
Scheme 1.
State of the art for remote alkene functionalization.
Recent advances in remote C(sp3)–H functionalization via chelating group-assisted metal-catalyzed chain-walking reaction
Jian Han , Li-Li Zeng , Qin-Yu Fei , Yan-Xiang Ge , Rong-Hui Huang , Fen-Er Chen

The direct functionalization of ubiquitous C(sp3)–H bonds constitutes a straightforward and environmentally benign approach for building various carbon–carbon or carbon–heteroatom bonds [1,2]. It streamlines the chemical synthesis of target compounds and allows for late-stage modification of complex structures [3-8]. Over the past decades, a broad spectrum of advanced tactics has been established for the activation and selective functionalization of inert alkyl C–H bonds [9-22]. Recently, the functionalization of alkenes has emerged as a powerful technique for the rapid assembly of aliphatic molecular architectures [23-34]. In particular, remote functionalization of unactivated alkenes involving metal migration enables diverse functional groups to be incorporated selectively at inert C(sp3)–H bonds that are distal from the original olefinic site, is appealing and meaningful (Scheme 1a) [35,36]. The alkyl-metal intermediate generated upon alkene insertion is prone to undergo rapid β-hydride elimination, owing to the lack of a polar group adjacent to the double bond. Subsequent metal migratory reinsertion leads to the generation of a more stable C(sp3)–[M] intermediate, followed by the formation of a new C(sp3)–X bond at specific site that is otherwise difficult to access. The site-selectivity predominantly relies on the steric or electronic effect as the driving force for olefin chain walking process [37-40]. Thus, these transformations tend to take place at the position α to a stabilizing group based on thermodynamic grounds, such as aryl [41-44], boryl [45], alkenyl [46], and other moieties [47-51], or kinetically preferred terminal position [52-54]. Nevertheless, migratory functionalization at other sites beyond the α- and terminal positions along the alkyl chain is underdeveloped. On the other side, precise control of multi-site-selectivity via regiodivergent functionalization of the same raw material to afford a variety class of compounds is another vast challenge [55,56].
A chelation-assisted strategy renders a powerful and compensatory toolkit for achieving regiodivergent functionalization of distinct C(sp3)–H bonds, providing a predicable synthetic method for the assembly of α-, β- and γ-functionalized alkane architectures (Scheme 1b) [57-60]. The directing group (DG) appending on the non-conjugated alkenes plays a vital role in facilitating olefin insertion and regioselective chain walking transposition [61-63]. Certainly, the ligand is also significant, stabilizing the metallacycles of different sizes and promoting a selective cross-coupling. The preferred formation of a stable five- or six-membered metallacycle intermediate is believed to terminate the chain-walking at a specific methylene site, which serves as the driving force for excellent site-selective migratory functionalization. This emerging strategy not only offers a concise and efficient pathway for the synthesis of regiodiverse functionalized value-added products, but also paves a novel avenue for the development of alkene migratory functionalization.
Several elegant reviews have summarized the major achievements in remote alkene functionalization in the absence of an auxiliary group [37-40,64,65]. It is highly desirable to stress the breakthrough in chelating-group assisted migratory functionalization of electronically unbiased alkenes. In this article, we have summarized the recent progress made in the area of metal-catalyzed controllable remote functionalization of unactivated alkenes, and highlight the crucial roles of mono- and bidentate directing groups. This review is organized by migratory reaction types, including hydrofunctionalization and difunctionalization, followed by a conclusion of the current situation and an outlook for future direction in this field.
In 2020, the Koh group established a Ni-catalyzed remote hydroalkylation of non-conjugated alkenes tethered to a classic 8-aminoquinoline (AQ) directing group, leading to the efficient construction of β-alkylated amides (Scheme 2) [61]. Radical clock experiment implied the formation of high-valent intermediate 4 and 8 go through a single-electron transfer with an alkyl halide and radical recombination process. A plausible mechanism suggests that this reaction is initiated by a Ni(0) species, which is coordinated with quinoline amide and reacts with alkyl halide to form complex 4. It then undergoes β-H elimination to generate a key Ni(Ⅱ)H intermediate 5. This species 5 inserts into the C=C bond of alkene to afford complex 6, followed by alkene isomerization to form a relatively more stabilized five-membered nickelacycle (7). The Ni(Ⅱ) complex 7 is reduced to produce Ni(Ⅰ) species 8, which undergoes the second oxidative addition with alkyl halide. Finally, reductive elimination of Ni(Ⅲ) complex 9 delivers the desired β-alkylated product and releases nickel(Ⅰ) halide, which is further reduced by Mn to regenerate complex LnNi(0). In this protocol, several readily accessible primary and secondary alkyl halides were employed as both the hydride source and alkyl donor. The AQ auxiliary could be easily removed to furnish the valuable functionalized carboxylic acids. The authors found that the formation of transient five-membered nickelacycle stabilized by strongly coordinating bidentate auxiliary (AQ) was critical to achieve high chemo- and regioselectivity control.
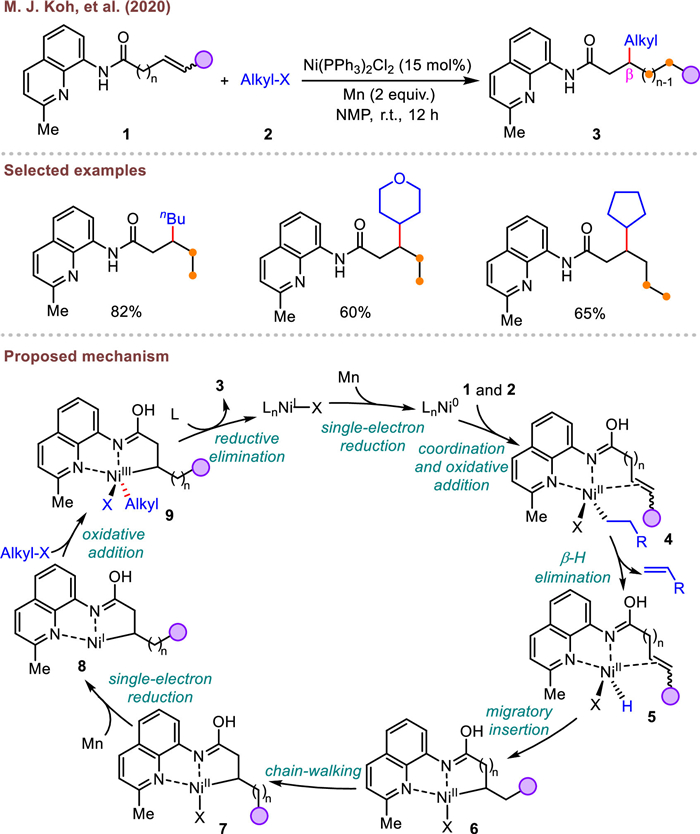
In 2022, Fu and co-workers described a concise and green method to realize a migratory hydroalkylation of unactivated alkenyl amide, resulting in the corresponding β-alkylated products in moderate yields and selectivities (Scheme 3) [66]. Different from Koh's work, the hydrogen source was from the silane. Besides, this approach benefited from the use of bench-stable and easy-to-handle silane regents, and devoid of stoichiometric metal additive. It should be noted that internal alkenes were also amenable to the optimal conditions, albeit with moderate efficiency.
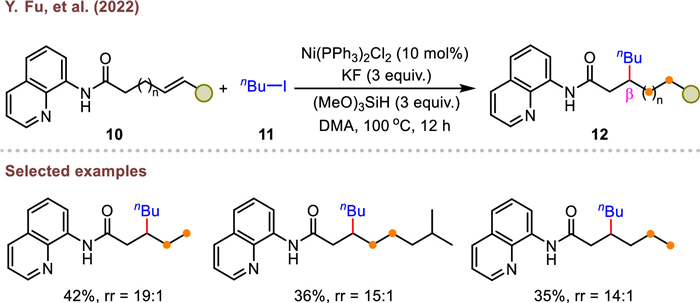
Enantiomerically pure cyclohexenyl esters were often recognized as prevalent scaffolds in bioactive molecules. Very recently, Zhu's group disclosed a NiH-catalyzed asymmetric migratory hydroalkylation of cyclic unactivated alkenes, which enabled expeditious assembly of multi-substituted chiral cyclohexyl esters (Scheme 4) [67]. By adopting a dynamic kinetic isomeric transformation (DYKAT) strategy, racemic and isomeric cycloalkenes were readily converted to challenging thermo-dynamically disfavored chiral 1,2-cis disubstituted cycloalkanes containing vicinal stereocenters with excellent regio-, diastereo- and enantioselectivity. Remarkably, cycloalkene bearing a racemic sterically congested quaternary stereocenter was also well accommodated to this catalytic framework. In this work, the crucial step is that the IL1NiIIH complex (16) in-situ generated undergoes a rapid and reversible migratory insertion and chain-walking process to afford a stable five-membered alkyl-nickel isomer (17) that is coordinated by the ester group and chiral bisoxazoline ligand (L1). Subsequent capture of alkyl radical and reductive elimination produces the desired product. Notably, the coordination effect of ester substituent gives rise to the preferred formation of intermediate 17 to other potential alkylnickel isomers.
Remote alkene functionalization with tunable regioselectivity from the identical alkene starting material provides a step-economic and cost-efficient route to prepare structurally diverse aliphatic molecules, which is a fascinating yet challenging task for the synthetic community.
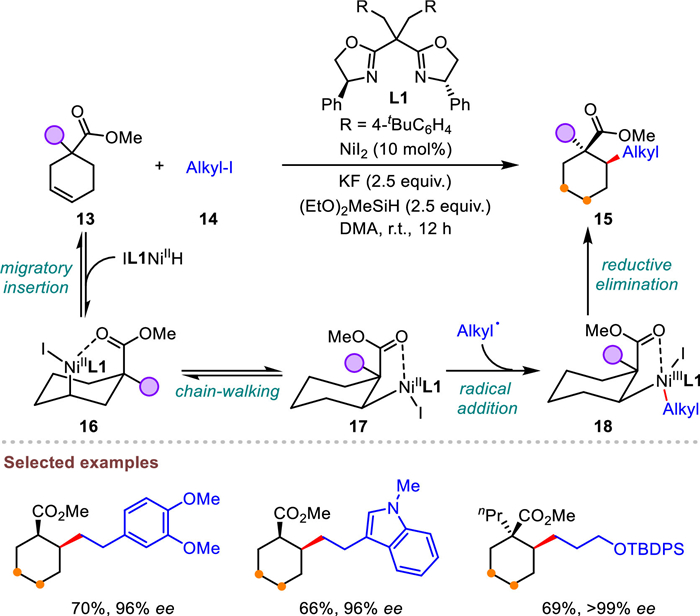
Inspired by the advancements in several examples of site-specific remote functionalizations assisted by strongly coordinating directing auxiliaries [61,67], Wang et al. envisioned that a clever combination of a suitable weak monodentate directing group and distinct optimized ligands could achieve regiodivergent remote alkene functionalization. In accordance with this idea, they developed a pioneering method to accomplish α- and β-selective hydroalkylation of electronically unbiased alkenes directed by simple amide (Scheme 5) [62]. Using either 1,10-phenanthroline (L2) or bisoxazoline (L3) as a ligand, a broad range of α- or β-alkylated products were synthesized with up to 94% yield and 99:1 regioselectivity. This innovative protocol demonstrated excellent tolerance towards diverse functional groups, as well as outstanding regioselectivity and stereoselectivity. The alkenes with different carbon-chain lengths reacted with alkyl iodides smoothly to yield the target products in high yields and regioisomeric ratios. In addition, when the alkenes containing either methyl or aryl groups bearing significant steric bulk at α-position were chosen as the substrate, they also proved to be effective coupling partners. Moreover, the mild nature of this approach made it suitable for late-stage modification of complex, pharmaceutically relevant compounds. Control experiments implied the irreplaceability of amide directing group and carbonyl coordination mode. Other mechanistic studies including competition, intermediate and deuterium labelling experiments were conducted seriously as well. These mechanistic observations serve as evidence for the formation of ligated 5- or 6-membered nickelacycle (23 and 25) via NiH migratory in the presence of distinct ligands constitutes the turnover-limiting step.
Around the same time, Lu and his colleagues reported a similar transformation via a temperature-dependent strategy, providing a straightforward method to access diverse α- and β-branched alkyl amines with good practicality and broad substrate scope (Scheme 6) [68]. As shown in Scheme 6, the α-alkylated products (27) were obtained at 100 ℃, and the β-alkylated products (28) were formed at a slightly low temperature (10 ℃). While switching the solvent to a mixture of DMF/tBuOH, maintaining the reaction temperature at 21 ℃, and replacing achiral ligand L4 with a C2-symmetric chiral bisoxazoline ligand L1, a wide array of enantioenriched β-branched alkyl amines (29) were synthesized in a catalytic asymmetric variant manner. Mechanistic investigations suggested that the formation of a stable, carbonyl-group-coordinated alkyl-nickel complex serves as the driving force of metal migration. The switchable site-selectivity is ascribed to the thermodynamic and kinetic properties of different reduction elimination intermediates, which is controlled through judicious chose of reaction temperature.
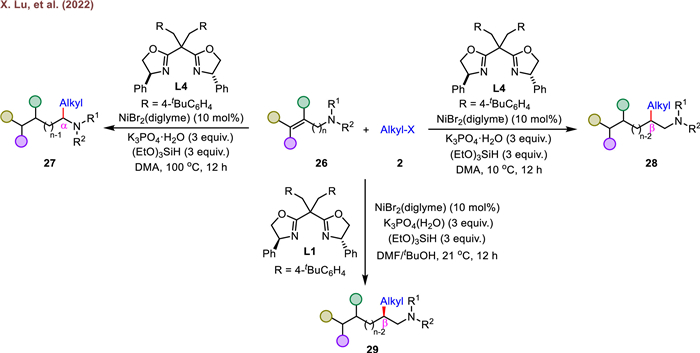
In the same year, the group of Shu investigated a Ni/Co-catalyzed regio- and site-selective hydroalkylation of alkenyl amines with alkyl halides in a similar fashion (Scheme 7) [69]. Through the ingenious catalyst-controlled strategy, a broad set of α-branched, β-branched and linear alkyl amines were successfully prepared in moderate to high yields with high levels of regioselectivities. They found the choice of a suitable metal catalysis and nitrogen-based ligand were crucial to achieve diverse regioselectivities. Notably, a wide variety of unactivated alkenes, including various N-acyl allylic amines, long-chain alkenes, 1,1-disubstituted and trisubstituted alkenes, all well performed to generate corresponding multi-substituted aliphatic amide derivatives. Afterwards, they further extended the reactivity to include stereocontrolled coupling of alkenes with alkyl halides dictated by the chiral bisoxazoline ligand (L8), delivering diverse enantiopure β-branched alkyl amines. Overall, this study showcases the versatility and potential of Ni/Co-catalyzed hydroalkylation for the synthesis of diverse alkyl amines, offering a valuable addition to the growing toolbox of metal-catalyzed synthetic methodologies.
Soon afterward, Martin and co-workers implemented a unique nickel-catalyzed interrupted deaminative chain-walking reaction of unactivated alkenes (Scheme 8) [70]. Through adjusting to the ligands (L9 for β-selectivity and L10 for γ-selectivity), a series of site-selective alkylated products were obtained in moderate to excellent yields. Alkyl pyridinium salts (Katritzky salts) have drawn considerable interest from chemists due to their important applications as alkyl radical precursors in nickel- or photoredox-catalyzed deaminative alkylation [71]. With the bench-stable alkyl pyridiniums, a wide range of alkyl groups were efficiently incorporated at β- or γ-position of the alkenyl amide in the presence of different ligands. It was worth noting both terminal and internal alkenes worked well in this protocol.

Enantiomerically pyrrolidine motifs are omnipresent in chemical and pharmaceutical industries. In 2023, Rong's team reported a straightforward Co/Ni-catalyzed enantioselective hydroalkylation of unactivated N-heteroendocyclic alkenes to achieve an array of chiral C2-/C3-alkylated pyrrolidines (Scheme 9) [72]. For the Co-catalyzed C3-selective hydroalkylation, the process is initiated by the generation of L11CoIH from the reaction between ligated CoII precursor with (MeO)2MeSiH. The L11CoIH reacts smoothly with an alkyl iodide (14) to produce an alkyl radical and IL11CoIIH, followed by a migratory insertion to the C=C bond of N-heteroendocyclic alkene 36 to deliver alkyl-cobalt intermediate (39). Then the intermediate 39 reacted with alkyl radical through the process of oxidative addition and subsequent reductive elimination to produce the desired C3-alkylated product (37). Turning the ligand framework to L12, C2-selective alkylated products 38 were obtained in moderate to good yields. In this catalytic cycle, IL12NiIIH (41) is generated equally from the corresponding L12NiIH via single-electron oxidation of the alkyl halide. The subsequent migratory insertion of IL12NiIIH species and chain-walking process produces a stable five-membered nickelacycle (42). Trapping of alkyl radical by the nickel center (42) sets the stage for following reductive elimination to deliver the anticipated C2-selective alkylated product (38) and complete the catalytic cycle. Both the two enantioselective reductive C(sp3)–C(sp3) couplings are distinguished by their modularity, mild conditions, wide scope, excellent regioselectivity and high enantioselectivity. This approach provides a preparatively useful route to access functionalized enantiopure alkylated pyrrolidine derivatives as well as structurally complex bioactive molecules.

Alkynes are essential structural moieties frequently found in many natural products, bioactive molecules, chemical probes, and functional materials, and also serve as important synthetic precursors in organic synthesis. Although remarkable progress has been made in the direct hydroalkynylation of activated alkenes, remote hydroalkynylation of nonconjugated aliphatic alkenes still lags behind.
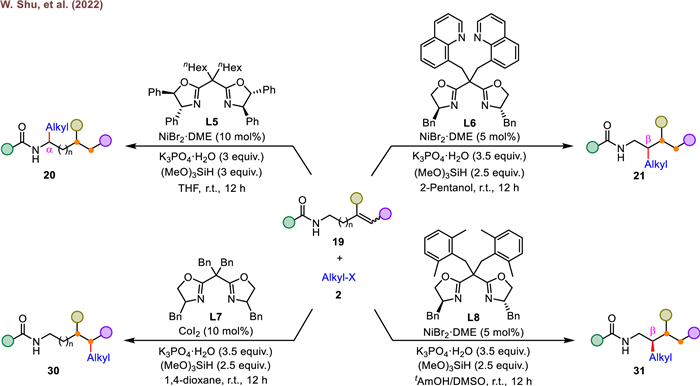
In 2016, the research group of Li described the first example of iridium-catalyzed enantioselective hydroalkynylation of α, β-unsaturated amides with complete γ-selectivity and high enantioselectivity (Scheme 10, top) [73]. They found weak-coordination-assisted iridium-catalyzed alkene isomerization could also provide a promising approach to achieve catalytic alkynylation of remote methylene C–H bond. As an extension of their interest in remote alkene hydrofunctionalization, they disclosed an elegant method for the synthesis of γ-alkynylated amides from the unactivated alkenyl amides and silyl alkynes (Scheme 10, bottom) [63]. Utilizing (R)-DM-Segphos (L13) as a ligand, a number of stereodefined alkynylated amides were obtained in good yields and enantioselectivities. To account for the observed γ-selectivity, a reliable mechanism was established [73]. First, oxidative addition of (triisopropylsilyl)acetylene to the bisphosphine-ligated iridium center generates an alkynyl iridium hydride 49. Then the complex 49 inserts into the alkene (47) to form an alkynyl-iridium intermediate (50), which undergoes chain walking and subsequent rapid alkene reinsertion to afford a stable five-membered metallacycle 52, in which the β-carbon of the amide is attached to the iridium center. Finally, the desired γ-desired product 48 is delivered upon the β-hydride elimination of complex 52, along with regeneration of Ir(Ⅰ) catalyst. This study significantly expanded the scope of iridium-catalyzed enantioselective reaction and provided an efficient method for the synthesis of enantioenriched γ-alkynylated alkyl amides, which are useful scaffolds in organic synthesis and drug development.
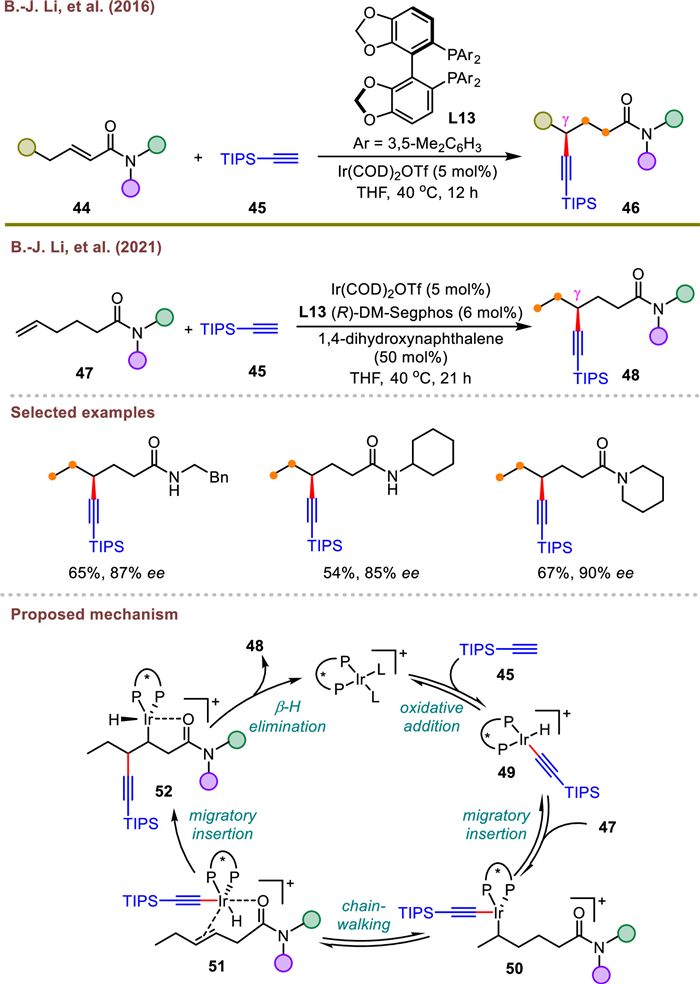
By means of ligand-controlled, directing group-assisted strategy, Wang's group recently conducted an extensive investigation of hydroalkynylation reaction of allyl amides at remote α-position adjacent to nitrogen with triisopropylsilyl alkynyl bromides 54 (Scheme 11) [74]. And the formation of a ligated five-member nickelacycle via NiH migration was believed to be the critical step in controlling the reaction regioselectivity. Simultaneously, the Markovnikov β-selective hydroalkynylation of allyl amides with aryl/alkyl substituted alkynyl bromides proceeded smoothly in the presence of dppf (L14) or chiral bisoxazoline ligand (L15), leading to the formation of racemic or enantioenriched β-alkynylated products with excellent yields, respectively. It merits mentioning that this transformation could also be carried out on gram-scale without any significant loss of its efficiency and selectivity. Moreover, the alkyne group in chiral product could undergo a series of stereospecific transformations without the loss of enantiopurity.
Over the past decades, the synthesis of amine derivatives has garnered sustainable attention due to their wide applications in pesticide chemistry, agrochemicals and pharmaceuticals. Direct hydroamination of simple and abundant alkenes with high regioselectivity has been well established, expanding the toolbox used for the synthesis of amine motifs. Recently, migratory hydroamination enables amino group to be installed at inert methyl C–H site that is distant from the initial olefinic site, offering a fascinating strategy to forge C–N bond skeleton.
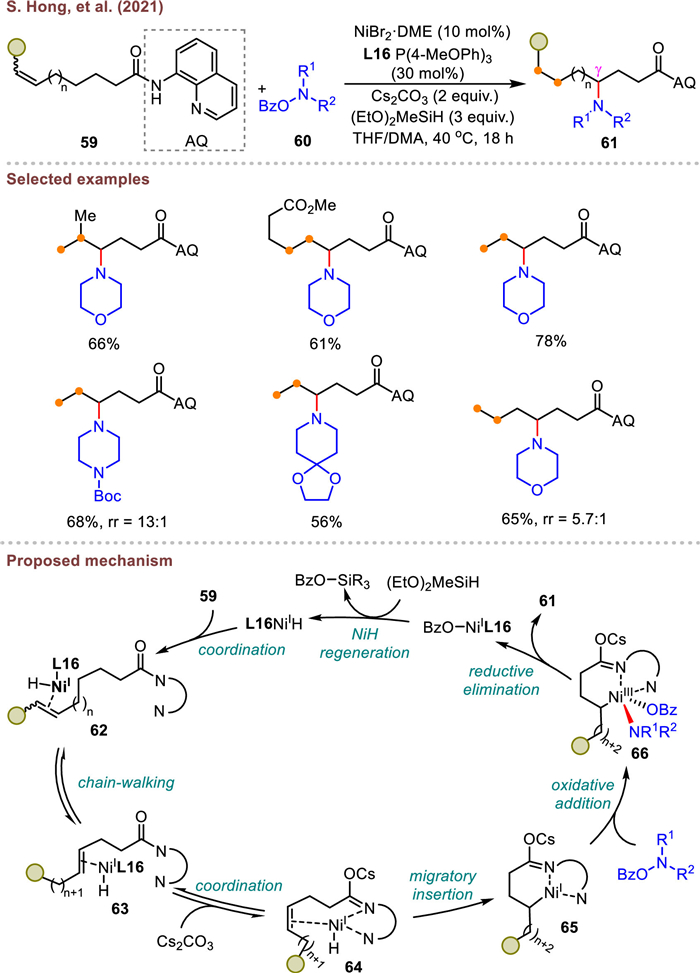
In 2021, Hong et al. introduced a NiH-catalyzed migratory hydroamination of unactivated alkenes via a chelation-assisted strategy, by using O-benzoylhydroxylamine as the aminating reagent (Scheme 12) [75]. This method offered a practical and highly versatile entry to the synthesis of high value-added γ-aminated products using L16 P(4-MeOPh)3 as a ligand. The researchers demonstrated that the internal alkenes and long-chain alkenes were also amenable to this transformation. Moreover, late-stage functionalization of hydroaminated product and upscaling to gram quantities is demonstrated, showcasing the feasibility and robustness of this protocol. The excellent regioselectivity was dictated by the formation of six-membered nickelacycle (65) stabilized by 8-aminoquinoline directing auxiliary.
Following the recent advances in the field of metal-catalyzed migratory functionalization, a significant breakthrough was achieved by Zhang's group. They demonstrated that the synergistic combination of a native amide directing group and a 1,10-phenanthroline ligand (L17) facilitated the nickel-catalyzed site-selective hydroamination of unbiased aliphatic alkenes (Scheme 13) [76]. Control experiments and computational studies suggested that the utilize of 2-iodo-2-methylpropane works as both a mild hydride source and radical precursor, which plays a vital role in accessing the remote functionalized products. Subsequently, they further extended the reaction scope by performing α-selective hydroetherification with alcohols as compatible coupling partners. Under the same set of reaction conditions, diversely substituted alkoxyl group were well installed on the C(sp3)–H position adjacent to the acylamino group. Remarkably, exclusive α-regioselectivity was observed in all cases.

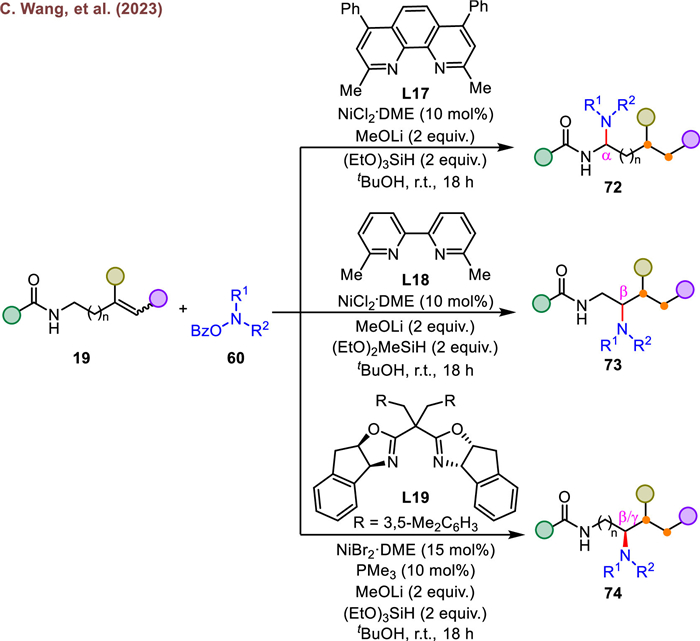
Shortly thereafter, the group of Wang further applied weak directing-group-assisted, ligand-controlled strategy to the regiodivergent coupling of alkenes with O-benzoylhydroxylamines (Scheme 14) [77]. By utilizing different ligands (L17 or L18), a rich variety of highly valuable 1,1-, and 1,2-diamines were obtained with good-to-excellent regioselectivity. In addition to the remarkable regioselectivity, an enantioselective variant of this transformation was also realized using chiral dibenzyl-substituted pH-Box ligand (L19). This approach leaded to the formation of chiral 1,2- and 1,3-diamineproducts with high efficiency, further demonstrating the versatility of this method. The amide directing group is indispensable and irreplaceable in facilitating the hydroamination of unactivated alkenes, as evidenced by the mechanistic outcomes. And the pyridyl and oxazoline moieties of the ligand have a significant effect on the reaction regio- and stereoselectivity.
Amides play a prominent role in various areas of chemistry given their ubiquitous nature in proteins, polymers, and pharmaceuticals. In recent years, the direct remote hydroamidation of readily available alkenes has gained wide attention from the synthetic community.
Dioxazolone, which is used as an effective source of electrophilic amide, has been witnessed as an important building block in various organic transformations [78]. In 2019, an efficient thioether-directed Ni-catalyzed remote γ-methylene C(sp3)–H amidation of alkene has been pioneered by Yu's team (Scheme 15) [79]. The mild conditions tolerated a diverse array of thioether substituted alkenes and dioxazolones, leading to the formation of various C–N bonds with good to excellent regiocontrol. Mechanistically, a six-membered nickelacycle (78) is generated from the migratory insertion of L20NiIH into C=C of alkene, followed by a spontaneous reversible β-hydride elimination/reinsertion process to deliver a more stable five-membered nickelacycle (79). Subsequent interception by an amidating reagent gives the target γ-amidated product. The formation of the preferred five-membered nickelacycle is believed to effectively terminate the chain-walking isomerization at a specific γ-methylene site. In this protocol, the weakly coordinating thioether group plays a key role in stabilizing the key nickelacycle intermediate (79).

gem‑Difluoroalkenes are not only valuable synthons for building blocks in organic synthesis, but also widely found in pharmaceuticals and pesticides as they usually work as stable bioisosteres for the metabolically susceptible carbonyl groups (aldehydes and ketones). Recently, the group of Wang described a NiH-catalyzed migratory and non-migratory hydrodifluoroallylation of alkenyl amide via an ingenious auxiliary-control strategy (Scheme 16) [80]. In the presence of common amide directing group, the migratory difluoroallylation happened at α-position exclusively. Interestingly, when the chelating group turned to phthalimide auxiliary, the Markovnikov-selective difluoroallylation proceeded smoothly and yielded various β-, γ-, δ-, and ε-selective difluoroallylated products using different chain lengths of alkenyl amide substrates. The authors speculated that the distinct site-selectivity primarily leveraged the difference in the electronic characteristics of the directing group, which affected the stability of the nickelacycle with different sizes.

Alkylboranes, known for their low toxicity and decent stability, are commonly utilized as versatile synthons for numerous synthetic transformations, as the C–B bonds can be readily converted into an array of C–C, C–O, or C–N bonds. Engle and co-workers recently presented a novel tungsten-catalyzed hydroboration of unactivated alkenes to realize the synthesis of β-borated amides under mild conditions (Scheme 17) [81]. This procedure had broad substrate scope and excellent reaction efficiency, in which both terminal olefins and internal aliphatic alkenes exhibit high compatibility. Besides, the chemoselectivity of this protocol was underscored by the preservation of the additional C=C bond in the product. The mechanistic proposal invokes a W(0)/W(Ⅱ) catalytic cycle. Initially, the LnW(0) coordinates with both carbonyl group and the C=C bond in the alkene substrate to form complex 89, which goes through an oxidative addition of allylic sp3 C–H to access intermediate 90. Subsequently, the LnW(0)H complex inserts into the π-allyl moiety, resulting in the formation of an isomerized alkene (91). The oxidative addition of H–Bpin to LnW(0) (92), followed by C–B reductive elimination then yields the final product 88, with simultaneous regeneration of the propagating LnW(0) catalyst. It was notable that the formation of a five-membered LnW(Ⅱ) species stabilized by carbonyl group acted as the driving force for the metal migration, resulting in the observed high regioselectivity. This approach provided a practical and compensatory platform for the synthesis of β-borated amide, which would otherwise be challenging to access.

Polysubstituted alkenes are widely present in bioactive compounds, natural product analogs, and drug candidates. As such, it is of great importance to develop a robust catalysis to access such compounds. Very recently, a convenient synthetic strategy was designed by Liang's group. They realized a ruthenium-catalyzed remote migration arylation of unactivated alkenes with excellent regio- and stereoselectivity (Scheme 18) [82]. Various aryl bromides bearing electron withdrawing or donating groups were found to be viable, affording corresponding β-arylated alkenes (96) in moderate to excellent yields. A proposed catalytic cycle for the β-selective arylation is outlined in Scheme 18. Notably, the formation of a pivotal five-membered rutheniumacycle intermediate (101) rationalizes the regio- and stereoselectivity. Control experiments further confirmed that both ruthenium catalysts, (RuHCl(CO)(PPh3)3 and [RuCl2(p-cymene)]2) were indispensable for the success of this transformation.

The multi-component reaction of alkenes is far difficult than the aforementioned two-component reaction, however, it can rapidly increase molecular complexity by coupling olefins with a series of electrophiles and nucleophiles. Considerable efforts have been dedicated to this field, nevertheless, migratory difunctionalization of unactivated alkene remains a rarity. This is primarily due to the inherent difficulties in controlling the chemo- and site-selectivity during multi-component coupling reactions. In recent examples, chelating strategy has emerged as a promising solution to address these issues.
In 2018, Zhao and co-workers disclosed a groundbreaking nickel-catalyzed dicarbofunctionalization of allyl amines containing a pendant weakly coordinating group of pyrimidine, which constitutes the first example of intermolecular 1,3-difunctionalization reaction of alkenes (Scheme 19) [83]. The use of excess amounts of the electrophile and nucleophile efficiently overcome the side reactions, such as two-component cross-coupling and homodimerization competing reactions. Additionally, alkenyl boronic acids were also found to be competent coupling partners, significantly expanding the substrate scope. As for the mechanism, it is postulated that aryl iodide initially undergoes oxidative addition to the LnNi(0) species involving a two-electron mechanism. Then the resulting LnNi(Ⅱ) intermediate inserts into the olefin, placing the electrophile to the position farther from pyrimidine, thus generates a six-membered nickelacycle species (107). The intermediate 107 tends to form a more stable five-membered chelation complex (109) via β-hydride elimination and reinsertion process. The subsequent transmetalation of 109 with organoboronic acid gives rise to complex 110, which then undergoes reductive elimination to furnish the α, γ-dicarbofunctionalized product (106) while regenerating the LnNi(0) species. Deuterium labelling experiments provided convincing evidence that the implementation of this transformation depends on a crucial chain-walking step. Moreover, the formation of a more stable five-membered chelation complex coordinated by pyridine group is considered to be a determining factor for excellent 1,3-regioselectivity. Despite the alkene scope being limited to terminal alkenes, this finding has opened up a new avenue for the migratory difunctionalization of unactivated alkenes.

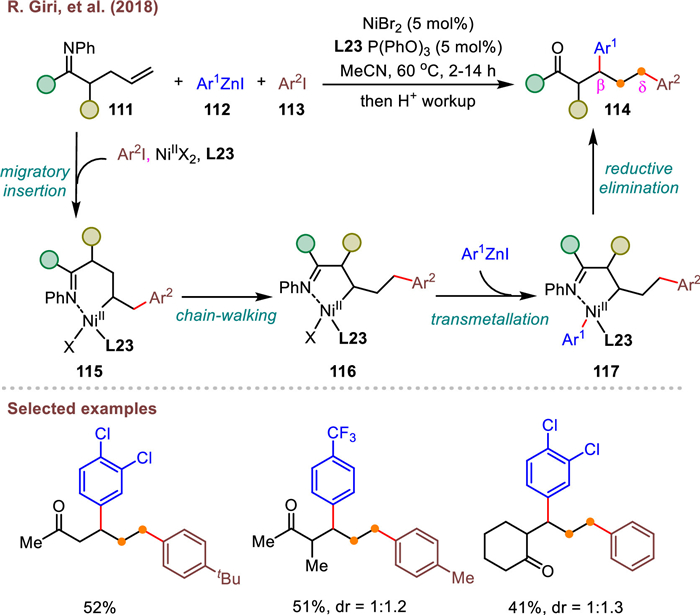
Following the success of Ni-catalyzed 1,3-dicarbofunctionalizan of common allyl amines, Giri et al. proposed an unprecedented nickel-catalyzed diarylation of unactivated alkene in ketimines as well in the same year (Scheme 20) [84]. A broad collection of aryl halides, arylzinc reagents and terminal alkenyl imines performed well to furnish synthetically useful β, δ-diaryl ketones (114) after hydrolysis workup. It is well-known that the structurally diversified β, δ-diaryl ketones are difficult to prepare by other ways. In this process, the key five-membered intermediate 116 is generated from a ring contraction of imine-ligated six-membered C(sp3)–NiX species 115, which contributes to defining the regiochemical outcome. It is noteworthy that the electron-deficient phosphine ligand (L23) plays an important role in promoting the β–H elimination and Ni–H reinsertion process to enable the success of ring contraction.
In 2019, Hong's group developed a cationic Pd(Ⅱ)-catalyzed 1,1-diarylation of unactivated alkenes directed by 8-aminoquinoline auxiliary group, affording a facile access to 1,1-diarylated products in moderate to excellent yields with excellent regioselectivities (Scheme 21) [85]. The alkene scope comprised α- or β-substituted alkenyl amides and cyclic internal alkenes. Besides, the coupling partners encompassed a wide range of nucleophiles, including indoles, azaindoles and electron-rich anilines. The addition of AgSbF6 was found to be key factor for the success of this transformation, as the weakly coordinating anion SbF6− could stabilize the dicationic potential surface in reaction solution. Control experiments revealed that the bidentate AQ directing group plays a crucial role in promoting the first regioselective nucleophilic attack on the alkene by forming the palladacycle.

The directing-group assisted migratory functionalization of easily accessible bulk chemical alkenes has emerged as a robust catalytic platform to achieve remote functionalization of inert C(sp3)–H bonds. The pendant chelating group is utilized to facilitate migratory insertion and chain walking transposition. Besides, take advantage of coordinating group stabilization, five- or six-membered metallacycle generates smoothly and subsequent cross-coupling proceeds at specific site (not limited to the position proximal to DG or terminal position). To a certain extent, this strategy represents a significant step-forward to expedite the construction of a rich library of functionalized alkane architectures. Despite intense endeavors, several preeminent challenges in chain-walking metal catalysis remain to be addressed:
1. The existing studies mainly focused on the migratory functionalization of terminal or 1,2-disubstituted alkenes. In fact, owing to the inherent low reactivity and sterically hinderance effect of tri- or tetrasubstituted olefins, the dynamic displacement of the metal catalyst throughout the alkyl chain is hindered, resulting in unfavorable chain-walking reaction. Consequently, there is a pressing need to innovate new catalytic system in tackling these reaction reactivity and selectivity issues.
2. The study of asymmetric remote hydrofunctionalization of unactivated alkenes with the assistance of directing auxiliary is in its early stage and other intriguing reactions remain unexplored. Besides, migratory difunctionalization of electronically unbiased alkenes with enantioselective variant at a remote site has never yet to be achieved. The key enantio‑determining step involves C(sp3)–[M] intermediate being intercepted by coupling partner to rapidly form a stereogenic center in the presence of chiral ligand, which is not easily realized [45]. No doubt, it is more difficult for the asymmetric internal alkenes to forge two skipped chiral centers [86].
3. The chelating strategy has displayed powerful ability in the promotion of migratory functionalization of unactivated alkenes and has permitted the control of chemo- and regioselectivity. However, the pre-install and removal of directing groups is not atom- and step-economic, which diminishes its practical application [87]. Employing common unactivated alkenes bearing native functional groups, such as free alkenyl amines, carboxylic acids and ketones, as substrates in combination with newly designed ligand scaffold, it would break through the substrate scope limitation in the remote functionalization.
Overall, the chelating-group directed transition-metal catalyzed remote functionalization of alkenes with predictable and tunable site-selectivity brings great opportunities for assembly of molecular complexity, which is otherwise difficult to access. This would pique the long-term interest of more researchers to study and more interesting work would come on the way.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the National Natural Science Foundation of China (No. 22001116), the Basic and Applied Basic Research Foundation of Guangdong Province (No. 2020A1515110816) and funds provided by Changzhou University (No. ZMF23020217).
X. Chen, K.M. Engle, D.H. Wang, J.Q. Yu, Angew. Chem. Int. Ed. 48 (2009) 5094–5115. doi: 10.1002/anie.200806273
Y. Qin, L. Zhu, S. Luo, Chem. Rev. 117 (2017) 9433–9520. doi: 10.1021/acs.chemrev.6b00657
J. Yamaguchi, A.D. Yamaguchi, K. Itami, Angew. Chem. Int. Ed. 51 (2012) 8960–9009. doi: 10.1002/anie.201201666
J. Wencel-Delord, F. Glorius, Nat. Chem. 5 (2013) 369–375. doi: 10.1038/nchem.1607
T. Cernak, K.D. Dykstra, S. Tyagarajan, et al., Chem. Soc. Rev. 45 (2016) 546–576. doi: 10.1039/C5CS00628G
D.J. Abrams, P.A. Provencher, E.J. Sorensen, Chem. Soc. Rev. 47 (2018) 8925–8967. doi: 10.1039/C8CS00716K
J.H. Docherty, T.M. Lister, G. Mcarthur, et al., Chem. Rev. 123 (2023) 7692–7760. doi: 10.1021/acs.chemrev.2c00888
Y. Li, B. Yuan, Z. Sun, W. Zhang, Green Synth. Catal. 2 (2021) 267–274. doi: 10.1016/j.gresc.2021.06.001
G. Rouquet, N. Chatani, Angew. Chem. Int. Ed. 52 (2013) 11726–11743. doi: 10.1002/anie.201301451
G. He, B. Wang, W.A. Nack, G. Chen, Acc. Chem. Res. 49 (2016) 635–645. doi: 10.1021/acs.accounts.6b00022
J. He, M. Wasa, K.S.L. Chan, et al., Chem. Rev. 117 (2017) 8754–8786. doi: 10.1021/acs.chemrev.6b00622
B. Liu, A.M. Romine, C.Z. Rubel, et al., Chem. Rev. 121 (2021) 14957–15074. doi: 10.1021/acs.chemrev.1c00519
S.K. Sinha, S. Guin, S. Maiti, et al., Chem. Rev. 122 (2022) 5682–5841. doi: 10.1021/acs.chemrev.1c00220
X.Q. Hu, J.R. Chen, W.J. Xiao, Angew. Chem. Int. Ed. 56 (2017) 1960–1962. doi: 10.1002/anie.201611463
J.C.K. Chu, T. Rovis, Angew. Chem. Int. Ed. 57 (2018) 62–101. doi: 10.1002/anie.201703743
X. Wu, Z. Ma, T. Feng, C. Zhu, Chem. Soc. Rev. 50 (2021) 11577–11613. doi: 10.1039/D1CS00529D
Y. Yang, W. Gao, Y. Wang, et al., ACS Catal. 11 (2021) 967–984. doi: 10.1021/acscatal.0c04618
Q. Zhang, B.F. Shi, Acc. Chem. Res. 54 (2021) 2750–2763. doi: 10.1021/acs.accounts.1c00168
Y. Han, B.F. Shi, Acta Chim. Sinica 81 (2023) 1522–1540. doi: 10.6023/A23070336
Y.J. Wu, G. Liao, B.F. Shi, Green Synth. Catal. 3 (2022) 117–136. doi: 10.1016/j.gresc.2021.12.005
D.Q. Dong, J.C. Song, S.H. Yang, et al., Chin. Chem. Lett. 33 (2022) 1199–1206. doi: 10.1016/j.cclet.2021.08.067
T. Li, L. Pan, Y. Zhang, et al., Chin. Chem. Lett. 35 (2024) 108897. doi: 10.1016/j.cclet.2023.108897
T. Pintauer, K. Matyjaszewski, Chem. Soc. Rev. 37 (2008) 1087–1097. doi: 10.1039/b714578k
W.T. Eckenhoff, T. Pintauer, Catal. Rev. 52 (2010) 1–59. doi: 10.1080/01614940903238759
S.W.M. Crossley, C. Obradors, R.M. Martinez, R.A. Shenvi, Chem. Rev. 116 (2016) 8912–9000. doi: 10.1021/acs.chemrev.6b00334
X.X. Wang, X. Lu, Y. Li, et al., Sci. China Chem. 63 (2020) 1586–1600. doi: 10.1007/s11426-020-9838-x
Z. Zhang, S. Bera, C. Fan, X. Hu, J. Am. Chem. Soc. 144 (2022) 7015–7029. doi: 10.1021/jacs.1c13482
R.M. Romero, T.H. Wöste, K. Muñiz, Chem. Asian J. 9 (2014) 972–983. doi: 10.1002/asia.201301637
G. Yin, X. Mu, G. Liu, Acc. Chem. Res. 49 (2016) 2413–2423. doi: 10.1021/acs.accounts.6b00328
Z.L. Li, G.C. Fang, Q.S. Gu, X.Y. Liu, Chem. Soc. Rev. 49 (2020) 32–48. doi: 10.1039/C9CS00681H
S.O. Badir, G.A. Molander, Chem 6 (2020) 1327–1339. doi: 10.1016/j.chempr.2020.05.013
J. Diccianni, Q. Lin, T. Diao, Acc. Chem. Res. 53 (2020) 906–919. doi: 10.1021/acs.accounts.0c00032
S. Zhu, X. Zhao, H. Li, L. Chu, Chem. Soc. Rev. 50 (2021) 10836–10856. doi: 10.1039/D1CS00399B
J. Han, R. He, C. Wang, Chem. Catal. 3 (2023) 100690. doi: 10.1016/j.checat.2023.100690
E. Larionov, H. Li, C. Mazet, Chem. Commun. 50 (2014) 9816–9826. doi: 10.1039/C4CC02399D
A. Vasseur, J. Bruffaerts, I. Marek, Nat. Chem. 8 (2016) 209–219. doi: 10.1038/nchem.2445
H. Sommer, F. Juliá-Hernández, R. Martin, I. Marek, ACS Cent. Sci. 4 (2018) 153–165. doi: 10.1021/acscentsci.8b00005
Y. Li, D. Wu, H.G. Cheng, G. Yin, Angew. Chem. Int. Ed. 59 (2020) 7990–8003. doi: 10.1002/anie.201913382
D. Janssen-Müller, B. Sahoo, S.Z. Sun, R. Martin, Isr. J. Chem. 60 (2020) 195–206. doi: 10.1002/ijch.201900072
S. Ghosh, S. Patel, I. Chatterjee, Chem. Commun. 57 (2021) 11110–11130. doi: 10.1039/D1CC04370F
Y. He, Y. Cai, S. Zhu, J. Am. Chem. Soc. 139 (2017) 1061–1064. doi: 10.1021/jacs.6b11962
J. Xiao, Y. He, F. Ye, S. Zhu, Chem 4 (2018) 1645–1657. doi: 10.1016/j.chempr.2018.04.008
X. Chen, Z. Cheng, J. Guo, Z. Lu, Nat. Commun. 9 (2018) 3939. doi: 10.1038/s41467-018-06240-y
W. Wang, C. Ding, Y. Li, et al., Angew. Chem. Int. Ed. 58 (2019) 4612–4616. doi: 10.1002/anie.201814572
Y. Zhang, B. Han, S. Zhu, Angew. Chem. Int. Ed. 58 (2019) 13860–13864. doi: 10.1002/anie.201907185
R. Yu, S. Rajasekar, X. Fang, Angew. Chem. Int. Ed. 59 (2020) 21436–21441. doi: 10.1002/anie.202008854
C. Zhang, C.B. Santiago, L. Kou, M.S. Sigman, J. Am. Chem. Soc. 137 (2015) 7290–7293. doi: 10.1021/jacs.5b04289
Y. Ebe, M. Onoda, T. Nishimura, H. Yorimitsu, Angew. Chem. Int. Ed. 56 (2017) 5607–5611. doi: 10.1002/anie.201702286
X. Yu, H. Zhao, S. Xi, et al., Nat. Catal. 3 (2020) 585–592. doi: 10.1038/s41929-020-0470-9
Q. Zhang, S. Wang, J. Yin, et al., Angew. Chem. Int. Ed. 61 (2022) e202202713. doi: 10.1002/anie.202202713
S. Ma, H. Fan, C.S. Day, et al., J. Am. Chem. Soc. 145 (2023) 3875–3881. doi: 10.1021/jacs.2c13054
F. Zhou, J. Zhu, Y. Zhang, S. Zhu, Angew. Chem. Int. Ed. 57 (2018) 4058–4062. doi: 10.1002/anie.201712731
S.Z. Sun, C. Romano, R. Martin, J. Am. Chem. Soc. 141 (2019) 16197–16201. doi: 10.1021/jacs.9b07489
Q. Wang, H. Jung, D. Kim, S. Chang, J. Am. Chem. Soc. 145 (2023) 24940–24951.
J. Gao, M. Jiao, J. Ni, et al., Angew. Chem. Int. Ed. 60 (2021) 1883–1890. doi: 10.1002/anie.202011231
Y. Zhang, J. He, P. Song, et al., CCS Chem. 3 (2021) 2259–2268.
J.A. Gurak Jr., K.S. Yang, Z. Liu, K.M. Engle, J. Am. Chem. Soc. 138 (2016) 5805–5808. doi: 10.1021/jacs.6b02718
B. Shrestha, P. Basnet, R.K. Dhungana, et al., J. Am. Chem. Soc. 139 (2017) 10653–10656. doi: 10.1021/jacs.7b06340
J. Derosa, V.T. Tran, M.N. Boulous, et al., J. Am. Chem. Soc. 139 (2017) 10657–10660. doi: 10.1021/jacs.7b06567
X. Zhao, H.Y. Tu, L. Guo, et al., Nat. Commun. 9 (2018) 3488. doi: 10.1038/s41467-018-05951-6
X. Chen, W. Rao, T. Yang, M.J. Koh, Nat. Commun. 11 (2020) 5857. doi: 10.1038/s41467-020-19717-6
L. Zhao, Y. Zhu, M. Liu, et al., Angew. Chem. Int. Ed. 61 (2022) e202204716. doi: 10.1002/anie.202204716
W.W. Zhang, B.J. Li, Tetrahedron Lett. 73 (2021) 153108. doi: 10.1016/j.tetlet.2021.153108
Y. Wang, Y. He, S. Zhu, Acc. Chem. Res. 55 (2022) 3519–3536. doi: 10.1021/acs.accounts.2c00628
Y. Li, G. Yin, Acc. Chem. Res. 56 (2023) 3246–3259. doi: 10.1021/acs.accounts.3c00505
X.X. Wang, Y.T. Xu, Z.L. Zhang, et al., Nat. Commun. 13 (2022) 1890. doi: 10.1038/s41467-022-29554-4
C. Chen, W. Guo, D. Qiao, S. Zhu, Angew. Chem. Int. Ed. 62 (2023) e202308320. doi: 10.1002/anie.202308320
J.W. Wang, D.G. Liu, Z. Chang, et al., Angew. Chem. Int. Ed. 61 (2022) e202205537. doi: 10.1002/anie.202205537
P.F. Yang, W. Shu, Angew. Chem. Int. Ed. 61 (2022) e202208018. doi: 10.1002/anie.202208018
J. Rodrigalvarez, H. Wang, R. Martin, J. Am. Chem. Soc. 145 (2023) 3869–3874. doi: 10.1021/jacs.2c12915
F.S. He, S. Ye, J. Wu, ACS Catal. 9 (2019) 8943–8960. doi: 10.1021/acscatal.9b03084
X. Wang, J. Xue, Z.Q. Rong, J. Am. Chem. Soc. 145 (2023) 15456–15464. doi: 10.1021/jacs.3c03900
Z.X. Wang, X.Y. Bai, H.C. Yao, B.J. Li, J. Am. Chem. Soc. 138 (2016) 14872–14875. doi: 10.1021/jacs.6b10415
M. Liu, H. Shi, X. Meng, et al., CCS Chem. (2023), 10.31635/ccschem. 023.202303288. doi: 10.31635/ccschem.023.202303288
C. Lee, H. Seo, J. Jeon, S. Hong, Nat. Commun. 12 (2021) 5657. doi: 10.1038/s41467-021-25696-z
T. Song, Y. Luo, K. Wang, et al., ACS Catal. 13 (2023) 4409–4420. doi: 10.1021/acscatal.3c00238
L. Xie, J. Liang, H. Bai, et al., ACS Catal. 13 (2023) 10041–10047. doi: 10.1021/acscatal.3c01845
K.M. van Vliet, B. de Bruin, ACS Catal. 10 (2020) 4751–4769. doi: 10.1021/acscatal.0c00961
B. Du, Y. Ouyang, Q. Chen, W.Y. Yu, J. Am. Chem. Soc. 143 (2021) 14962–14968. doi: 10.1021/jacs.1c05834
L. Zhu, J. Huang, F. Meng, et al., Org. Chem. Front. 10 (2023) 5623–5630. doi: 10.1039/D3QO01053H
T.C. Jankins, R. Martin-Montero, P. Cooper, et al., J. Am. Chem. Soc. 143 (2021) 14981–14986. doi: 10.1021/jacs.1c07162
Y.Y. Luan, J.Y. Li, X.Y. Gou, et al., Org. Lett. 25 (2023) 5111–5116. doi: 10.1021/acs.orglett.3c01844
W. Li, J.K. Boon, Y. Zhao, Chem. Sci. 9 (2018) 600–607. doi: 10.1039/C7SC03149A
P. Basnet, R.K. Dhungana, S. Thapa, et al., J. Am. Chem. Soc. 140 (2018) 7782–7786. doi: 10.1021/jacs.8b03163
J. Jeon, H. Ryu, C. Lee, et al., J. Am. Chem. Soc. 141 (2019) 10048–10059. doi: 10.1021/jacs.9b04142
Y. Xi, W. Huang, C. Wang, J. Am. Chem. Soc. 144 (2022) 8389–8398. doi: 10.1021/jacs.2c03411
H. Wang, M.J. Koh, Cell Rep. Phys. Sci. 3 (2022) 100901. doi: 10.1016/j.xcrp.2022.100901

Scheme 2 Nickel-catalyzed β-selective hydroalkylation of alkenyl amides under reductive condition.

Scheme 3 Migratory β-selective hydroalkylation of alkenyl amides without extra ligand.

Scheme 5 Ligand-controlled NiH-catalyzed regiodivergent chain-walking hydroalkylation of alkenes.

Scheme 6 Nickel-catalyzed switchable site-selective hydroalkylation of alkenes by temperature regulation.

Scheme 8 Nickel-catalyzed site-selective deaminative hydroalkylation of alkenyl amides.

Scheme 9 Co/Ni-catalyzed regio- and enantioselective hydroalkylation of N-heteroendocyclic alkenes.

Scheme 11 Nickel-catalyzed regio- and enantioselective of hydroalkynylation of alkenes.

Scheme 13 Nickel-catalyzed α-selective hydroamination and hydroetherification of unactivated alkenes.

Scheme 14 Ligand-controlled NiH-catalyzed regiodivergent and enantioselective hydroamination of alkenes.

Scheme 15 Ni-catalyzed thioether directed γ-selective hydroamidation of unactivated alkenes.

Scheme 16 Auxiliary-controlled NiH-catalyzed regiodivergent hydrodifluoroallylation of alkenes.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:









 下载:
下载: