terminal alkene with RhCl(PPh3)3 under a hydrogen atmosphere gave a high yield of reduction products (92%) and 1.2:1 diastereoselectivity, leading to bicyclic ketone 19 in 50% isolated yield. The structure of 19 was verified by X-ray crystallographic analysis (see Supporting information for details). Then the subjection of ketone 19 to radical reaction conditions (n-Bu3SnH and AIBN) gave tetracyclic ketone 16 in 83% yield. It should be noted that tetracyclic ketone 16 was prepared on a gram scale, which guaranteed the material supply for the gram-scale synthesis of dysideanone G (6).
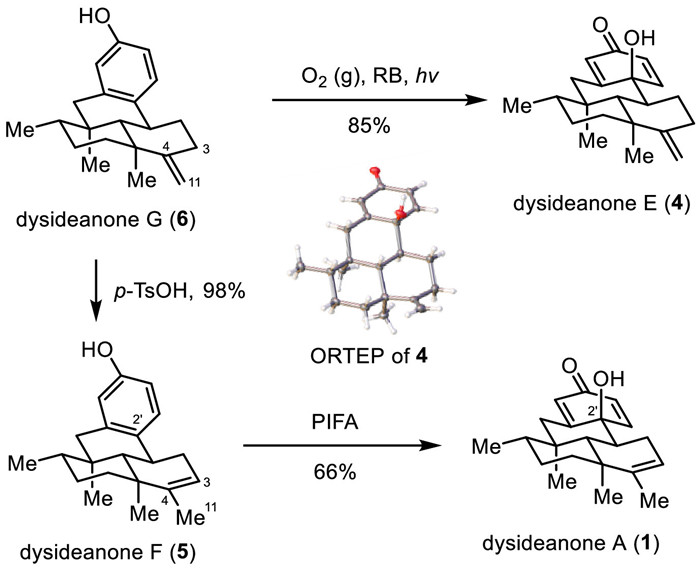
With gram-scale dysideanone G (6) in hand, we switched our attention to the synthesis of other members of dysideanones. As depicted in Fig. 6, light-promoted oxidation of dysideanone G (6) using O2 as oxidant and RB (Rose Bengal) as sensitizer rendered dysideanone E (4) in 85% yield [14,15]. The structure of dysideanone E (4) was verified by X-ray crystallographic analysis. Acidic-promoted double bond migration (p-TsOH, AcOH) of dysideanone G (6) gave dysideanone F (5) in very high yield (98%) [16]. However, light-promoted oxidation of dysideanone F (5) using O2 as oxidant and Rose Bengal as sensitizer only led to dysideanone A (1) in a very low yield (<10%). Much to our delight, this challenge was successfully solved by using PIFA [(bis(trifluoroacetoxy)iodo)benzene] as an oxidant, rendering dysideanone A (1) in 66% yield [17-19].
The spectroscopic data of synthetic dysideanones A and E–G (1 and 4–6) perfectly matched with those reported for the natural products. The absolute configuration of the synthetic dysideanones A and E–G (1 and 4–6) were ambiguously determined by the absolute configuration of the starting material Wieland–Miescher ketone derivative 9 and X-ray crystallographic analysis of dysideanone E (4) and other advanced intermediates. However, the optical rotation data (i.e., the signs and values) of dysideanones A (1) and E (4) are not identical to those reported for the natural samples, as shown in Table 1. To make things worse, through personal communication, we learned that the natural samples of dysideanones A (1) and E (4) had been completely consumed for structure characterization and bioactivity evaluation. Thus, we could not go further to determine the specific rotation and the absolute configuration of natural dysideanones A (1) and E (4). But we would like to note that the measured optical rotation, including the value and the sign, of a specific compound could be dramatically affected by the contamination of impurities, isomers, and even sample concentrations [20].
 DownLoad:
CSV
DownLoad:
CSV

|
In conclusion, we have accomplished the first enantioselective and divergent total synthesis of marine sesquiterpene (hydro)quinone meroterpenoids dysideanones A and E–G (1 and 4–6). The key reactions of our synthetic route included a site-selective and diastereoselective intermolecular alkylation of Wieland–Miescher ketone derivative 9 and benzyl bromide 10 to efficiently connect the sesquiterpene fragment and the aromatic moiety and an intramolecular radical cyclization reaction to construct the core 6/6/6/6-fused backbone of dysideanones. Dysideanone G (6) was prepared on a gram-scale and dysideanones A, E, and F (1, 4, and 5) were transformed from dysideanone G (6) divergently and easily.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Qunlong Zhang: Writing – review & editing, Writing – original draft, Methodology, Investigation, Formal analysis, Data curation. Jingyi Kang: Writing – review & editing, Methodology, Investigation, Formal analysis, Data curation. Jingwen Wang: Methodology, Investigation, Formal analysis, Data curation. Tiancheng Tan: Methodology, Investigation, Formal analysis, Data curation. Zhaoyong Lu: Writing – original draft, Supervision, Project administration, Funding acquisition, Conceptualization.
Financial support for this work was provided by the National Natural Science Foundation of China (Nos. 22171146, 21971121, and 22188101), the Fundamental Research Funds for the Central Universities, Nankai University (No. 63231199), and the State Key Laboratory of Medicinal Chemical Biology. We thank Drs. Qingxin Cui and Yiming Zhang (Nankai University) for NMR spectroscopic assistance and Dr. Quanwen Li (Nankai University) for X-ray crystallographic analysis.
Supplementary material associated with this article can be found, in the online version, at doi:
X.H. Tian, L.L. Hong, W.H. Jiao, H.W. Lin, Nat. Prod. Rep. 40 (2023) 718–749. doi: 10.1039/d2np00045h
M. Nazir, M. Saleem, M.I. Tousif, et al., Biomolecules 11 (2021) 957–1017. doi: 10.3390/biom11070957
P.A. García, Á.P. Hernández, A.S. Feliciano, Ma Á. Castro, Mar. Drugs 16 (2018) 292–343. doi: 10.3390/md16090292
S.N. Sunassee, M.T. Davies-Coleman, Nat. Prod. Rep. 29 (2012) 513–535. doi: 10.1039/c2np00086e
I.S. Marcos, A. Conde, R.F. Moro, et al., Mini-Rev. Org. Chem. 7 (2010) 230–254. doi: 10.2174/157019310791384128
W.H. Jiao, T.T. Xu, H.B. Yu, et al., J. Nat. Prod. 77 (2014) 346–350. doi: 10.1021/np4009392
T.Y. Zhou, G.T. Zhou, Patent, CN 201410678842, 2015.
W.H. Jiao, G.H. Shi, T.T. Xu, et al., J. Nat. Prod. 79 (2016) 406–411. doi: 10.1021/acs.jnatprod.5b01079
H.Y. Liu, M. Zhou, R.Y. Shang, et al., Chin. J. Nat. Med. 20 (2022) 148–154.
M.A. Haque, C.K. Jana, Chem. Eur. J. 23 (2017) 13300–13304. doi: 10.1002/chem.201703094
M.A. Haque, B.L. Sailo, G. Padmavathi, A.B. Kunnumakkara, C.K. Jana, Eur. J. Med. Chem. 160 (2018) 256–265. doi: 10.1016/j.ejmech.2018.08.088
X. Wang, S. Zhang, P. Cui, S. Li, Org. Lett. 22 (2020) 8702–8707. doi: 10.1021/acs.orglett.0c03294
C. Chong, Q. Zhang, J. Ke, et al., Angew. Chem. Int. Ed. 60 (2021) 13807–13813. doi: 10.1002/anie.202100541
S.P. Pitre, L.E. Overman, Chem. Rev. 122 (2022) 1717–1751. doi: 10.1021/acs.chemrev.1c00247
A.A. Ghogare, A. Greer, Chem. Rev. 116 (2016) 9994–10034. doi: 10.1021/acs.chemrev.5b00726
Q. Zhang, Y. Kuang, L. Chang, et al., Chin. Chem. Lett. 35 (2024) 108338. doi: 10.1016/j.cclet.2023.108338
L. Fang, Y. Chen, J. Huang, et al., J. Org. Chem. 76 (2011) 2479–2487. doi: 10.1021/jo102202t
G. Tong, Z. Liu, P. Li, Org. Lett. 16 (2014) 2288–2291. doi: 10.1021/ol5008263
A. Kimishima, H. Umihara, A. Mizoguchi, S. Yokoshima, T. Fukuyama, Org. Lett. 16 (2014) 6244–6247. doi: 10.1021/ol503175n
H.J. Chen, C.Y. Chen, P. Gao, Y. Wu, Tetrahedron 69 (2013) 9848–9851. doi: 10.1016/j.tet.2013.09.002

Figure 2 Plausible biosynthetic transformations of dysideanones A and D–G (1 and 3–6).

Figure 5 Alternative and gram-scale synthesis of tetracyclic ketone 16 from alkene bromide 12.

Figure 6 Divergent total synthesis of dysideanones A, E, and F (1, 4, and 5) from dysideanone G (6).
Table 1. Comparison of the optical rotation data of natural and synthetic dysideanones A and E–G (1 and 4–6).
|
|
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


