Figure 1.
Representative examples for one-dimensional PAHs and heteroaromatics.
Nonalternant isomer of pentacene fusing two azulene units
Bo Yu , Pengchen Du , Jianwen Guo , Hanshen Xin , Jianhua Zhang

Polycyclic aromatic hydrocarbons (PAHs) have long been a significant research topic in the fields of chemistry and materials science [1-5]. Therefore, understanding the structural characteristics, physicochemical properties, and performance of one-dimensional higher-order benzene molecules becomes crucial in studying PAHs [1,3,6-13]. For example, fully benzenoid fused ring pentacene and indenofluorenes-based fused ring indeno[1,2-b]fluorene are the high-performance performing five-ring-fused organic semiconductors for field-effect transistors [2,6]. However, as the number of benzene rings increases (> 5), the solubility and stability of the compounds decrease significantly. Currently, all-benzene fused ring compounds, such as pentacene, still face inherent issues including low solubility and poor photostability. These drawbacks greatly restrict their potential applications [14,15]. Therefore, there has been a strong emphasis on researching new non-benzene fused ring structures to overcome these limitations [8,16-19].
Azulene, a nonbenzenoid 10π electron isomer of naphthalene, has a dipole moment of 1.08 D [20,21]. Recently, azulene has attracted considerable attention in the design, synthesis, and application of PAH/heteroaromatic compounds, owing to its distinctive structure and electronic properties [3,17,22-31]. The synthesized PAH/heteroaromatic compounds exhibited different properties and performances by replacing two benzenoid rings with one nonbenzenoid azulene unit. For example, Takai and coworkers have reported an azulene-fused linear PAH PhenAz2 (Fig. 1) with small bandgap, good solubility and high stability [32]. Chi and coworkers synthesized azulene-fused acenes compound 3 (Fig. 1) that are much more stable than their respective benzene hydrocarbon isomers [33]. In 2020, we reported an unexpected cyclization reaction that resulted in the synthesis of an azulene-pyridine-fused heteroaromatic compound Az-Py-1 (Fig. 1). Az-Py-1 exhibited notable hole mobilities of up to 0.29 cm2 V−1 s−1 [34]. To further explore the potential application of the azulene-fused PAHs [17,35], as in that of the linear π-extension isomers of pentacene, a novel derivative dicyclohepta[b, h]-as-indacene (Az-PH1, Fig. 1) was synthesized. This derivative consists of two symmetrically fused azulene rings and was created using rhodium(II)- or nickel(0)-catalyzed ring-closing techniques [36].
Here, we compared the energy gap, aromaticity, and photo-oxidative stability of Az-PH1 with that of pentacene using density functional theory, electrochemical, and photophysical tests. Notably, Az-PH1 exhibited significantly higher photo-oxidative stability compared to pentacene. This bottom-up synthesis of linear materials incorporating azulene is of great significance for expanding the application of non-benzene PAHs in carbon materials and supplementing one-dimensional fused ring materials.
The synthetic route for Az-PH1 was initially designed and presented in Scheme 1a, Path A. Via Vilsmeier-Haack reaction, compound 1a was prepared from 2-bromoazulene (S1) [37] in 89% yield. Compound 1a underwent a self-coupling reaction by using Ni(cod)2 as a catalyst in DMF to give product 1b with 61% yield [16]. Then we treated 1b with p-toluenesulfonyl hydrazide in methanol, affording the intermediate 1c in 79% yield. The rhodium-catalyzed intramolecular condensation reaction of 1c successfully afforded Az-PH1 in 18% yield [36]. Through the synthetic route A, the total yield of Az-PH1 synthesized from S1 was 7.7%.

In order to improve the overall synthesis yield of Az-PH1, we explored alternative synthetic routes in addition to optimizing each reaction step in Path A. As shown in Scheme 1b, compound 1a was treated with p-toluenesulfonyl hydrazide in methanol before self-coupling reaction, giving compound 1d with 89% yield. During our attempt to synthesize intermediate 1c from 1d using Ni(cod)2 catalyzed self-coupling reaction, we unexpectedly obtained Az-PH1 in 23% yield. Upon further investigation, we propose that Ni(cod)2, a catalyst for the self-coupling reaction, can also serve as a catalyst for the cyclization reaction of N-tosylhydrazones [38,39]. The proposed mechanism for the formation of Az-PH1 is illustrated in Scheme S2 (Supporting information). Starting from S1, the total yield of Az-PH1 prepared by Path B was 18.2%, which was significantly higher than that of Path A. Besides, Path B has shorter synthesis steps and avoids the use of rhodium catalysts, making it a more economical and efficient route.
To enhance the solubility of Az-PH1 (2.34 mg/mL in chloroform), we introduced alkyl chains and synthesized Az-PH2 and Az-PH3, as depicted in Scheme 1c. Compounds 2a and 3a, substituted by butyryl and octanoyl groups, were obtained through a Friedel-Crafts acylation reaction, with 57% and 62% yields, respectively. The carbonyl group was then reduced by BH3 [40]. Following Path B for the synthesis of Az-PH1 from S1, 13,14-dibutyldicyclohepta[b, h]-as-indacene (Az-PH2) and 13,14-dioctyldicyclohepta[b, h]-as-indacene (Az-PH3) were obtained at a higher yield 41% and 46%, respectively, due to the increase in solubility (see Supporting information for the details).
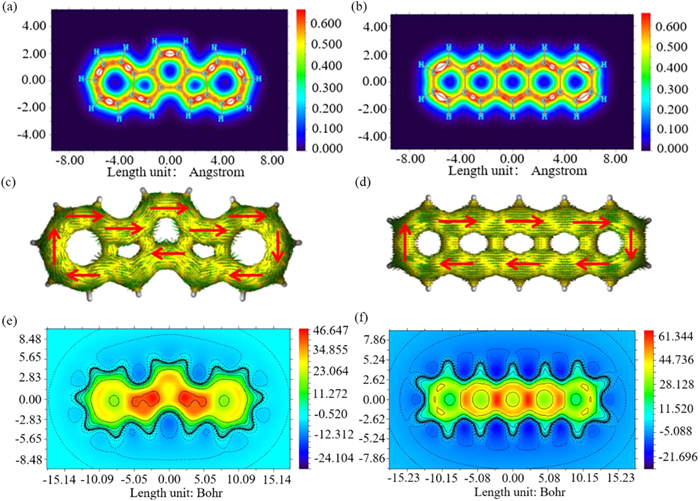
To compare the electronic structure of Az-PH1 with its benzenoid isomer pentacene, we calculated (ⅰ) the localized orbital locator contributed from π electrons only (LOL) [41,42], (ⅱ) the anisotropy of the induced current density (ACID) [43,44], and (ⅲ) the two-dimensional isochemical shielding surface (2D-ICSS) [19] as shown in Fig. 2. The LOL was a function reflecting the localization of electrons in different regions of a molecule. The LOL image of Az-PH1 (Fig. 2a) indicates that the main delocalization path of π electrons is in the left and right azulene outer and middle six-membered rings. This leads to the development of a conjugated π ring on the outer side of Az-PH1, resembling pentacene (Fig. 2b) and exhibiting a trend towards global aromaticity. The aromatic character of Az-PH1 is evidenced by the ACID plots, which exhibit a clockwise ring current flow similar to that of pentacene (Figs. 2c and d). The 2D-ICSS map of Az-PH1 indicates a stronger shielding effect on the five-membered ring side, which differs from the characteristic observed in pentacene (Figs. 2e and f). Additionally, the nucleus independent chemical shift (NICS) analysis (Fig. S1 in Supporting information) also supports the notion that the five-membered ring side of Az-PH1 exhibits good aromaticity, as indicated by its highest NICS value of -13.442 ppm.
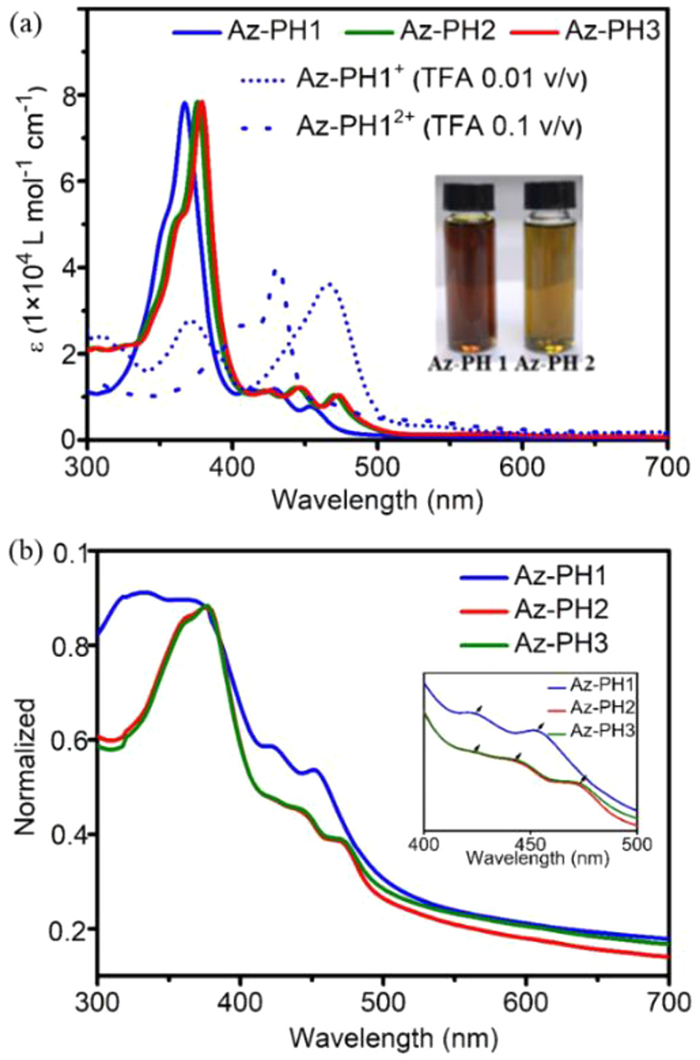
To evaluate these nonalternant isomers of pentacene fusing azulene units as new molecular material candidates, their photophysical and electrochemistry properties were determined, and the details were summarized in Table 1. The UV–vis absorption spectra of Az-PH1/2/3 in dichloromethane (DCM) solution were compared in Fig. 3a and Fig. S3 (Supporting information). Az-PH1/2/3 have the same conjugated skeleton and exhibit strong absorptions at 365 nm, 371 nm and 374 nm, respectively, and three moderate absorptions peaks in the range of 400–500 nm. A weak absorption centered at 576 nm for Az-PH1,582 nm for Az-PH2, and 586 nm for Az-PH3 was observed (Fig. S2 and Tables S2–S4 in Supporting information), deriving from the symmetrically forbidden S0 to S1 transition. The yellowish green color observed in the dichloromethane solution of Az-PH2 and Az-PH3 is attributed to their ability to transmit a higher amount of green light compared to Az-PH1 (Fig. 3a). When the trifluoroacetic acid (TFA) was added in DCM solution of Az-PH1, a two-step absorption change that may correspond to the generation of single and double protonated species, was detected (Fig. 3a and Fig. S3) [17]. Besides, Az-PH12+ displays a strong blue fluorescence under 365 nm light irradiation (Figs. S4 and S5 in Supporting information). Furthermore, we drop-coated Az-PH1/2/3 on glass slides, and the absorption profile of the Az-PH1 film did not resemble its solution state, as shown in Fig. 3b. Az-PH1 showed a strong π→π* peak at 300–350 nm, along with only two moderate absorption peaks of the n→π* in the range 400–500 nm (Fig. 3b, inset). Nevertheless, the characteristic absorption peaks of Az-PH2 and Az-PH3 are almost similar to those in DCM solution. The results demonstrated that the introduction of alkyl chains has a significant effect on the stacking behavior of conjugated skeleton. On the other hand, Az-PH1/2/3 exhibit a weak emission spectrum that originates from S2 or higher excited states. Three main emission peaks were observed at 475 nm, 490 nm, and 489 nm, respectively (Fig. S6 in Supporting information).
Thermogravimetric analysis (TGA) was performed under a nitrogen atmosphere to investigate the thermal properties of pentacene, picene, Az-PH1. TGA measurement demonstrates that pentacene, picene and Az-PH1 was thermally stable upon the onset decomposition temperatures of 336 ℃, 338 ℃, 312 ℃, respectively (Fig. S7 in Supporting information). The electrochemical properties of Az-PH1/2/3 were investigated by cyclic voltammetry and shown in Fig. S8 (Supporting information). All potentials reported were quoted with reference to the ferrocene-ferrocenium (Fc/Fc+) couple at a scan rate of 0.05 V/s [45]. From the onset of the oxidation and reduction potentials, the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energy levels of Az-PH1/2/3 are calculated and listed in Table 1. All three molecules displayed similar quasi-reversible reduction peaks (Eonsetred1 = -1.82 V, -1.74 V, -1.73 V). Az-PH2 and Az-PH3 showed reversible oxidation peaks, while Az-PH1 exhibited an irreversible wave. Possibly this can be attributed to the effect of the alkyl chains.
To gain insight into the structural and electronic properties, density functional theory (DFT) calculations were conducted on Az-PH1/2/3 and pentance with the B3LYP/6–311G(d) level of theory. The entire backbone of Az-PH1 possesses a highly planar geometry (dihedral angle < 1°), and the alkyl chains of Az-PH2 and Az-PH3 cross on both sides of the parent nuclear plane (Fig. S9 in Supporting information, side view). The calculated HOMO-LUMO gap of Az-PH1 (2.23 eV) is much narrower than that of picene (4.23 eV) and is comparable to pentacene (2.19 eV). The electron density distribution of HOMO orbital of Az-PH1 is effectively delocalized over the entire backbone, whereas its LUMO is relatively localized on the azulene ring. Furthermore, owing to the introduction of electrondonating alkyl chains, the energy gaps of Az-PH2 and Az-PH3 can be reduced due to an increase in their HOMO energy levels, and the HOMO energy levels were determined to be -4.82 eV for Az-PH1, -4.66 eV for Az-PH2 and Az-PH3. This increase aligns with the non-mirrorrelated frontier molecular orbital (FMO) geometry of azulene.
To investigate the photo-oxidative stability of the materials, we tested the absorption spectral changes of Az-PH1 in DCM under 365 nm light irradiation and measured its half-life time (t1/2). Compound Az-PH1 exhibits fine photo-stability under 365 nm light irradiation (Fig. 4a) and has a moderate t1/2 of 18.8 days in PAHs (Fig. 4b), strongly depending on the mode of ring annulation and the topology of their π-electron system [32]. In contrast to Az-PH1, pentacene is unstable since three double bonds only 1 Clar's sextet can be drawn in only one ring of its π-system without duplication (t1/2 < 13 min, in tetrahydrofuran solution) [46].

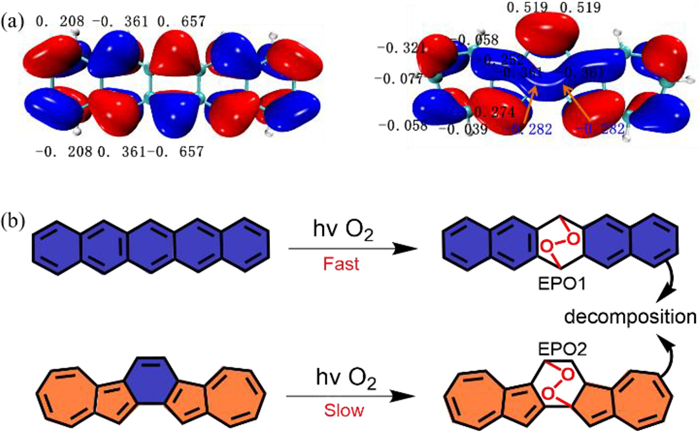
To further analyze the decomposition pathways of Az-PH1 and pentacene, the FMO theory and the theoretical evaluation of the energy changes (∆Eo-a) was studied [47-49]. In FMO theory the second-order perturbation energy ∆Ed-a (detailed information about the calculation of ΔEd-a can be found in the Supporting Information) can predict well that the addition of 1O2 is preferred at the innermost ring in higher acenes where the atom orbitals have the largest coefficients [50]. When considering Az-PH1 as isoelectronic structures of pentacene, the most reactive position should be the central ring, which was confirmed by the hydrogen spectrum of Az-PH1* (Fig. S10 in Supporting information). As shown in Fig. 5a, the highest atomic coefficients of Az-PH1 are also on the center ring (0.519, -0.282, respectively), which is an asymmetric atomic coefficient. Therefore, acenes (pentacene, Az-PH1) preferentially produce two endoperoxides (EPOs) EPO1 and EPO2 (Fig. 5b) when they were exposed to light.
The obtained data from the oxygenations of acenes, as shown in Table S1 (Supporting information), indicate that the ΔEo-a of EPO2 (0.147 Hartree) is greater than that of EPO1 (-0.067 Hartree). This implies that the reaction rate of Az-PH1 is slower compared to pentacene when reacting with 1O2. From a thermodynamic standpoint, this implies that the formation of the azulene-fused ring is more favored energetically when compared to the benzene-fused ring.
In conclusion, three non-alternating isomers of pentacene, named Az-PH1/2/3, have been constructed by fusing two azulene units. Az-PH1 was initially synthesised by rhodium(II)-catalyzed cyclization of bis(N-tosylhydrazone). Interestingly, Az-PH1 was also accidentally obtained in a one-step tandem reaction catalysed by Ni(cod)2. Az-PH1 displays a comparable global aromaticity flow to pentacene when analyzing LOL, ACID and 2D-ICSS. DFT calculations confirm that the entire backbone of Az-PH1 possesses a remarkably planar structure. The introduction of alkyl chains significantly alters the stacking behavior of Az-PH1. Meanwhile, Az-PH2 and 3 show a narrow bandgap by increasing the HOMO energy level. Besides, Az-PH1 is more stable to photooxygen than pentacene, which is consistent with the thermodynamic calculations. These results suggest that the PHA combining azulene will achieve more attractive electronic and photophysical properties. The π-extended system Az-PH1 with highly planar geometry provides promising carrier transport materials for the organic electronics neighbourhood.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This research was financially supported by the National Natural Science Foundation of China (No. 22101170).
Supplementary material associated with this article can be found, in the online version, at doi:
A. Pron, P. Gawrys, M. Zagorska, et al., Chem. Soc. Rev. 39 (2010) 2577–2632. doi: 10.1039/b907999h
K. Takimiya, S. Shinamura, I. Osaka, et al., Adv. Mater. 23 (2011) 4347–4370. doi: 10.1002/adma.201102007
J. Mei, Y. Diao, A.L. Appleton, et al., J. Am. Chem. Soc. 135 (2013) 6724–6746. doi: 10.1021/ja400881n
B. Hou, J. Li, X. Yang, et al., Chin. Chem. Lett. 33 (2022) 2147–2150. doi: 10.1016/j.cclet.2021.08.079
T. Yang, Q. Wu, F. Dai, et al., Adv. Funct. Mater. 30 (2019) 1903889.
H. Jiang, S. Zhu, Z. Cui, et al., Chem. Soc. Rev. 51 (2022) 3071. doi: 10.1039/D1CS01136G
C. Sutton, C. Risko, J.L. Brédas, Chem. Mater. 28 (2015) 3–16.
T.G. Lohr, J.I. Urgel, K. Eimre, et al., J. Am. Chem. Soc. 142 (2020) 13565–13572. doi: 10.1021/jacs.0c05668
P.M. Ajayan, Nature 575 (2019) 49–50.
K.S. Novoselov, V.I. Fal'ko, L. Colombo, et al., Nature 490 (2012) 192–200. doi: 10.1038/nature11458
X. Li, X. Wang, L. Zhang, et al., Science 319 (2008) 1229–1232. doi: 10.1126/science.1150878
R. Dorel, A.M. Echavarren, Acc. Chem. Res. 52 (2019) 1812–1823. doi: 10.1021/acs.accounts.9b00227
H. Okamoto, N. Kawasaki, Y. Kaji, et al., J. Am. Chem. Soc. 130 (2008) 10470–10471. doi: 10.1021/ja803291a
C. Tonshoff, H.F. Bettinger, Chemistry 27 (2021) 3193–3212. doi: 10.1002/chem.202003112
J.I. Urgel, S. Mishra, H. Hayashi, et al., Nat. Commun. 10 (2019) 861. doi: 10.1038/s41467-019-08650-y
H. Xin, C. Ge, X. Jiao, et al., Angew. Chem. Int. Ed. 57 (2018) 1322–1326. doi: 10.1002/anie.201711802
S. Wang, M. Tang, L. Wu, et al., Angew. Chem. Int. Ed. 61 (2022) e202205658. doi: 10.1002/anie.202205658
J.X. Dong, H.L. Zhang, Chin. Chem. Lett. 27 (2016) 1097–1104. doi: 10.1016/j.cclet.2016.05.005
P. Liu, X.Y. Chen, J. Cao, et al., J. Am. Chem. Soc. 143 (2021) 5314–5318. doi: 10.1021/jacs.1c01826
L. Ou, Y. Zhou, B. Wu, et al., Chin. Chem. Lett. 30 (2019) 1903–1907. doi: 10.1016/j.cclet.2019.08.015
D.L. Wang, S.F. Li, W. Li, et al., Chin. Chem. Lett. 22 (2011) 789–792. doi: 10.1016/j.cclet.2011.01.003
H. Xin, B. Hou, X. Gao, Acc. Chem. Res. 54 (2021) 1737–1753. doi: 10.1021/acs.accounts.0c00893
Y. Wang, Z. Jiang, L. Liao, Chin. Chem. Lett. 27 (2016) 1293–1303. doi: 10.1016/j.cclet.2016.07.004
F. Ullah, H. Chen, C.Z. Li, Chin. Chem. Lett. 28 (2017) 503–511. doi: 10.1016/j.cclet.2016.11.009
L.C. Murfin, M. Weber, S.J. Park, et al., J. Am. Chem. Soc. 141 (2019) 19389–19396. doi: 10.1021/jacs.9b09813
H. Nishimura, N. Ishida, A. Shimazaki, et al., J. Am. Chem. Soc. 137 (2015) 15656–15659. doi: 10.1021/jacs.5b11008
X. Zhang, T. Li, Chin. Chem. Lett. 28 (2017) 2058–2064. doi: 10.1016/j.cclet.2017.09.008
Y. Fei, J. Liu, Adv. Sci. 9 (2022) e2201000. doi: 10.1002/advs.202201000
H. Luo, J. Liu, Angew. Chem. Int. Ed. 62 (2023) e202302761. doi: 10.1002/anie.202302761
Y. Liang, S. Wang, M. Tang, et al., Angew. Chem. Int. Ed. 62 (2023) e202218839. doi: 10.1002/anie.202218839
K. Wang, L. Chen, Y.Y. Huang, et al., Chin. J. Chem. 41 (2023) 2589–2596. doi: 10.1002/cjoc.202300149
M. Murai, S. Iba, H. Ota, et al., Org. Lett. 19 (2017) 5585–5588. doi: 10.1021/acs.orglett.7b02729
A. Ong, T. Tao, Q. Jiang, et al., Angew. Chem. Int. Ed. 61 (2022) e202209286. doi: 10.1002/anie.202209286
H. Xin, J. Li, R.Q. Lu, et al., J. Am. Chem. Soc. 142 (2020) 13598–13605. doi: 10.1021/jacs.0c06299
Y. Shibuya, K. Aonuma, T. Kimura, et al., J. Phys. Chem. C 124 (2020) 4738–4746. doi: 10.1021/acs.jpcc.9b11214
Y. Xia, Z. Liu, Q. Xiao, et al., Angew. Chem. Int. Ed. 51 (2012) 5714–5717. doi: 10.1002/anie.201201374
J. Xiang, W.L. Tan, J. Zhang, et al., Macromolecules 55 (2022) 8074–8083. doi: 10.1021/acs.macromol.2c01101
T. Yao, K. Hirano, T. Satoh, et al., Angew. Chem. Int. Ed. 51 (2012) 775–779. doi: 10.1002/anie.201106825
J. Radolko, P. Ehlers, P. Langer, Adv. Synth. Catal. 363 (2021) 3616–3654. doi: 10.1002/adsc.202100332
J. Xiang, W. Tan, J. Zhang, et al., Macromolecules 55 (2022) 8074–8083. doi: 10.1021/acs.macromol.2c01101
S.N. Steinmann, Y. Mo, C. Corminboeuf, Phys. Chem. Chem. Phys. 13 (2011) 20584–20592. doi: 10.1039/c1cp21055f
Z. Liu, T. Lu, Q. Chen, Carbon 165 (2020) 468–475. doi: 10.1016/j.carbon.2020.04.099
D. Geuenich, K. Hess, F. Köhler, et al., Chem. Rev. 105 (2005) 3758–3772. doi: 10.1021/cr0300901
L. Qin, Y.Y. Huang, B. Wu, et al., Angew. Chem. Int. Ed. 62 (2023) e202304632. doi: 10.1002/anie.202304632
D.T. Chase, A.G. Fix, S.J. Kang, et al., J. Am. Chem. Soc. 134 (2012) 10349–10352. doi: 10.1021/ja303402p
A. Maliakal, K. Raghavachari, H. Katz, et al., Chem. Mater. 16 (2004) 4980–4986. doi: 10.1021/cm049060k
W. Fudickar, T. Linker, J. Am. Chem. Soc. 134 (2012) 15071–15082. doi: 10.1021/ja306056x
C.J.M. van den Heuvel, J.W. Verhoeven, T.J. de Boer, Recl. des Trav. Chim. des Pays-Bas 99 (1980) 280–284. doi: 10.1002/recl.19800990908
S. Chien, M. Cheng, K. Lau, et al., J. Phys. Chem. A 109 (2005) 7509. doi: 10.1021/jp0503009
A.R. Reddy, M. Bendikov, Chem. Commun. 11 (2006) 1179–1181.

Figure 2 The localized orbital locator (contribution from π electrons only) of (a) Az-PH1, (b) pentacene. Calculated ACID plots (contribution from π electrons only) of (c) Az-PH1, (d) pentacene. Calculated 2D-ICSS values of (e) Az-PH1 and (f) pentacene. The images are mapped at 1 Å above the XY planes.

Figure 3 (a) UV–vis spectra of Az-PH1/2/3, and protonation by TFA in Az-PH1/2/3 solution (inset: the picture of Az-PH1 and 2). (b) UV–vis spectra of Az-PH1/2/3 drop-coated on glass (inset: the magnified absorptions in 400–500 nm).

Figure 4 (a) Absorption spectral changes under 365 nm light irradiation for Az-PH1 in DCM. (b) The change of the optical density at 365 nm of Az-PH1 under ambient conditions.

Figure 5 (a) The atom orbital coefficients of the HOMOs of pentacene and Az-PH1. Orbiso= 0.02. (b) Pathways of oxygenations of pentacene and Az-PH1 in irradiation calculated with B3LYP/6–311(d, p) (top: pentacene; bottom: Az-PH1).

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:
