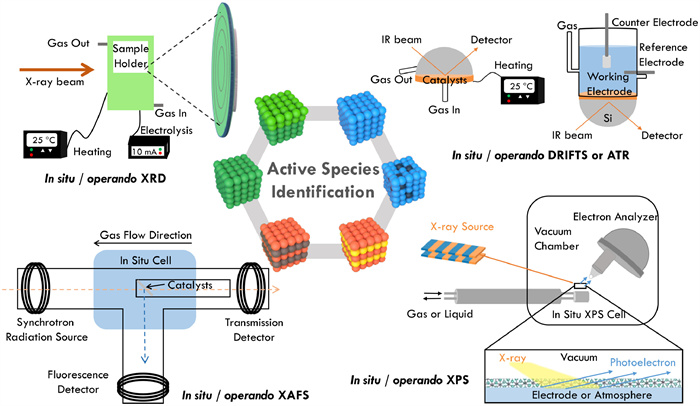
Figure 1.
Schematic diagram of the in situ/operando techniques for active species identification.
Understanding of the structural evolution of catalysts and identification of active species during CO2 conversion
Li Li , Fanpeng Chen , Bohang Zhao , Yifu Yu

Over the years, economic growth driven by the development and utilization of fossil fuels has promoted the living standards of people all over the world, however, the crises of the soaring consumption of energy and ever-increasing concentration of the atmospheric CO2 is becoming increasingly serious simultaneously [1–6]. A series of ecological problems arose as a consequence, and the most severe among these problems is the continuous rise of the global average temperature and sea level, which have brought about multiple constraints on economic development and human existence [7–10]. It is therefore recognized that attenuating the excessive CO2 emissions and lowering the CO2 concentration in the atmosphere is a long-term and imperative task [11–15]. To date, a series of strategies have been adopted to control the concentration of atmospheric CO2, replacing fossil fuels with alternative clean fuels without carbon exhausts and capture and storage of CO2 [3,6,16–20]. In addition, it is a promising approach to utilize CO2 as a C1 building block that turns the waste CO2 into value-added fuels (e.g., carbon monoxide (CO) [12,21], methane (CH4) [8,22], methanol (CH3OH) [23–26], and formate (HCOOH) [27–30, [31]) and downstream chemicals (ethylene (C2H4) [3,7], ethanol (C2H5OH) [32], propane (C3H8) [33], and gasoline (C5-C11) [1,34,35]) for the sake of economic and ecological consideration.
However, as a linear molecule, CO2 is too stable (C=O bond energy: 750 kJ/mol) to be activated without hydrogen sources as a reductant and energy input as a driven force [4,9,36–38]. Taking H2, H2O, or CH4 as the hydrogen source, the CO2 conversion can be divided into thermal, electro, and photocatalytic reactions, depending on the form of energy input (heat, electricity, irradiation) [5,39–42]. Note that, the structural evolution of catalytic material with time on stream is inevitably due to the integration of reactants and the driven force [43–46]. Generally speaking, the intrinsic activity of the catalyst is determined by the crystal phase restructuring of catalytic materials during the reaction on stream [46–48]. Thus, the identification of real active species in the process of CO2 conversion is vital to the rational design of catalysts and significantly enhances the conversion efficiency [27,28]. Recently, with the development of in situ and operando characterization techniques [49–51], some pioneer researchers and our group have observed the structural restructuring of catalytic materials in real-time and unveiled the inherent mechanism of the reconstruction process toward real active species [52–54], which provide new strategies to explore the structure-activity relationship and reaction mechanism, finally guiding the rational design of catalysts with high efficiency [36,55].
For more than a decade, plenty of excellent reviews about CO2 conversion have emerged, the focus has always been on the different catalytic ability (activity and selectivity) to convert CO2 due to the unique properties of materials [4,13,17,56]. Despite the tremendous research efforts in the field, many scientific questions on the effect of various active sites in governing CO2 conversion activity, selectivity, and stability are still unclear [52,53]. Moreover, only a few research have focused on the structural evolution of catalytic materials and the identification of real active species in the process of CO2 conversion [57,58]. However, it is the evolution, reconstruction, and acquisition of active sites that matter when in-depth exploring the mechanism and reaction pathway of the catalytic process [46,47,52]. In light of such a significant impact on the understanding of the complicated process, a timely review that tracks the structural evolution of catalytic material and identification of real active species during the conversion of CO2 in different catalytic fields is highly desirable to empower the future research and accelerate the industrialization. Very recently, Li et al. [57] summarized the recent developments of in situ studies on probing the catalyst evolution, and monitoring the reaction intermediates and catalytic products, during the CO2 reduction process, but the detailed discussion was limited to the electrocatalytic system. In fact, the phenomenon of that activity and selectivity influenced by dramatic phase transformation during the CO2 conversion was common and urgent to be explored [49,59–61]. As a consequence, adopting in situ/operando characterization techniques to explore the dynamic reconstruction of catalytic materials and clarify the real structure-activity relationships is of great significance, but highly challenging.
Herein, the aim of this review is to center on summarizing and understanding the state-of-the-art in situ restructuring of catalytic materials under various realistic catalytic conditions. In addition, identifying the structure evolution and real active species, and establishing the tight relationship between structure and activity are illustrated for the rational design of catalysts with high efficiency. In this paper, nowadays available in situ/operando characterization techniques and how to utilize these methods to find out the thermal, electro, and photo-induced structural evolution and identify the real active species will be comprehensively reviewed (Fig. 1). Besides, the challenges and opportunities of the integration of advanced theoretical simulation and in situ experiments will also be addressed.
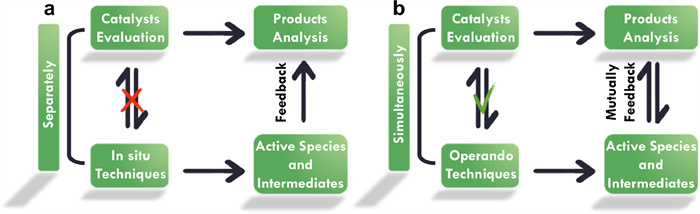
In order to probe the dynamic evolution of catalytic materials during reactions, various in situ/operando characterization techniques have been developed by researchers in the past few years [62–64], including the design and manufacture of specific reactors [65,66]. As illustrated in Fig. 2a, the definition of "in situ" is collecting the spectra or images of catalysts under similar conditions and/or the same atmosphere compared to the catalytic operation without evaluation of catalysts [59,67]. Whereas, "operando" means combining the catalytic reaction and product analysis with the simultaneous characterization of catalysts to gain the realistic status of the working catalyst (Fig. 2b) [59,67]. It has to be admitted that each of these techniques owns its unique capabilities, limitations, and special demands [68–70]. Thus, a comprehensive understanding of the catalytic materials evolution and catalytic process requires the combination of several in situ/operando characterization techniques. A brief introduction of in situ/operando X-ray diffraction patterns (XRD), X-ray absorption spectroscopy (XAS), X-ray photoelectron spectroscopy (XPS), transmission electron microscopy (TEM), infrared (IR), and Raman spectroscopy, which are closely related to the structure-activity relationship, mechanism, and pathway of the reaction process, will be demonstrated below. The denoted references give more detailed information about the respective techniques and their application.
XRD is a kind of technique most frequently used to certify the crystalline phases of materials with long-range structural order because the principle of diffraction occurs when light is scattered by a periodic array with long-range order [62,70,71]. A common diffractogram plots the reflection intensity against the angle of the detector, which is determined by the general relationship between the crystal lattice plane spacing of atoms and the wavelength and angle of the incident X-rays deduced from Bragg's Law [72]. To gain more information on catalytic materials during reaction on stream, special in situ cells were developed to satisfy the specific reaction conditions (e.g., gas or liquid environment; thermal or electricity input) [62,73]. In situ XRD has numerous advantages including non-destructive nature, easy preparation, high speed and sensitivity, and wide range of applications, but was limited by the indistinguishableness of long-range disorder or amorphous materials. In addition, it is only the information about the average structure of the bulk materials that was obtained from in situ XRD.
XAS is a technique always known as Near-edge X-ray absorption spectroscopy (XANES) for the oxidation state estimating of an element in a solid sample and Extended X-ray absorption fine structure (EXAFS) for the quantification of coordination number between metal atoms and other atoms within the catalyst. For the sake of a detailed relationship between activity and structure as well as the quick change of catalysts, in situ XAS and Quick XAS (QXAS) techniques based on the accelerated scanning speed were studied and employed [74,73]. In situ QXAS has become a powerful characterization technique in identifying the true catalytic active species under real-time conditions because there is no sample crystallinity requirement with rich structural information obtained [73]. However, there is a lack of a standard database for the comparison and analysis, with low surface sensitivity, and can only proceed under the synchrotron radiation source.
Conventional XPS is a wildly applied method for elements and surface structure characterization under ultrahigh vacuum conditions [75–77]. The inherent surface sensitivity is caused by that only the electron from a few nanometers below the surface can escape after being stimulated by the X-ray beams [78–80]. It has great potential to probe the active species if XPS could be adopted under in situ conditions. However, the need for accurate measurements of the energy of ejected photoelectrons, which is hindered by the gas phase electron scattering, makes it difficult to apply under high gas pressure. Fortunately, with the breakthroughs in differential pumping, X-ray source, and electron energy analyzer, ambient pressure XPS (APXPS), which could achieve working pressure up to tens of mbar, has gained an improved understanding of reaction mechanisms on model surfaces and interfaces [81–85]. Though shortcomings like non-precise reaction conditions control and lack of reactive species information still exist, APXPS has become a powerful technique among the catalysis community due to the highly sensitive qualitative and quantitative surface analysis, as well as the depth profile to study the catalyst composition during the reaction on stream.
IR and Raman spectrum are similar and complementary methods to analyze and confirm the structure relying on the vibration and rotation of molecules. In the case of the former, it mainly reflects the information of surface adsorbed species based on absorption signal, while the latter enables the detection of transition metal oxide vibrations in the low spectral range coming from the signal of scattering [64,86,87]. Nowadays, to get more information on catalysts under realistic conditions, diffuse reflectance infrared Fourier transform spectroscopy (DRIFTs) and attenuated total reflection infrared (ATR-IR) spectroscopy that could be done under working conditions developed rapidly [88,89]. In situ DRIFTs and ATR-IR are apt for monitoring the adsorbed species on the catalyst surface, but less for the transformation of catalysts owing to the low resolution in the low spectral range [90]. The premise of water has a low Raman scattering cross-section and can be used as the solvent. Surface-enhanced Raman scattering (SERS) technique which provokes the enhancement of the Raman signal on the metal surface overcomes the obstacle of low sensitivity and makes in situ SERS a suitable complementary to in situ/operando IR counterparts, especially for the identification of the real active species to some extent as it can monitor the catalyst surface in higher oxidation states [91,92]. As for IR and Raman spectroscopy, although they are highly accessible and simple, the limited spatial resolution and low sensitivity to changes on the catalyst surface led to the requirement of other techniques to complement.
In brief, in situ/operando XRD and XAS with less surface sensitivity, are suitable for exploring the phase transformation, changes in valence states, and the coordination numbers in the bulk phase of catalytic material during reaction on stream. For surface analysis and depth profile in real-time, APXPS comes into sight of researchers. To further figure out the formation mechanism of active sites and the reaction pathway, the reaction intermediates probed by in situ/operando IR and SERS are significant [47,68,93]. In addition, short-lived free radicals also can be detected by electron paramagnetic resonance (EPR) spectroscope which can probe unpaired electrons, to elucidate reaction catalytic mechanisms [94,95]. Thus, it is the combination of these techniques that is necessary for the in-depth understanding of in situ restructuring towards the active species under realistic conditions.
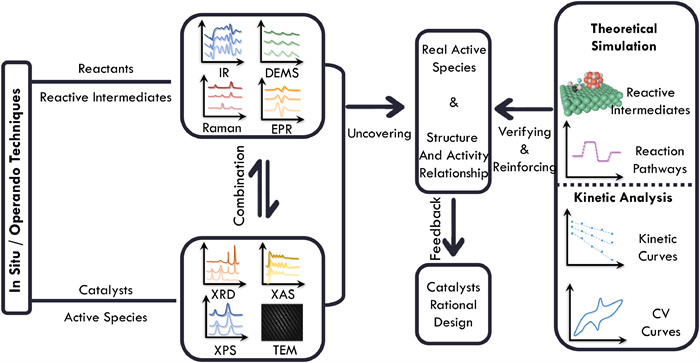
CO2 conversion is a multi-electron-transfer process with various possible products and reaction pathways. Uncovering the accurate structure-activity relationship by identifying the structural evolution and active species with time on-stream is necessary for the rational design of catalysts with high efficiency. Thus, recognizing the structural evolution and veritable active species of catalytic materials during the reaction process by in situ/operando characterization techniques (e.g., XRD, XAS, XPS, and TEM) is a primary premise. Moreover, the key intermediates for specific products, always unstable and instantaneous, also play a pivotal role in enhancing activity and altering selectivity. The surface detection techniques with high time resolution (e.g., DRIFTS, ATR-IR, SERS, and EPR) applied under similar or real reaction conditions are employed to unveil these intermediates such as *CHO to boost the C-C coupling process in electrocatalytic CO2 reduction and *H coverage in the hydrogenation process [96,97]. In addition, the capabilities of various characterization methods are summarized in Table 1.
Combining the dynamic structural evolution and reactant intermediates, the real active species and tight relationship between structure and activity are uncovered and established. Furthermore, the theoretical simulation which can provide an in-depth exploration of the interaction between reactive intermediates and the real active species, as well as the kinetic analysis is used to verify and reinforce the mechanism for the overall reaction. Finally, the real active species for specific products of CO2 conversion and the accurate structure-activity relationship feedback to the rational design for efficient catalysts (Fig. 3). The specific sequence and methods for thermo-, electro-, and photocatalytic CO2 conversion process are discussed in detail in the next chapter.
Heat, electricity, and irradiation are the commonly used energy input forms in the CO2 conversion process, which all possess the ability to convert CO2 into various reduction products. Although the mechanisms are different, they are all closely related to the structure evolution of the catalyst under working conditions. This section generalized the improved understanding of in situ generated active sites of CO2 conversion from the aspects of different energy input forms.
Thermocatalytic CO2 conversion including CO2 hydrogenation and reforming has been well established over the last half century [24,34,92]. Typically, these two processes proceed at around 300 ℃ or higher temperature, at which the catalytic materials inevitably undergo a structural evolution due to the interaction with reactant gasses [2,98]. Thus, it is significant to unveil the dynamic structural evolution process and identify the veritable active sites for the rational design of CO2 conversion catalysts. Recently, with the development of in situ/operando characterization techniques, lots of literature produced by pioneer researchers has been focused on identifying the real active sites induced by the inputted thermal energy [52,53,99,100].
The mechanism of CO2 hydrogenation to methanol over commercial copper/zinc oxide/aluminum oxide (Cu/ZnO/Al2O3) has been the subject of intense debate for decades. However, the question of the active sites, the ZnCu bimetallic sites or ZnO-Cu interfacial sites, remains open to discussion. Recently, a comprehensive work reported by Zhao et al. [53] highlighted the synergy of Cu and ZnO at the interface rather than ZnCu alloy facilitated methanol synthesis through the XPS spectra, discrete fourier transform (DFT) calculation, and kinetic monte carlo (KMC) simulations. The activity (Fig. 4a) of ZnCu (111) was initially low and increased with the reaction on stream until reaching the similar value exhibited in Fig. 4b for the relevant ZnO-Cu (111). From the XPS spectra in Fig. 4c, such an activity enhancement was accompanied by the shift in the corresponding binding energy of Zn 2p3/2. The obtained Zn to ZnO transformation along with the increasing methanol activity indicated that the real active site for CO2 conversion to methanol was ZnO-Cu (111). The results from KMC simulations under the experimental conditions were consistent with the conclusion, the rate for the methanol production on ZnCu (211) is much lower than the ZnO-Cu (111) and quickly decays (Fig. 4d). Furthermore, Wang et al. [99] demonstrated that there is no reduction of the ZnO or formation of ZnCu alloy which can be observed in the Cu/ZnO/Faujasite (FAU) even under highly pressure of pure hydrogen atmosphere (15 bar) at 260 ℃, which was monitored by operando time-resolved XAS (Figs. 4e and f). Thus, the conclusion that a ZnCu alloy phase is not required for high methanol yields and selectivity was obtained and the superior reactivity towards methanol synthesis over copper-zinc based system could be attributed to the interplay between Cu and ZnO phases.

Recently, a strategy to boost the activity of CO2 hydrogenation to methanol by incorporating N atoms into Co nanosheets (denoted as Co4N) was proposed [99]. Several studies have manifested that Co-based catalysts are promising materials for CO2 conversion due to their excellent hydrogenation ability, lower costs, and tunable selectivity via the electronic structure adjustment [42–44]. Besides, transition metal nitride nanostructures have been proven to be good catalytic materials for various hydrogenation reactions because of their excellent hydrogen dissociation ability. The turnover frequency (TOFs) value of Co4N nanosheets reached 25.6 h−1 under 32 bar pressure at 150 ℃ in a slurry reactor, which increased approximately 64-fold than that of Co nanosheets counterpart (Fig. 5a).

For the rational design of high-efficiency and stable catalysts, it is significant to probe the essential issues of structure, molecular function, and reactivity in detail. The Müller group has recently made progress by combining operando XAS-XRD and in situ transmission electron microscopy (TEM) to identify the three distinct catalytic stages of In2O3 nanoparticles (NPs) during CO2 hydrogenation with time on stream (TOS) including activation, stable performance, and deactivation [101]. The three stages could be distinguished by the catalytic performance with TOS (Fig. 6a): The first 48 min named activation (ⅰ); the following 42 min attributed to stable performance (ⅱ), and deactivation marked by a gradual decline of activity was from 90 min to 210 min (ⅲ). The coordination numbers (CN) and ΔE0 (E0(TOS) − E0(In2O3)) obtained from the fitting of the EXAFS FTs (Figs. 6b and c) decreased and shifted to In2+ from In3+, respectively, indicating the formation of oxygen vacancy sites. The constant tendency of CN and ΔE0 during stage (ⅱ) demonstrated the In2O3-x phase as the real active sites.
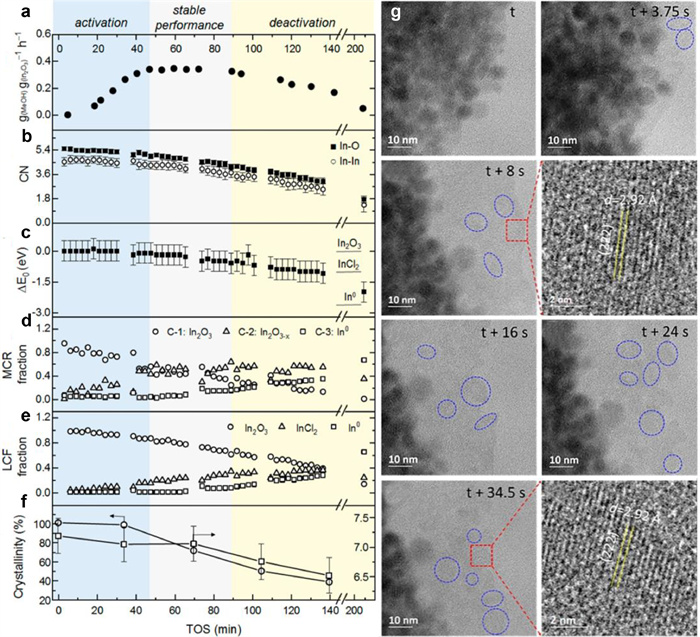
Multivariate curve resolution constrained (MCR)-alternating least squares algorithm (ALS) and linear combination fitting (LCF) aimed at separating the spectral components allowing to identification of the chemical changes with TOS (Figs. 6d and e) further confirmed the aforementioned conclusion that the most active catalyst is composed of 50%−40% In2O3 and 50%−60% In2O3-x. Moreover, the over-reduction to molten In0 was related to the deactivation stage of the catalysts. The decrease in crystallinity and the lowered average crystallite size corresponding to the formation of molten metallic indium were characterized by operando XRD (Fig. 6f), which coincide with the deactivation stage, also affirming the In0 as the deactivation phase. In situ TEM images in Fig. 6g confirmed that the catalysts were under a dynamic interconversion situation between amorphous/molten and crystalline In2O3-x domains during the reaction process. In general, the operando and in situ characterization forecasted that there exists an inherent relationship between the CO2 hydrogenation to methanol performance and the oxidation state, local structure, and crystallinity of In2O3, which could be summarized as In2O3-x acts as the active phase, while the over-reduction state to In0 leads to the deactivation.
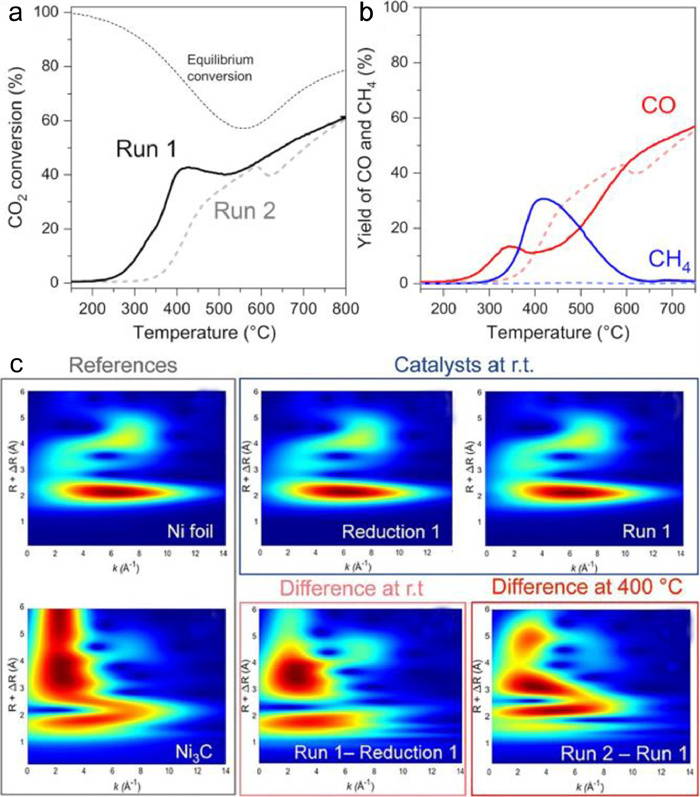
Generally, product selectivity is highly related to the active sites. Thus, the identification of real active sites of catalytic materials during reaction on stream is also of great importance in altering the product distributions of CO2 conversion. Very recently, Rossi and co-workers reported a total selectivity change from CH4 to CO after a 100 ℃ to 800 ℃ CO2 conversion process over the traditional Ni-SiO2 prepared by the impregnation method [102]. The conversion in the temperature region lower than 500 ℃ was slightly higher over Run 1 than Run 2 (Fig. 7a), which may be due to the exothermic nature of methanation. Interestingly, as shown in Fig. 7b, the formation of CH4 was completely depressed and CO was almost the only product over the spend catalysts (Run 2), comparing to the fresh catalyst (Run 1). To further understand the nature of the selectivity change, the continuous Cauchy wavelet transform (WT) was applied to the operando EXAFS spectrums. After subtracting the Ni-Ni scattering contribution, the difference spectrums of Run 1-Reduction 1 and Run 2-Run 1 were similar to the reference Ni3C (Fig. 7c), indicating the existence of Ni3C species during Run 2 process. The conclusion they obtained was that the Ni3C-like phase formed after Run 1 is the real active site for the boosted CO selectivity.
To sum up, the catalytic materials may undergo a phase restructuring, which relates tightly with the activity and selectivity, due to the interaction with the reactants under the thermal driven force. The real active sites in commercial Cu/ZnO/Al2O3 catalysts were identified to be the interface of Cu and ZnO [52,99]. A series of in situ characterization revealed that Co4N and Fe2N could be in situ restructured into Co4NHx and FexCy active sites during the reaction on stream, leading to the outperformance corresponding to the metallic counterparts [53,100]. Moreover, the three stages including: activation, stable performance, and deactivation during the CO2 hydrogenation process of In2O3-x were disclosed for the rational design of efficient and stable catalytic materials [101]. Furthermore, the relationship of selectivity and active sites over the classical Ni-based catalysts was clarified by operando XAS [102].
Electrocatalysis has emerged as an environment-friendly technology for synthesis, energy storage, and conversion over the past decades [103]. However, the inevitable reconstruction of electrode surfaces due to the use of electricity with high energy density as a driving force leads to poor stability or misunderstanding of the real active sites [27,104]. In this regard, it is of great significance to explore the real active sites derived from the pre-catalysts in the electrocatalytic system. For example, the in situ formed metal oxyhydroxides that are identified as the real active sites for electrocatalytic oxygen evolution reaction (OER), accelerate the development of OER catalysts with high efficiency [105]. Thus, monitoring the structural evolution of electrodes and identifying the real active sites for electrocatalytic CO2 reduction reaction (CO2RR) by the in situ/operando characterization techniques are vital to the rational design of highly efficient and stable catalytic materials.
The Wang group utilized in situ and operando XAS to disclose a reversible structural transformation and oxidation state change under the applied potential over copper(Ⅱ) phthalocyanine (CuPc) catalyst, which is responsible for the CO2 to CH4 conversion [106]. The maximum 66% Faradaic efficiency (FE) together with a 13 mA/cm2 partial current density of CH4 could be obtained under −1.06 V vs. reversible hydrogen electrode (RHE) (Figs. 8a and b). For in situ and operando XAS, after depositing catalysts on -100 µm-thick carbon fiber paper, the as-prepared catalyst electrode was mounted onto a custom-designed in situ XAS fluorescence cell, with other conditions unchanged comparing with the custom electrochemical measurements. As the potential applied, the peaks for Cu(Ⅱ) and Cu(0) showed a downtrend and uptrend, respectively, both from the Cu K-edge XANES spectra (Fig. 8c) and first-order derivatives of the XANES spectra (Fig. 8d). The Cu(0) peaks disappeared, and the spectrum return almost the same as the initial CuPc, when the applied potential switched back to 0.64 V, indicating a reversed electro-induced oxidation state change. The in situ EXAFS at −1.06 V vs. RHE further verified the existence of Cu(0) due to the appearance of metallic Cu–Cu bond (Fig. 8e). The XRD after electrolysis showed no existence of Cu or Cu2O (Fig. 8f), which is consistent with the results of reversible structure transformation by in situ XAS. DFT calculation revealed the reversible restructuring process based on the intrinsic instability of the small nanoclusters as observed from the scanning electron microscope (SEM) image in Fig. 8g.

Intermetallic compounds (IMCs), such as transition metal carbides, nitrides, or hydrides, are widely used in various catalytic reduction reactions [107]. It is reasonable to deduce that the metallic catalysts on the electrode will undergo a hydrogen atom insertion process due to the interaction with the active hydrogen in H2O under the applied potential. Gao et al. recently revealed the phase transformation from metallic Pd towards palladium hydride (β-PdH) [108] during CO2RR by in situ XRD and XAS over a Pd nanoparticle with -4 nm supported on carbon (Pd/C) (Fig. 9a). Syngas with H2/CO ratio between 0.5–1.0 could be obtained at the potential range of −0.5 V to −1.0 V vs. RHE (Fig. 9b). In situ XANES revealed the formation of β-PbH by the negative shift of peaks A and B with the increasing applied potential (Fig. 9c). As for the in situ XRD measurement, the spectrum was acquired under a 1 mV/s linear sweep voltammetry. An obvious shift towards a lower angle value was observed in Fig. 9d, indicating a lattice expansion due to the hydrogen atom insertion into Pd to form β-PdH. Further DFT calculation demonstrated that both the binding energy of CO and H were lower on β-PdH than Pd, accounting for the excellent performance stability (Fig. 9e). A similar conclusion was also obtained by Bie et al. [109], the formation of β-phase Pd hydride from metallic Pd during CO2RR facelifted the CO production.

Mostly, the CO2 reduction electrocatalysts containing metals face the unavoidable problem of self-reduction to the metallic phase, leading to the enhanced hydrogenation evolution reaction (HER). Thus, developing stable catalysts with high selectivity toward CO2RR and strong resistance to self-reduction is of great significance. Recently, our group found hydrocerussite, which formed via in situ conversion of tannin-lead(Ⅱ) (TA-Pb) complex with 6-coordinated Pb(Ⅱ) ions revealed by EXAFS (Fig. 10a), as a stable and active site for electrocatalytic CO2RR [27]. The FE of formate reached above 90% in the potential range of −0.82 V to −1.12 V vs. RHE (Fig. 10b). The phase transformation from TA-Pb to cerussite (PbCO3 denoted as t-PCO) and then hydrocerussite (Pb3(CO3)2(OH)2 denoted as t-PCOH) was monitored by in situ XRD (Fig. 10c). Quasi-in situ XAFS (Fig. 10d) was conducted to identify the intermediates of the restructuring process and the XANES (Fig. 10e) of different stage verified the unchanged valence state, indicating the resistance of self-reduction. The time-dependent in situ Raman spectra (Fig. 10f) clearly showed that there is a small peak located at 1065 cm−1, which corresponds to the C–O symmetric stretching vibration of CO32− appeared after applying potential for 210 s, indicating the formation of t-PCO and further affirm the restructuring process revealed by XAFS. Finally, the appearance of PbO and Pb after electrolysis without CO2 suggested that the self-reduction of the electrocatalyst and the following enhanced HER were effectively hindered by the preference of CO2RR on hydrocerussite (Figs. 10g and h).

To summarize, as an environment-friendly and powerful driven force, electricity could induce a phase transformation process through ligand dissociation or atom insertion, or lead to poor selectivity because of the enhanced HER caused by the self-reduction of catalysts. The excellent activity and selectivity of CuPc were attributed to a reversible restructuring process depending on the applied potential by in situ XAS [105]. Also, a similar restructuring process was observed between Pd/C and β-PdH [107,108]. Note that, hydrocerussite, a stable active species for producing formates, is generated from the TA-Pb precursor under the existence of CO2 and bias [27].
Photocatalysis driven by renewable solar energy has become popular among researchers because of its advantages in alleviating the world energy crisis without bringing extra environmental problems [41]. Honestly speaking, the catalytic materials usually undergo an electron transfer or crystallization/decrystallization process to generate a built-in electric field rather than experience a total phase restructuring during the photoinduced activation process due to the mild nature of photo energy [4]. For all this, it is also important to monitor these behaviors and unveil the impact on building the active sites during the reaction for an in-depth understanding mechanism of reactions driven by photo energy and the preparation of catalysts with high efficiency. However, the in situ/operando techniques for photo-driven type reactions are limited by the construction of a built-in irradiation source and the uncertainties introduced by the illumination.
For photocatalysis, revealing the electron transfer behavior under illumination is vital to the active sites' construction, better activity, selectivity, and understanding of the reaction mechanism. In these regards, Yu and co-workers employed in situ irradiated XPS (ISI-XPS) to confirm the way of electron transfer over the nitrogen-doped graphene (NG) wrapped CdS hollow spheres (denoted as CdG2) [110]. As displayed in Figs. 11a and b, a positive shift of the Cd 3d and S 2p peaks was observed after NG coating on the CdS, following a further move toward higher energy under illumination, indicating a decreased electron density in CdS. In the meantime, the electron transfer from CdS to NG was further confirmed by the decreased intensity of the Cd 3d and S 2p peaks. Furthermore, the peaks of C 1s and N 1s exhibited a negative shift under the irradiated conditions (Figs. 11c and d). Note that the large shift in the binding energy of pyrrolic N indicated that the nitrogen atoms act as the sites to accept the transferred electrons from CdS and meanwhile the real active sites for CO2 reduction. Interestingly, the DFT calculations made by Ghuman et al. [111] further revealed the upshift of the graphene Fermi level induced by the enriched electrons transfer from TaON to graphene (Figs. 11e and f), led to a high filling state of the antibonding orbitals that facilitate the activation of CO2. These results theoretically accounted for the formation of real active sites because of photoinduced electron transfer.

Welch et al. reported a hydroxylated indium oxide nanostructure (In2O3-x(OH)y) synthesized by a thermal dehydration method for a photo-enhanced reverse water-gas shift (RWGS) reaction [112]. The CO production rate under illumination reached 4.3 folds higher than in the dark. Coupling in situ spectroscopic and kinetic experiments, they deduced a mechanism that is similar to the frustrated Lewis pairs (FLPs) catalysis (Fig. 12a) [113,114]. The hydroxide groups and indium atoms proximal to the oxygen vacancy over In2O3-x(OH)y acted as Lewis basic and Lewis acidic sites, respectively, facilitating the H2 dissociation and CO2 activation during the RWGS process. Moreover, their further work theoretically explained the reason for the photo-enhanced activity by using time-dependent DFT calculations [115]. As shown in Fig. 12b, under excited state, the charge transfer from highest occupied molecular orbital (HOMO) to lowest unoccupied molecular orbital (LUMO), LUMO+1, and LUMO+2 respectively, to form the transition densities, and then the charge redistributed from these transitions made the unsaturated In atoms more electron-deficient, meanwhile -OH electron-rich, leading to the stronger Lewis alkalinity and acidity on the surface of In2O3-x(OH)y. This kind of highly activated FLPs with an excited state interacted with the reactants and upshifted the total energy of the system, accounting for the enhanced RWGS activity (Fig. 12c).

Our recent work proposed metallic Mo2N nanobelts (Figs. 13a and b) for photo-assisted thermal catalytic CO2 to CO conversion [116]. A 26.7-fold CO production than that in the dark was obtained under illumination. As the in situ DRIFTS and quasi-in situ electron spin resonance shown in Figs. 13c and d, the greatly enhanced production under illumination was attributed to the formation of highly active species Mo2NHx via the photoinduced H2 heterolysis. Furthermore, the ISI-XPS in Fig. 13e demonstrated a negative shift of N 1s peaks with an unchanged binding energy of Mo 3p, indicating the electron-rich environment among the surface N atoms [12,117]. The non-equivalent electron distribution on Mo and N led to a broken symmetry of electron densities, resulting in the strengthened local electric field between Mo and N atoms, which could induce the H2 heterolysis to form the active Mo2NHx species (Fig. 13f).

In general, the development of in situ techniques in the photo-activation process was hindered by the obstacle of building the inside irradiation source and the uncertain influence of the driving illumination on the spectrum results. Photo irradiation, as a kind of mild and renewable driving force, could not lead to a total phase restructuring during the activation stage. Commonly, the photoinduced construction of active sites was caused by the built-in electric field. Such as the N active sites formed by the charge transfer from CdS towards NG in the CdG2 [110]. The enhanced activity due to the photoexcited FLPs was influenced by the electric field essentially [112,115]. Finally, our work highlighted the strengthened local electric field caused by photoinduced broken symmetry of electron densities, leading to the formation of active Mo2NHX species [116].
On the grounds of these cases mentioned above, the CO2 conversion activity, selectivity, and stability of certain catalytic materials are highly related to the structure evolution under working conditions driven by heat, electricity, or irradiation. In other words, the identification of real active sites during reaction on stream is the premise of regulating and controlling the efficiency, product distribution, and practicability of the current stage of CO2 conversion. However, the in situ/operando techniques are still in their infancy stage and there exist various complicated parameters that impact the apparent performance. These methods still need to be further developed and improved to accurately supervise the structure evolution and identify real active sites, for the rational design of efficient catalysts with high selectivity and stability.
Beginning with the brief introduction of a series of state-of-the-art in situ/operando characterization techniques, we summarized various catalytic material evolution behaviors to real active species in real-time driven by heat, electricity, or illumination. The multifaceted catalytic materials evolution forms in this review include: phase transformation due to the reactants-evolved pre-activation process; valence state changes caused by the driven force or the reaction atmosphere; and the oriented electron transfer induced built-in electrical field. The phase transformation phenomenon comprises two kinds of scenarios. One is reduction metallization/alloying under a reduction atmosphere. Another type is that there is a strong interaction between pre-catalysts and reactants under the powerful driven force, such as the heat or electricity-driven cases. Valence state changes are driven by electron transfer, which could both caused through reducing reagents, electricity, and photogenerated electrons. The oriented electron transfer, which always occurs in photo-driven cases, is the gentlest reconstruction way because of the relatively lower energy of the photo-driven force. Thus, the capability and extent of electron transfer of different driven forces determines the reconstruction manners and results. Moreover, through the timely probing of the catalytic materials as well as the product distribution, the dynamic process of structure-performance relationship in the CO2 conversion is unveiled, which is of great significance to the in-depth understanding of reaction mechanism and rational design of CO2 catalytic systems with high efficiency. However, despite the pioneer research in this field, there still exist lots of challenges along with opportunities for future development as follows:
(Ⅰ) Further progress of in situ characterization techniques still necessary. Unlike the mature thermocatalysis system, the design of in situ/operando reactors is complicated on account of the interference of liquid electrolyte or solvent which is unavoidable in electrocatalysis and photocatalysis systems [46,47]. Especially for the photo-driven reaction process, it was further hindered by the obstacle of building the inside irradiation source. Taking the in situ IR spectrum in a photocatalysis system for example, in the most reported literature, diffuse reflection mode coupling with the outside irradiation source is preferred to be selected. However, the drawbacks of outside illumination included not only inefficient light absorption because only the outermost layer of the stacked catalyst could obtain the irradiation, but also the imported disturbance on the essential infrared signal by the incident light due to the mechanism of diffuse reflection mode. Recently, a kind of flexible cyclic annular light emitting diode light surrounding the quartz reactor coupling with the transmission mode IR enhanced the light absorption efficiency, meanwhile, the incident light perpendicular to the infrared signal avoided the interference to the maximum extent [7]. Besides, other techniques that require relatively rigorous conditions such as ultrahigh vacuum are more powerful in intuitively explaining the reaction mechanism. Such as the visualized H2 emission from the water absorbed on the electron-rich TiO2 surface that is detected by in situ TEM. Thus, it is possible to acquire the dynamic process of the interaction between catalytic materials and reactants during CO2 conversion in real-time with further progress of in situ characterization techniques.
(Ⅱ) A tightly relationship between structural evolution and adsorbed intermediates with high temporal-spatial resolution should be built. According to these cases listed in the current review and the relevant studies our group has done, we have noticed that the structural evolution of catalysts will not merely result in the phase or electronic changes, but also affect the adsorption manner of the reactants or the key intermediates on the catalyst surface. In this regard, clarifying the intrinsic interaction from the restructured real active species to the altered adsorption behavior, and then to the apparent performance enhancement and selectivity change is vital to understand the mechanism of CO2 conversion better and promote industrialization. Therefore, in situ ultra-fast spectroscopic techniques should be developed to monitor the reaction intermediates change in real-time, simultaneously, accompanied with the morphological and phase characterization techniques with high time resolution. The results of in situ APXPS in our ongoing work reveals the initial hydrogenation of CO2 to *HOCO intermediates over the electron-rich Co surface, causing the reaction pathway change. In a word, it will be meaningful to relate the catalytic material evolution behaviors tightly with the modified adsorption manner of the key intermediates with high temporal-spatial resolution.
(Ⅲ) More close combination of theoretical analysis and in situ/operando studies should be established. With the fast development of DFT calculation theory and more advanced algorithms, it is common to use theoretical models to simulate the reaction process in terms of the kinetic energy barriers and Gibbs free energy of the specific catalytic process, and further reflect the configuration on the catalysts of various intermediates or products by the calculated adsorption energy and the charge density difference. In the meantime, the results from in situ/operando techniques along with the dynamic performance measurements provide the experimental evidence for the catalyst structural evolution-induced performance enhancement or selectivity change. Thus, the combination of theoretical analysis and in situ/operando studies endows the deep exploration of the CO2 conversion mechanism from the aspects of dynamic structural evolution. However, currently, the majority of research including the incorporation of theoretical and in situ/operando studies is restricted to the stage of mutual authentication of correctness. In the long run, a series of experimental results should be fed into the process of theoretical simulation for the rational prediction of the real active species that transformed during the reaction on stream. Moreover, the prediction from DFT calculation could also be put into effect in practical experiments in return. For example, the metastable state phases predicted by theoretical analysis with remarkable performance (e.g., specific nitrides, carbides, high-entropy alloy, or high exponent plane), which are difficult to synthesize or not stable in the air, could be obtained through the delicate design in situ restructuring process. Further endeavors of CO2 conversion mechanism exploration rely on the mutual promotion of theoretical and in situ/operando results are bound to be a hot topic by the reason of the improved investigation efficiency.
In summary, the current review emphasizes the understanding and identification of the real active species during reaction on-stream through a brief introduction of the state-of-the-art in situ/operando characterization techniques and systematic summarization of the research among thermo-, electro-, and photo-catalytic CO2 conversion. Noting that the frequent and inevitable structural evolution behaviors of catalytic materials will occur no matter under which kind of driven force (e.g., thermal, electricity, or irradiation). Besides, the future challenges and opportunities in technique modification, mechanism exploration, and theoretical feedback are also addressed. There must be more exciting results occurring in the field of CO2 conversion with the advancement of in situ/operando studies.
The authors declare no conflict of interest.
We appreciate the National Natural Science Foundation of China (No. 22209120) and the China Postdoctoral Science Foundation (No. 2022M722364) for financial support.
P. Gao, S.G. Li, X.N. Bu, et al., Nat. Chem. 9 (2017) 1019–1024. doi: 10.1038/nchem.2794
X. Jiang, X.W. Nie, X.W. Guo, et al., Chem. Rev. 120 (2020) 7984–8034. doi: 10.1021/acs.chemrev.9b00723
F.P. García De Arquer, C.T. Dinh, A. Ozden, et al., Science 367 (2020) 661–666. doi: 10.1126/science.aay4217
M.Y. Sun, B.H. Zhao, F.P. Chen, et al., Chem. Eng. J. 408 (2021) 127280. doi: 10.1016/j.cej.2020.127280
T.T. Kong, Y.W. Jiang, Y.J. Xiong, Chem. Soc. Rev. 49 (2020) 6579–6591. doi: 10.1039/c9cs00920e
F.W. Li, A. Thevenon, A. Rosas-Hernandez, et al., Nature 577 (2020) 509–513. doi: 10.1038/s41586-019-1782-2
L. Wang, B.H. Zhao, C.H. Wang, et al., J. Mater. Chem. A 8 (2020) 10175–10179. doi: 10.1039/d0ta01256d
L. Luo, M. Wang, Y. Cui, et al., Angew. Chem. Int. Ed. 59 (2020) 14434–14442. doi: 10.1002/anie.201916032
G.B. Chen, G.I.N. Waterhouse, R. Shi, et al., Angew. Chem. Int. Ed. 58 (2019) 17528–17551. doi: 10.1002/anie.201814313
C.Q. Song, X. Liu, M. Xu, et al., ACS Catal. 10 (2020) 10364–10374. doi: 10.1021/acscatal.0c02244
J.G. Yu, J. Jin, B. Cheng, M. Jaroniec, J. Mater. Chem. A 2 (2014) 3407–3416. doi: 10.1039/c3ta14493c
B.H. Zhao, Y. Huang, D.L. Liu, et al., Sci. China Chem. 63 (2019) 28–34.
M. Marszewski, S.W. Cao, J.G. Yu, M. Jaroniec, Mater. Horiz. 2 (2015) 261–278. doi: 10.1039/C4MH00176A
L. Ji, L. Li, X. Ji, et al., Angew. Chem. Int. Ed. 59 (2020) 758–762. doi: 10.1002/anie.201912836
H. Zhang, T. Wang, J. Wang, et al., Adv. Mater. 28 (2016) 3703–3710. doi: 10.1002/adma.201505187
S. Chu, Y. Cui, N. Liu, Nat. Mater. 16 (2017) 16–22. doi: 10.1038/nmat4834
Z.J. Wang, H. Song, H.M. Liu, J.H. Ye, Angew. Chem. Int. Ed. 59 (2020) 8016–8035. doi: 10.1002/anie.201907443
Q.L. Wu, M.D. Pan, S.K. Zhang, et al., Energies 15 (2022) 6666. doi: 10.3390/en15186666
D.P. Sun, Y. Zheng, D. Chen, Energy Environ. Prot. 37 (2023) 117–124.
J. He, K.E. Dettelbach, D.A. Salvatore, et al., Angew. Chem. Int. Ed. 56 (2017) 6068–6072. doi: 10.1002/anie.201612038
J. Gu, C.S. Hsu, L.C. Bai, et al., Science 364 (2019) 1091–1094. doi: 10.1126/science.aaw7515
D.L. Liu, C.H. Wang, Y.F. Yu, et al., Chem 5 (2019) 376–389. doi: 10.1504/ijcse.2019.103942
L. Wang, M. Ghoussoub, H. Wang, et al., Joule 2 (2018) 1369–1381. doi: 10.1016/j.joule.2018.03.007
C. Wu, L. Lin, J. Liu, et al., Nat. Commun. 11 (2020) 5767. doi: 10.1038/s41467-020-19634-8
H.P. Yang, Y. Wu, G.D. Li, et al., J. Am. Chem. Soc. 141 (2019) 12717–12723. doi: 10.1021/jacs.9b04907
S.Y. Kong, X.M. Lv, X. Wang, et al., Nat. Catal. 6 (2023) 6–15.
Y. Shi, Y. Ji, J. Long, et al., Nat. Commun. 11 (2020) 3415. doi: 10.1038/s41467-020-17120-9
Y. Liang, W. Zhou, Y.M. Shi, et al., Sci. Bull. 65 (2020) 1547–1554. doi: 10.1016/j.scib.2020.04.022
S.Y. Yang, M.H. Jiang, W.J. Zhang, et al., Adv. Funct. Mater. 33 (2023) 2301984. doi: 10.1002/adfm.202301984
H. Liu, B.Y. Li, Z.H. Liu, et al., ACS Catal. 13 (2023) 5033–5042. doi: 10.1021/acscatal.2c06135
H.F. Shen, H.Y. Jin, H.B. Li, et al., Nat. Commun. 14 (2023) 2843. doi: 10.1038/s41467-023-38497-3
W. Xia, Y.J. Xie, S.Q. Jia, et al., J. Am. Chem. Soc. 145 (2023) 17253–17264. doi: 10.1021/jacs.3c04612
S. Wang, L. Zhang, P.F. Wang, et al., Nat. Catal. 5 (2022) 1038–1050. doi: 10.1038/s41929-022-00871-7
Z. He, M. Cui, Q. Qian, et al., Proc. Natl. Acad. Sci. U. S. A. 116 (2019) 12654–12659. doi: 10.1073/pnas.1821231116
J. Wei, Q.J. Ge, R.W. Yao, et al., Nat. Commun. 8 (2017) 15174. doi: 10.1038/ncomms15174
X.D. Li, Y.F. Sun, J.Q. Xu, et al., Nat. Energy 4 (2019) 690–699. doi: 10.1038/s41560-019-0431-1
H.L. Li, J.K. Zhao, L.H. Luo, et al., Acc. Chem. Res. 54 (2021) 1454–1464. doi: 10.1021/acs.accounts.0c00715
S. Liu, L.R. Winter, J.G. Chen, ACS Catal. 10 (2020) 2855–2871. doi: 10.1021/acscatal.9b04811
V. Kumaravel, J. Bartlett, S.C. Pillai, ACS Energy Lett. 5 (2020) 486–519. doi: 10.1021/acsenergylett.9b02585
L. Zhang, Z.J. Zhao, J.L. Gong, Angew. Chem. Int. Ed. 56 (2017) 11326–11353. doi: 10.1002/anie.201612214
J. Albero, Y. Peng, H. García, ACS Catal. 10 (2020) 5734–5749. doi: 10.1021/acscatal.0c00478
D. Goud, R. Gupta, R. Maligal-Ganesh, S.C. Peter, ACS Catal. 10 (2020) 14258–14282. doi: 10.1021/acscatal.0c03799
B.M. Wyvratt, J.R. Gaudet, D.B. Pardue, et al., ACS Catal. 6 (2016) 5797–5806. doi: 10.1021/acscatal.6b00936
L. Liu, A. Corma, Chem. Rev. 118 (2018) 4981–5079. doi: 10.1021/acs.chemrev.7b00776
M.A. Newton, Chem. Soc. Rev. 37 (2008) 2644–2657. doi: 10.1039/b707746g
L.C. Liu, A. Corma, Nat. Rev. Chem. 5 (2021) 256–276. doi: 10.1038/s41570-021-00255-8
Y.P. Zhu, J.L. Wang, H. Chu, et al., ACS Energy Lett. 5 (2020) 1281–1291. doi: 10.1021/acsenergylett.0c00305
A. Beck, X. Huang, L. Artiglia, et al., Nat. Commun. 11 (2020) 3220. doi: 10.1038/s41467-020-17070-2
A.D. Handoko, F. Wei, Jenndy, et al., Nat. Catal. 1 (2018) 922–934. doi: 10.1038/s41929-018-0182-6
B.W. He, Y.X. Zhang, X. Liu, L.W. Chen, ChemCatChem 12 (2020) 1853–1872. doi: 10.1002/cctc.201902285
J.H. Park, K.M. Choi, D.K. Lee, et al., Sci. Rep. 6 (2016) 25555. doi: 10.1038/srep25555
S. Kattel, P.J. Ramírez, J.G. Chen, et al., Science 355 (2017) 1296–1299. doi: 10.1126/science.aal3573
B.H. Zhao, M.Y. Sun, F.P. Chen, et al., Angew. Chem. Int. Ed. 60 (2021) 4496–4500. doi: 10.1002/anie.202015017
F.F. Wang, J. Tian, M.Z. Li, et al., Angew. Chem. Int. Ed. 59 (2020) 8203–8209. doi: 10.1002/anie.201916049
X. Li, L. Liang, Y.F. Sun, et al., J. Am. Chem. Soc. 141 (2019) 423–430. doi: 10.1021/jacs.8b10692
A. Álvarez, A. Bansode, A. Urakawa, et al., Chem. Rev. 117 (2017) 9804–9838. doi: 10.1021/acs.chemrev.6b00816
X. Li, S. Wang, L. Li, et al., J. Am. Chem. Soc. 142 (2020) 9567–9581.
R. Daiyan, W.H. Saputera, H. Masood, et al., Adv. Energy Mater. 10 (2020) 1902106. doi: 10.1002/aenm.201902106
A. Dutta, A. Kuzume, M. Rahaman, et al., ACS Catal. 5 (2015) 7498–7502. doi: 10.1021/acscatal.5b02322
T.J. Lin, K. Gong, C.Q. Wang, et al., ACS Catal. 9 (2019) 9554–9567. doi: 10.1021/acscatal.9b02513
Y.L. Zhang, D.L. Fu, X.L. Liu, et al., ChemCatChem 10 (2018) 1272–1276. doi: 10.1002/cctc.201701779
X.N. Li, X.F. Yang, J.M. Zhang, et al., ACS Catal. 9 (2019) 2521–2531. doi: 10.1021/acscatal.8b04937
A. Gurlo, R. Riedel, Angew. Chem. Int. Ed. 46 (2007) 3826–3848. doi: 10.1002/anie.200602597
D.Q. Liu, Z. Shadike, R. Lin, et al., Adv. Mater. 31 (2019) 1806620. doi: 10.1002/adma.201806620
W. Li, D.M. Lutz, L. Wang, et al., Joule 5 (2021) 77–88. doi: 10.1504/ijetm.2021.115730
B.M. Weckhuysen, Chem. Soc. Rev. 39 (2010) 4557–4559. doi: 10.1039/c0cs90031a
H. Topsøe, J. Catal. 216 (2003) 155–164. doi: 10.1016/S0021-9517(02)00133-1
C.W. Jones, F. Tao, M.V. Garland, ACS Catal. 2 (2012) 2444–2445. doi: 10.1021/cs3006692
L.E. Mundt, L.T. Schelhas, Adv. Energy Mater. 10 (2020) 1903074. doi: 10.1002/aenm.201903074
P.P. Paalanen, S.H. Van Vreeswijk, B.M. Weckhuysen, ACS Catal. 10 (2020) 9837–9855. doi: 10.1021/acscatal.0c01851
D.A. Svintsitskiy, T.Y. Kardash, O.A. Stonkus, et al., J. Phys. Chem. C 117 (2013) 14588–14599. doi: 10.1021/jp403339r
Y. Zhou, D.E. Doronkin, Z.Y. Zhao, et al., ACS Catal. 8 (2018) 11398–11406. doi: 10.1021/acscatal.8b03724
S. Bordiga, E. Groppo, G. Agostini, et al., Chem. Rev. 113 (2013) 1736–1850. doi: 10.1021/cr2000898
C. Vogt, E. Groeneveld, G. Kamsma, et al., Nat. Catal. 1 (2018) 127–134. doi: 10.1038/s41929-017-0016-y
V. Bon, E. Brunner, A. Pöppl, S. Kaskel, Adv. Funct. Mater. 30 (2020) 1907847. doi: 10.1002/adfm.201907847
E.J. Crumlin, E. Mutoro, W.T. Hong, et al., J. Phys. Chem. C 117 (2013) 16087–16094. doi: 10.1021/jp4051963
U. Bentrup, Chem. Soc. Rev. 39 (2010) 4718–4730. doi: 10.1039/b919711g
Y. Takagi, T. Uruga, M. Tada, et al., Acc. Chem. Res. 51 (2018) 719–727. doi: 10.1021/acs.accounts.7b00563
Y. Han, H. Zhang, Y. Yu, Z. Liu, ACS Catal. 11 (2021) 1464–1484. doi: 10.1021/acscatal.0c04251
L. Trotochaud, A.R. Head, O. Karslioglu, et al., J. Phys. Condens. Matter 29 (2017) 053002. doi: 10.1088/1361-648X/29/5/053002
D.E. Starr, Z. Liu, M. Hävecker, et al., Chem. Soc. Rev. 42 (2013) 5833–5857. doi: 10.1039/c3cs60057b
K. Roy, L. Artiglia, J.A. Van Bokhoven, ChemCatChem 10 (2018) 666–682. doi: 10.1002/cctc.201701522
C.J. Zhang, M.E. Grass, A.H. Mcdaniel, et al., Nat. Mater. 9 (2010) 944–949. doi: 10.1038/nmat2851
B.M. Auer, J.L. Skinner, J. Chem. Phys. 128 (2008) 224511. doi: 10.1063/1.2925258
A.C. Ferrari, S.E. Rodil, J. Robertson, Phys. Rev. B 67 (2003) 155306. doi: 10.1103/PhysRevB.67.155306
X. Wang, Y. Liu, T.H. Zhang, et al., ACS Catal. 7 (2017) 1626–1636. doi: 10.1021/acscatal.6b03547
M.F. Baruch, J.E. Pander, J.L. White, A.B. Bocarsly, ACS Catal. 5 (2015) 3148–3156. doi: 10.1021/acscatal.5b00402
F.N. Ajjan, M.J. Jafari, T. R ˛ ebis, et al., J. Mater. Chem. A 3 (2015) 12927–12937. ´ doi: 10.1039/C5TA00788G
T. Geisler, L. Dohmen, C. Lenting, M.B.K. Fritzsche, Nat. Mater. 18 (2019) 342–348. doi: 10.1038/s41563-019-0293-8
Y.L. Deng, B.S. Yeo, ACS Catal. 7 (2017) 7873–7889. doi: 10.1021/acscatal.7b02561
F.G. Baddour, E.J. Roberts, A.T. To, et al., J. Am. Chem. Soc. 142 (2020) 1010–1019. doi: 10.1021/jacs.9b11238
E. Gomez, B.H. Yan, S. Kattel, J.G. Chen, Nat. Rev. Chem. 3 (2019) 638–649. doi: 10.1038/s41570-019-0128-9
X.L. Zheng, B. Zhang, P. De Luna, et al., Nat. Chem. 10 (2018) 149–154. doi: 10.1038/nchem.2886
S.A. Bonke, T. Risse, A. Schnegg, A. Brückner, Nat. Rev. Methods Prim. 1 (2021) 33. doi: 10.1038/s43586-021-00031-4
R.D. Webster, Curr. Opin. Electrochem. 40 (2023) 101308. doi: 10.1016/j.coelec.2023.101308
B.H. Zhao, F.P. Chen, C.Q. Cheng, et al., Adv. Energy Mater. 13 (2023) 2204346. doi: 10.1002/aenm.202204346
B.H. Zhao, F.P. Chen, M.K. Wang, et al., Nat. Sustain. 6 (2023) 827–837. doi: 10.1038/s41893-023-01084-x
M. Zabilskiy, V.L. Sushkevich, M.A. Newton, J.A. Van Bokhoven, ACS Catal. 10 (2020) 14240–14244. doi: 10.1021/acscatal.0c03661
L.B. Wang, W.B. Zhang, X.S. Zheng, et al., Nat. Energy 2 (2017) 869–876. doi: 10.1038/s41560-017-0015-x
A. Tsoukalou, P.M. Abdala, D. Stoian, et al., J. Am. Chem. Soc. 141 (2019) 13497–13505. doi: 10.1021/jacs.9b04873
T.S. Galhardo, A.H. Braga, B.H. Arpini, et al., J. Am. Chem. Soc. 143 (2021) 4268–4280. doi: 10.1021/jacs.0c12689
Z.W. Seh, J. Kibsgaard, C.F. Dickens, et al., Science 355 (2017) eaad4998. doi: 10.1126/science.aad4998
H.L. Jiang, Q. He, Y.K. Zhang, L. Song, Acc. Chem. Res. 51 (2018) 2968–2977. doi: 10.1021/acs.accounts.8b00449
F. Zhang, Y.M. Shi, T. Xue, et al., Sci. China Mater. 60 (2017) 324–334. doi: 10.1007/s40843-017-9017-6
Z. Weng, Y. Wu, M. Wang, et al., Nat. Commun. 9 (2018) 415. doi: 10.1038/s41467-018-02819-7
K. Gschneidner, A. Russell, A. Pecharsky, et al., Nat. Mater. 2 (2003) 587–591. doi: 10.1038/nmat958
W.C. Sheng, S. Kattel, S.Y. Yao, et al., Energy Environ. Sci. 10 (2017) 1180–1185. doi: 10.1039/C7EE00071E
D.F. Gao, H. Zhou, F. Cai, et al., Nano Res. 10 (2017) 2181–2191. doi: 10.1007/s12274-017-1514-6
C. Bie, B. Zhu, F. Xu, et al., Adv. Mater. 31 (2019) e1902868. doi: 10.1002/adma.201902868
L. Pei, Y.J. Yuan, W.F. Bai, et al., ACS Catal. 10 (2020) 15083–15091. doi: 10.1021/acscatal.0c03918
K.K. Ghuman, T.E. Wood, L.B. Hoch, et al., Phys. Chem. Chem. Phys. 17 (2015) 14623–14635. doi: 10.1039/C5CP02613J
G.C. Welch, R.R. San Juan, J.D. Masuda, D.W. Stephan, Science 314 (2006) 1124–1126. doi: 10.1126/science.1134230
D.W. Stephan, Chem 6 (2020) 1520–1526. doi: 10.1016/j.chempr.2020.05.007
K.K. Ghuman, L.B. Hoch, P. Szymanski, et al., J. Am. Chem. Soc. 138 (2016) 1206–1214. doi: 10.1021/jacs.5b10179
M.Y. Sun, B.H. Zhao, R. Yang, et al., ACS Energy Lett. 6 (2021) 2024–2029. doi: 10.1021/acsenergylett.1c00645
J.X. Low, B.Z. Dai, T. Tong, et al., Adv. Mater. 31 (2019) 1807920. doi: 10.1002/adma.201807920
Y. Lu, W.J. Yin, K.L. Peng, et al., Nat. Commun. 9 (2018) 2752. doi: 10.1038/s41467-018-05144-1

Figure 1 Schematic diagram of the in situ/operando techniques for active species identification.

Figure 3 The general sequence and methods for identifying active species and establishing a relationship between structure and activity.

Figure 4 (a) The time-dependent methanol production rate over ZnCu (111). (b) The ZnO surface coverages dependent methanol production rate over Cu (111). (c) The binding energies measurement of Zn 2p on Zn/Cu (111) after CO2 hydrogenation. (d) The time-dependent methanol production rate of ZnCu (211) and ZnO/Cu (111). (e) The XANES spectra evolution of Zn K-edge as well as the difference spectrum between final and initial spectra for the Cu/ZnO/FAU sample during operando conditions. (f) Relative fractions of zinc formate, ZnO, and Zn2+ species monitored by operando time-dependent XAS upon cycling feed gas composition at 260 ℃ and 15 bar. (a-d) Reproduced with permission [52]. Copyright 2020, Science. (e, f) Reproduced with permission [99]. Copyright 2020, American Chemical Society.

Figure 5 (a) Products and TOFs values of Co, Co4N nanosheets, and commercial Cu/ZnO/Al2O3. (b) Temperature-dependent kinetic analysis of Co and Co4N. (c) In situ DRIFTs spectra after H2 treated at 30 ℃ for 30 min over Co and Co4N nanosheets. (d) The quasi-in situ XPS spectra for N 1s of Co4N treated at 30 ℃ for 30 min in an H2 atmosphere. Reaction conditions: 150 ℃, 3.2 MPa (CO2/H2 = 1:3). (e) Proposed mechanism of structure evolution progress through carbonyl iron. (a-d) Reproduced with permission [100]. Copyright 2017, Springer Nature. (e) Reproduced with permission [53]. Copyright 2021, Wiley.

Figure 6 (a) Time-dependent methanol production rate. (b) The In-O and In-In coordination numbers fitted by operando EXAFS. (c) The energy shift (ΔE0) of absorption edge versus the In2O3 reference during reaction. (d) MCR-ALS and (e) LCF analyses of the In K-edge XANES data. (f) The crystalline fraction and particle size of the In2O3 during the operando CO2 hydrogenation. (g) The transformation from crystalline In2O3 into the amorphous or molten phase. Conditions: 300 ℃, 800 mbar (CO2/H2 = 1:3). Reproduced with permission [101]. Copyright 2019, American Chemical Society.

Figure 7 The CO2 conversion (a) and selectivity (b) as a function of temperature from 100 ℃ to 800 ℃ over 15 wt% Ni/SiO2 (Run 1: solid line; Run 2: dashed line). Reaction conditions: Wcat = 15 mg; total gas flow 100 mL/min; CO2/H2 = 1/4. (c) 2D contour plots wavelet transform results obtained from the temperature-dependent operando XAFS. Reproduced with permission [102]. Copyright 2021, American Chemical Society.

Figure 8 The FE (a) and partial current density (b) of different products for electrocatalytic CO2 reduction by CuPc. (c) Cu K-edge XANES spectra under the in situ CO2 electroreduction conditions. The first-order derivatives (d) and Fourier-transformed Cu K-edge EXAFS spectra (e) derived from the in situ XAS results. (f) XRD patterns of CuPc before and after electrocatalytic CO2 reduction. (g) SEM images of CuPc after the reaction process. Scale bar: 100 nm. Reproduced with permission [106]. Copyright 2018, Springer Nature.

Figure 9 (a) High resolution transmission electron microscope (HR-TEM) and particle size distribution of Pd/C. (b) The FE of CO and H2 as well as the CO/H2 ratio under different potentials. (c) Pd K-edge XANES spectra acquired by in situ XAS under the electrolysis conditions. (d) In situ XRD patterns of Pd/C catalyst under LSV from 0.68 V to −0.56 V vs. RHE at the scan rate of 1 mV/s. (f) The calculated binding energy of *H, *CO, and *HOCO over Pd (111), Pd-NP, and PdH (111), PdH-NP, respectively. Reproduced with permission [108]. Copyright 2017, The royal society of chemistry.

Figure 10 (a) EXAFS spectrum of Pb L3-edge over TA-Pb. (b) The potential-dependent FE of HCOOH, CO, and H2. Quasi-in situ XRD pattern (c), EXAFS (d), and XANES spectra (e) of Pb L3-edge of TA-Pb, t-PCO, and t-PCOH. (f) In situ Raman spectra of TA–Pb as a function of time under CO2 bubbling. (g) The XRD pattern comparison between the used catalysts under −0.92 V for 5 h in CO2 and Ar. (h) Schematic diagrams illustrating the behaviors of hydrocerussite as the cathodic electrocatalyst. Reproduced with permission [27]. Copyright 2020, Springer nature.

Figure 11 High-resolution XPS spectra of Cd 3d (a), S 2p (b) in CdS and CdG2 under dark and UV illumination; C 1s (c), N 1s (d) in NG and CdG2 under dark and UV illumination. (e) The CO2 adsorption is caused by the upshifting Ef of graphene by the formation of heterojunction. (f) Schematic diagram illustrating the electron transfer pathway for CO2 photoreduction into CH4 over the hierarchical TaON@G heterostructures. (a-d) Reproduced with permission [110]. Copyright 2019, Wiley-VCH. (e, f) Reproduced with permission [111]. Copyright 2020, American Chemical Society.

Figure 12 (a) The graphical illustration of the proposed overall mechanism over In2O3-x(OH)y for RWGS reaction based on the FLPs theory. (b) The DFT-revealed main contributing molecular orbitals and transition densities for the S1 (HOMO to LUMO), S2 (HOMO to LUMO+1) and S3 (HOMO to LUMO+2) excited states (ES) of the defected In2O3-x(OH)y surface. (c) The computational analyses of the ground state (GS) and ES of the In2O3-x(OH)y cluster model, explaining for the experimentally observed difference in activation energy under dark and light conditions. (a) Reproduced with permission [112]. Copyright 2015, The Royal Society of Chemistry. (b, c) Reproduced with permission [115]. Copyright 2016, American Chemical Society.

Figure 13 (a) SEM images of the as-prepared Mo2N nanobelts. (b) The band structure and density of state (DOS) of bulk Mo2N were obtained by DFT. (c) In situ DRIFTS spectra of Mo2N catalyst under a pulse-intake of H2 or CO2 and dark or illumination conditions. D-H2: under dark, 175 ℃, H2 atmosphere; L-H2 or CO2: 1.0 W/cm2 UV–visible (UV–vis) illumination, 175 ℃, H2 or CO2 atmosphere. (d) Quasi-in situ EPR spectra under dark and UV–vis light conditions in H2 atmosphere. (e) In situ irradiated XPS spectra of N 1s and Mo 3p3/2 over Mo2N nanobelts. (f) Schematic diagram illustrating the H2 heterolysis caused by photoinduced broken symmetry of electron density of Mo-N pairs. Reproduced with permission [116]. Copyright 2021, American Chemical Society.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:

 下载:
下载: