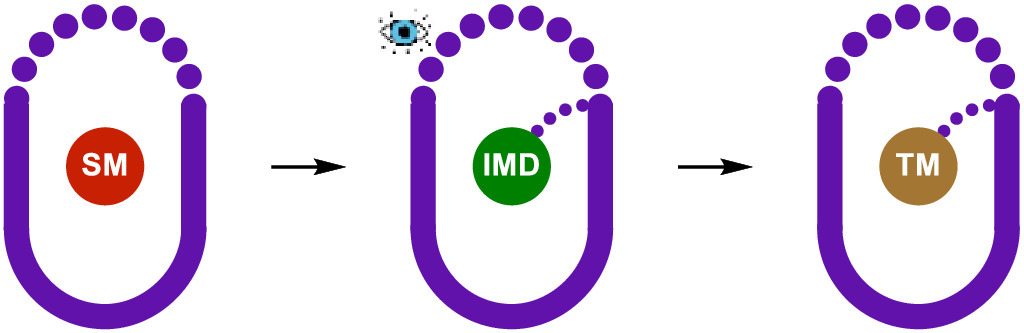
Scheme 1.
Stabilization and observation of labile reaction intermediates mediated by supramolecular containers.

Stepwise reactions involve the appearance and disappearance of reactive intermediates. Determining the sequence of steps – the reaction mechanism – involves identifying the intermediates, an undertaking made difficult by their short lifetimes. Typically, intermediates are not observed but their presence can be deduced from kinetic data [1] and identified by chemical reasoning. In other words, the steady-state concentrations of intermediates are low and makes detection, characterization, and investigation difficult. One way to increase the steady-state concentration of the intermediates is to increase their rate of formation and decrease their rate of disappearance. Application of container compounds can be used to accomplish this [2,3] in low concentrations.
The original container molecules of modern supramolecular chemistry – crown ethers (Pederson) and cryptands (Lehn) – have long been celebrated for their practical applications as hosts for ionic guests [4–7]. Organic synthesis rapidly led to containers of greater sophistication and capacity so that entire molecules could be sequestered. Applications expanded accordingly. Cram "tamed" cyclobutadiene in a carcerand [8], Mock captured a dipolar addition transition state in a cucurbituril [9] and Breslow [10] accelerated Diels–Alder reactions in a cyclodextrin. These applications are not practical, instead, they refer to the science of chemistry itself: Mechanisms, reactive intermediates, catalysis and intermolecular forces. Container compounds are now a tool for studying reactivity and in this review, we show how different containers are used in recent academic studies.
The stabilization of labile reactive intermediates by supramolecular containers has gradually developed in the past three decades [11]. The inner cavity (inner phase) of a molecular container provides an isolated microenvironment that protects the guest molecule(s) within from the influence of the bulk solution [2,12–17]. The encapsulated guest usually interacts with the inside of the host or other co-encapsulated guest(s). In bulk solution, the molecule can interact with the entire environment formed from both the solvent and other reactive molecules, which results in rapid transformation or decomposition of the molecule thus makes it unstable and hard to detect. Therefore, the inner cavity of the supramolecular container acts as an ideal environment for the stabilization and characterization of various labile reactive intermediates (Scheme 1). Here we mainly focus on the unstable guest species – key intermediates in organic transformations – that were detected and stabilized within supramolecular containers in chronological order.
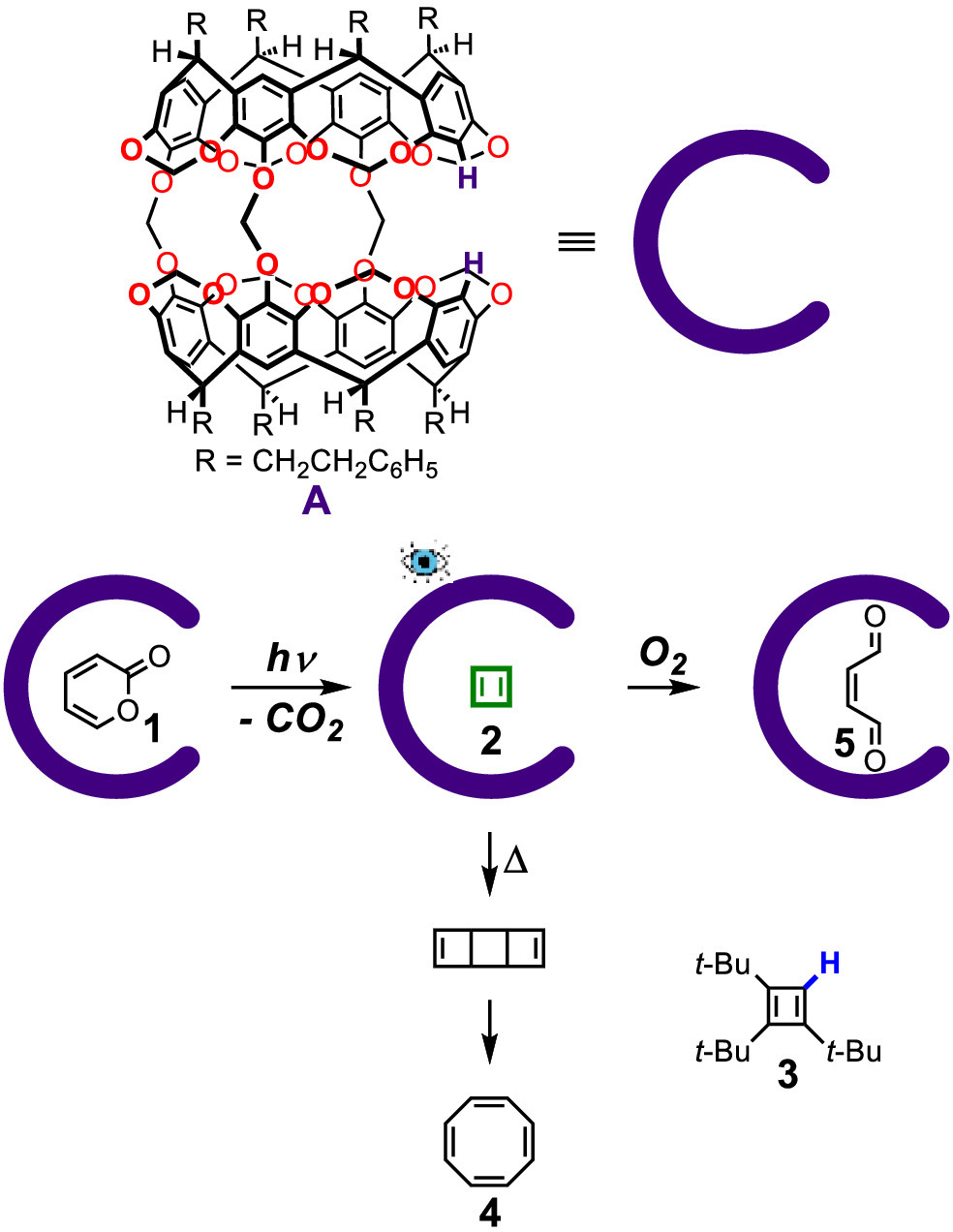
The term carcerand was coined by Cram from the Latin word of carcer (means prison) to describe the first kind of molecular container compound synthesized from the covalent connection of two resorcin[4]arenes [18,19]. The hollow inner space of carcerand could accommodate various guest molecules whose properties are different from those in bulk solutions. In 1991, Cram group reported the first example of the stabilization of cyclobutadiene intermediate in the inner cavity of carcerand A (Fig. 1) [8]. Cyclobutadiene is a very reactive species that undergoes rapid dimerization and ring expansion to afford cyclooctatetraene [20–25]. It is only stable under certain conditions and was detected in an argon matrix at 8 K [26–28]. As for characterization, only FT-IR spectroscopy was investigated under the abovementioned conditions. In Cram's work, by refluxing the mixture of carcerand, cyclobutadiene precursor α-pyrone (1) and solvent chlorobenzene, the carceplex A·1 was formed and then precipitated from the resulting cooled mixture with hexanes. The irradiation of carceplex A·1 in THF-d8 with a 75 W xenon lamp for 30 min resulted in the formation of encapsuled cyclobutadiene (2) and CO2, which was subsequently expelled from the cavity of carcerand A. The 1H NMR spectrum of the resulting solution showed a sharp signal at δ = 2.35 ppm, which is Δδ = −3.03 ppm upfield compared with that of the ring proton of 3 because of the shielding effect from the aromatic walls of the molecular container. The sharp signals of both the encapsulated cyclobutadiene protons and the inward-pointing protons of the host indicated that the guest cyclobutadiene rapidly rotates along all axes inside the cavity on the NMR time scale and possesses a singlet ground state. The encapsulated cyclobutadiene was proved to be stable even at 60 ℃ in the absence of oxygen. When the solution of carceplex A·1 was heated in the sealed NMR tube under vacuum at 220 ℃ for 5 min, the bound cyclobutadiene exchanged with the solvent THF, escaping from the host and subsequently dimerizing to cyclooctatetraene (4). However, in the presence of oxygen, the encapsulated cyclobutadiene was quickly ring-opened and oxidized to malealdehyde (5) inside the cavity of carcerand A, since oxygen is small enough to pass through the host and reacts with the encapsulated cyclobutadiene. The formation of 5 from 1 and 2 was first observed in this work. In solution, only 4 was formed under argon at 8 K. In this specific example, the stabilization of cyclobutadiene intermediate in supramolecular container A prevents its decomposition to form product 5. This shows supramolecular containers have good ability in regulating and controlling reaction mechanism and selectivity.
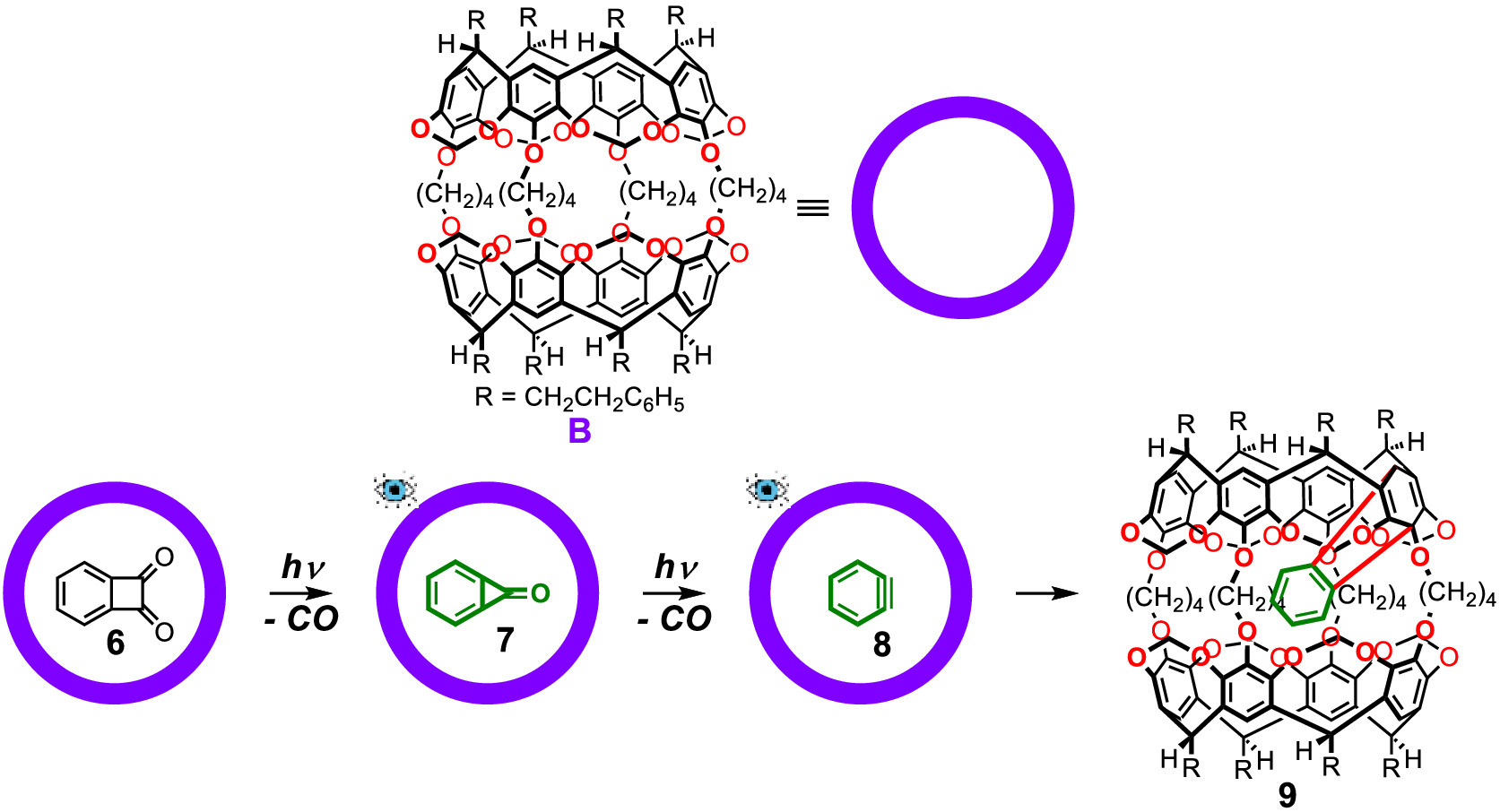
Following Cram's pioneering work, the Warmuth group has also reported a series of labile reactive intermediates stabilized through encapsulation. The first species discussed is o-benzyne [29,30]. Its earlier detection and characterization were conducted under low temperature (<−150 ℃) and limited spectra were obtained including FT-IR, UV–vis, gas-phase microwave spectroscopy and solid-state 13C NMR [31–34]. As shown in Fig. 2, the stabilization of o-benzyne by Warmuth started from the encapsulation of its precursor benzocyclobutenedione (6) by molecular container B. And after irradiation with λ > 400 nm light upon the dry carceplex solution, the highly strained benzocyclopropenone (7), which is also a labile intermediate that was previously studied at low temperature (< −78 ℃), was generated inside the container proved by X-ray crystal structure analysis. Further photolysis of 7 with filtered UV light at 280 ± 10 nm generated the o-benzyne intermediate (8) inside the pocket of B. Solution 1H and 13C NMR spectroscopies were characterized at −75 ℃. However, at room temperature, the encapsulated 7 underwent rapid "innermolecular" Diels–Alder reaction with one aryl unit of the host from the inside at the 1,4-position affording the adduct 9. In another calculation study presented by Warmuth and Houk, according to the above-reported characterization data, it was shown that o-benzyne is aromatic but with more acetylenic than cumulenic character [35].
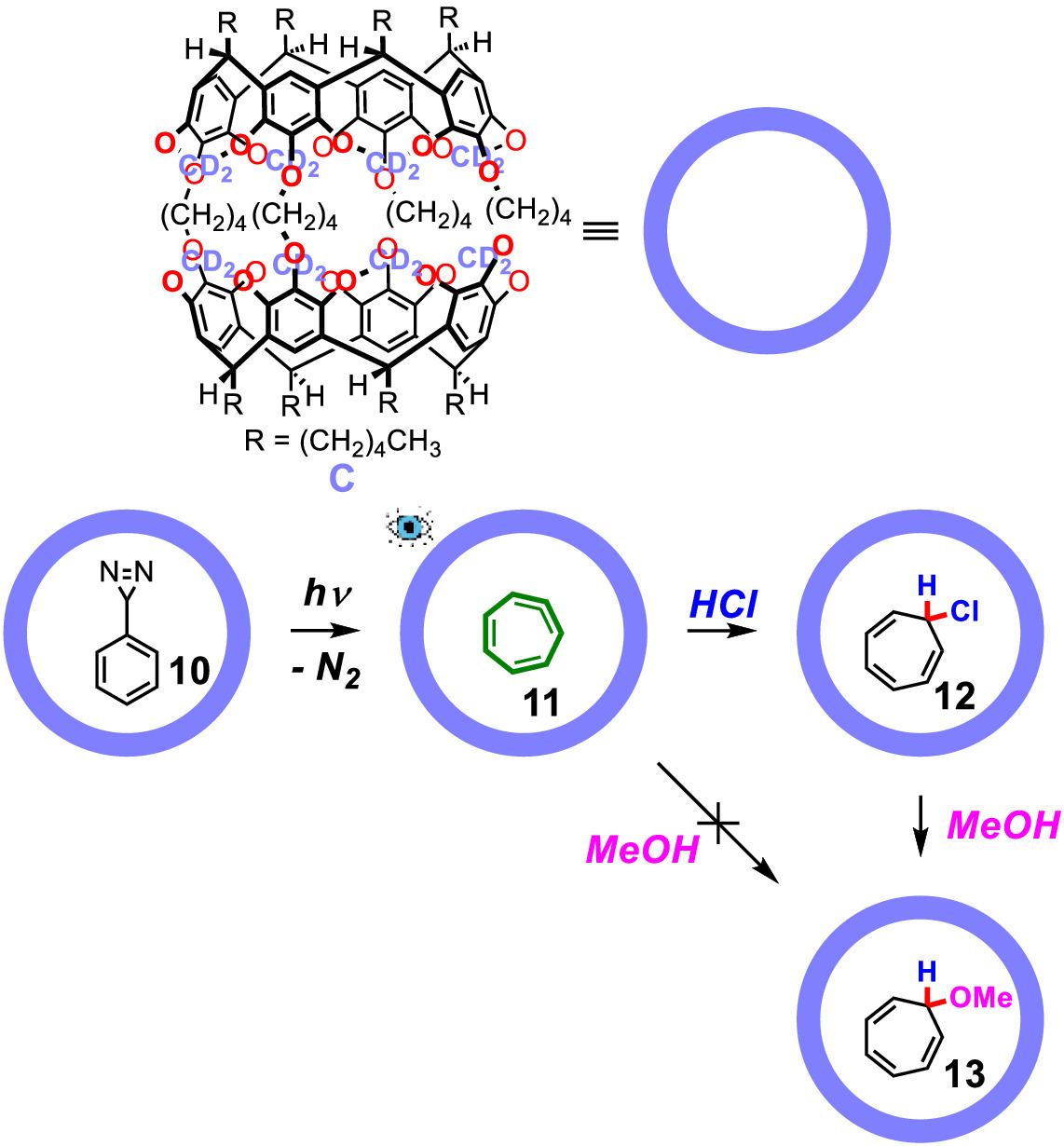
In 2000, Warmuth investigated another important intermediate generated from the rearrangement of phenylcarbene, 1,2,4,6-cycloheptatetraene [36], which was previously detected at 15 K under argon atmosphere [37,38]. In a previous report, limited spectroscopic study showed the existence of the highly strained allene part of 1,2,4,6-cycloheptatetraene, but rapid dimerization at higher temperature prevented further investigation. However, once incarcerated, the dimerization of this highly reactive species would be suppressed because only one 1,2,4,6-cycloheptatetraene was allowed in the cavity of the carcerand host owing to size limitation. In Warmuth's report, 1,2,4,6-cycloheptatetraene (11) was generated from phenyldiazirine (10) via photochemical rearrangement of phenylcarbene inside carcerand C (Fig. 3). The eight acetal carbons were fully deuterated to prevent the phenylcarbene from "innermolecularly" inserting to the acetal C—H bonds of the carcerand host, together with the addition of triplet sensitizer acetophenone-d8. Characterization including IR and NMR of the encapsulated 11 was then conducted, and in the FT-IR spectrum, it shows a weak band at 1810 cm−1, which could be assigned to the allenic carbon–carbon stretching frequency of 11 compared to 1824 cm−1 and 1823 cm−1 in the argon-matrix isolation experiments. With 1H NMR, homonuclear 1H-1H COSY, NOESY spectra and cycloheptatriene as a model compound, the authors calculated the chemical shifts of the protons of 11. This was further confirmed by the 1H—1H coupling constants and the sharpness of the resonances of the acetal protons in the 1H NMR spectrum of the corresponding undeuterated carceplex. The incarcerated 11 reacted with HCl to afford 7-chlorocyclohepta-1,3,5-triene (12), which was further transformed to 7-methoxycyclohepta-1,3,5-triene (13) with the addition of methanol. However, direct treatment of the incarcerated 11 with methanol didnot afford 13, even at 60 ℃ for 3 h, which indicated that the reactivity of 11 in the nonpolar inner phase differs significantly from that of the bulk phase. This example also demonstrates the ability of supramolecular containers in controlling reactivity and stabilization of certain reactive intermediates by their microenvironment. The encapsulation of the stabilized 11 prevents the dimerization because they are protected inside the host and cannot approach each other as in solution where 11 would rapid undergo dimerization even at 10 K. In a following study, the authors further investigated the chemistry and properties of the encapsulated 11 in detail [39]. The similarly stabilization and investigation of its aza-analogue, 1-azacyclohepta-1,2,4,6-tetraene, generated from the rearrangement of phenylnitrene, was also reported [40,41].
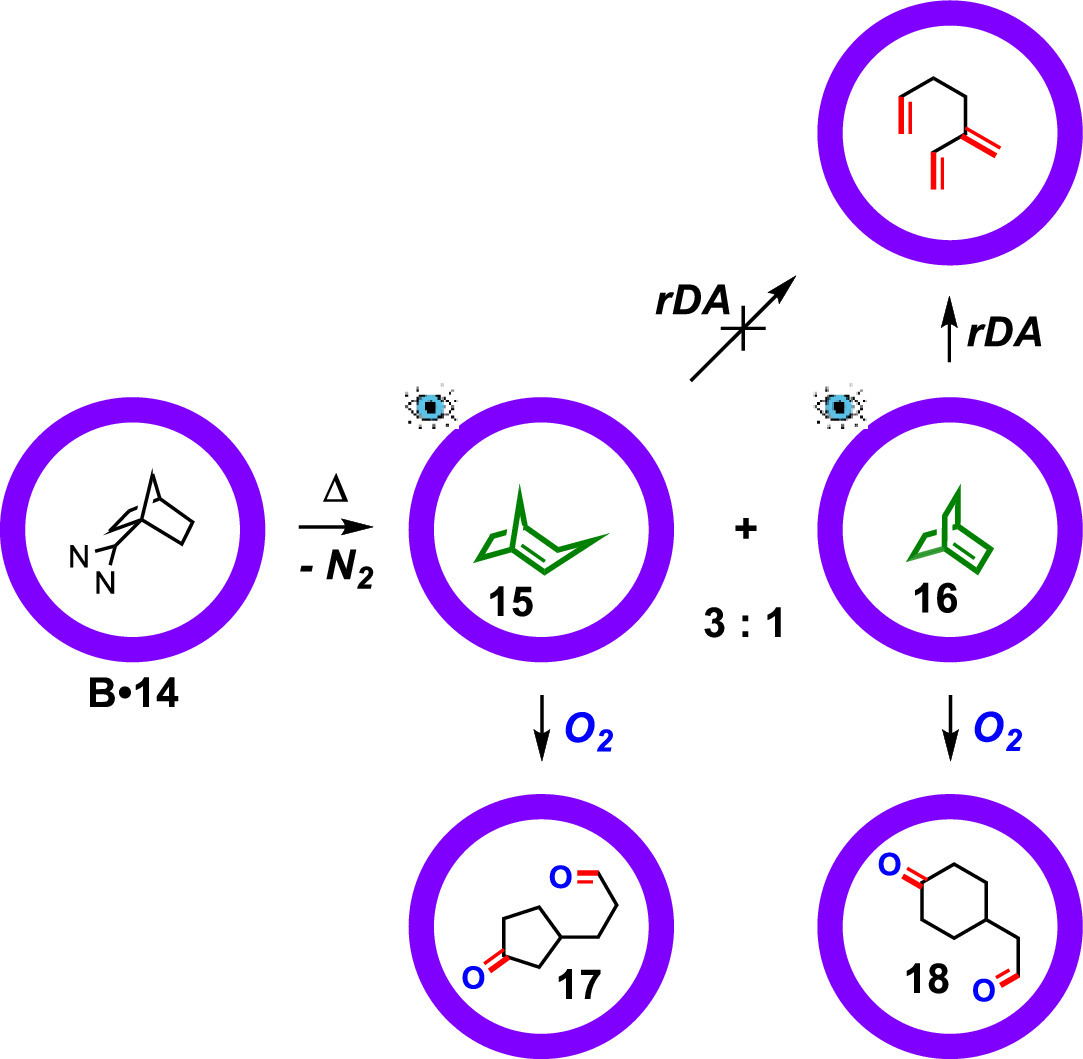
When a double bond appears at a bridgehead position in bridged-ring compounds, it will produce huge ring strain, which makes it very unstable. This is Bredt's Rule [42]. An anti-Bredt's olefin containing a double bond in the bridgehead site would undergo rapid dimerization or retro Diels-Alder reaction. The stabilization, detection and characterization of anti-Bredt's olefins was also reported by Warmuth [43]. The carbene precursor 1-bicyclo[2.2.1]heptyldiazirine (14) was first incarcerated in the cavity of carcerand B, which underwent photo- and thermolysis to produce different products through various pathways. As shown in Fig. 4, upon heating, the encapsulated diazirine precursor 14 turned into (Z)-bicyclo[3.2.1]oct-1-ene (15) and bicyclo[2.2.2]oct-1-ene (16) in a 3:1 ratio. Inside carcerand B, 15 is very thermally stable in the absence of O2 even at 95 ℃ for one week. In the presence of O2, 15 was oxidized to 17 quantitively at 60 ℃ for two days. The thermal stability of the incarcerated 16 is also remarkable, the rate constant for the retro Diels-Alder reaction of which is 2.2 × 10−5 s−1 at 61.7 ℃, which is at least 2000-fold more stable compared to the corresponding free species. The bound 16 also reacted with O2 and rapidly afford 18. The generation of reactive anti-Bredt's olefin species (15 and 16) and their controllable oxidation to diacyl products (17 and 18), which could not be detected in solution, further reveal the role of supramolecular containers in stabilizing reactive intermediates and the accompanied ability in controlling reactivity and selectivity.
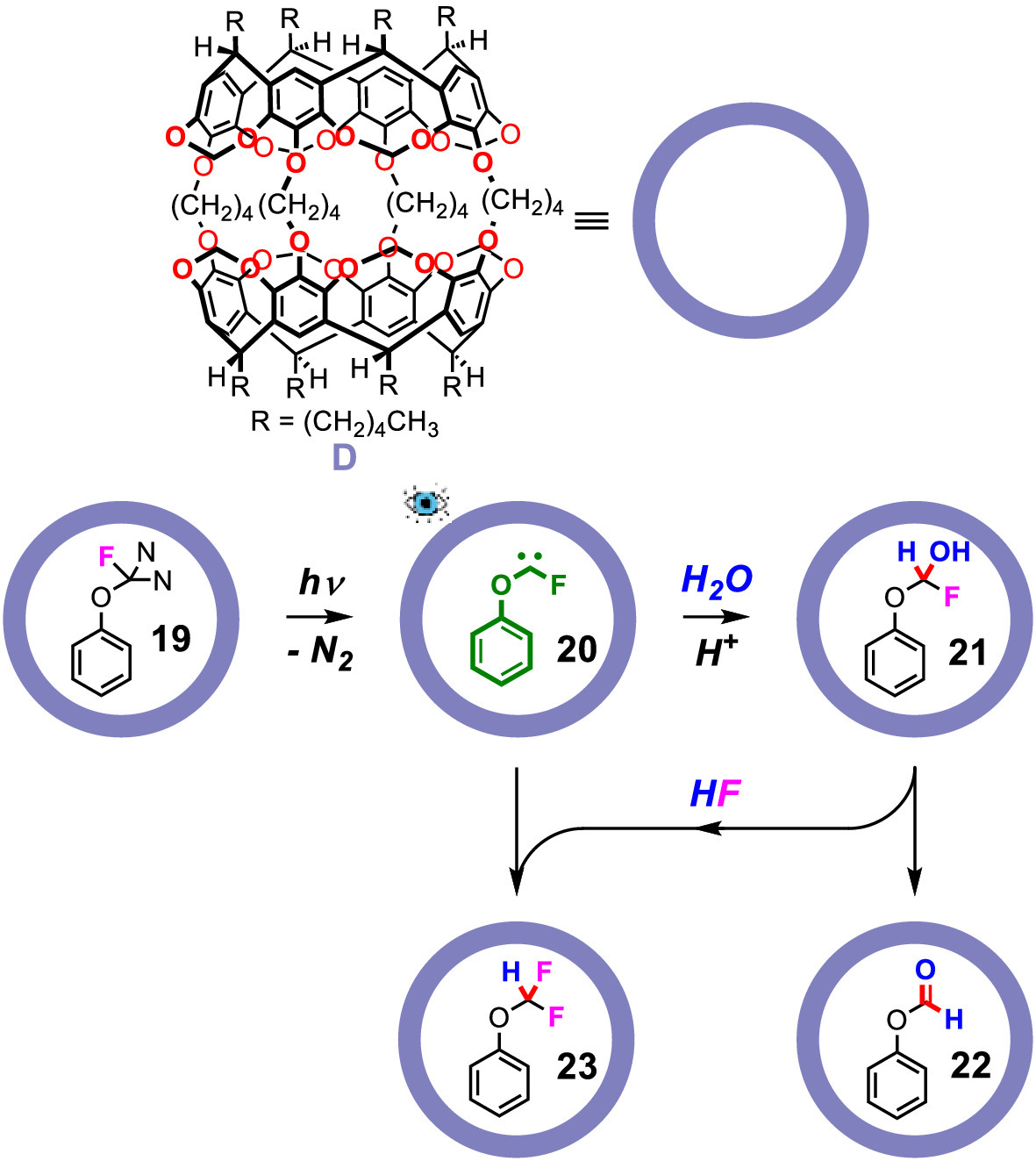
In the above examples, carbene species were frequently encountered but was not detected and characterized, even in the inner phase of carcerands. The stability of carbene species strongly depends on its substituents [44–47], and intrinsic stability can be achieved by electron donating substituents and steric effects [48–53]. Confinement might also be used in the stabilization of certain carbene species. The Warmuth group chose fluorophenoxycarbene as the model compound to investigate the stabilization of carbene species confined to carcerand D, because it was stabilized by its substituents and its size would fit the carcerand. The 3-fluoro-3-phenoxydiazirine (19) was used as a precursor and incarcerated (Fig. 5). After photolysis, fluorophenoxycarbene (20) was observed and its 1H NMR, 13C NMR, 19F NMR, 1H[19F] NOE difference spectra were thoroughly investigated for the first time. The spectra showed that the cis isomer of fluorophenoxycarbene is more favored inside the carcerand, which was contrary to the DFT calculation results. After several days, 20 slowly converted to phenyl formate (22) and phenyl-(difluoromethyl)ether (23) in about a 1:1 ratio. The authors concluded that the transformation goes through a hemiacetal intermediate (21). Addition of pyridine-d5 to the system before photoirradiation would prevent the decomposition of 20 for days, which implied that the addition of water to 20 forming 21 is acid-catalyzed, rather than through an ylide pathway. The stabilization of the fluorophenoxycarbene arises from three main factors: (1) The encapsulation of fluorophenoxycarbene prevents its dimerization; (2) The inner phase of the carcerand is hydrophobic that prevents water from approaching fluorophenoxycarbene; (3) The carbene-π interaction between fluorophenoxycarbene and the carcerand may also be involved.
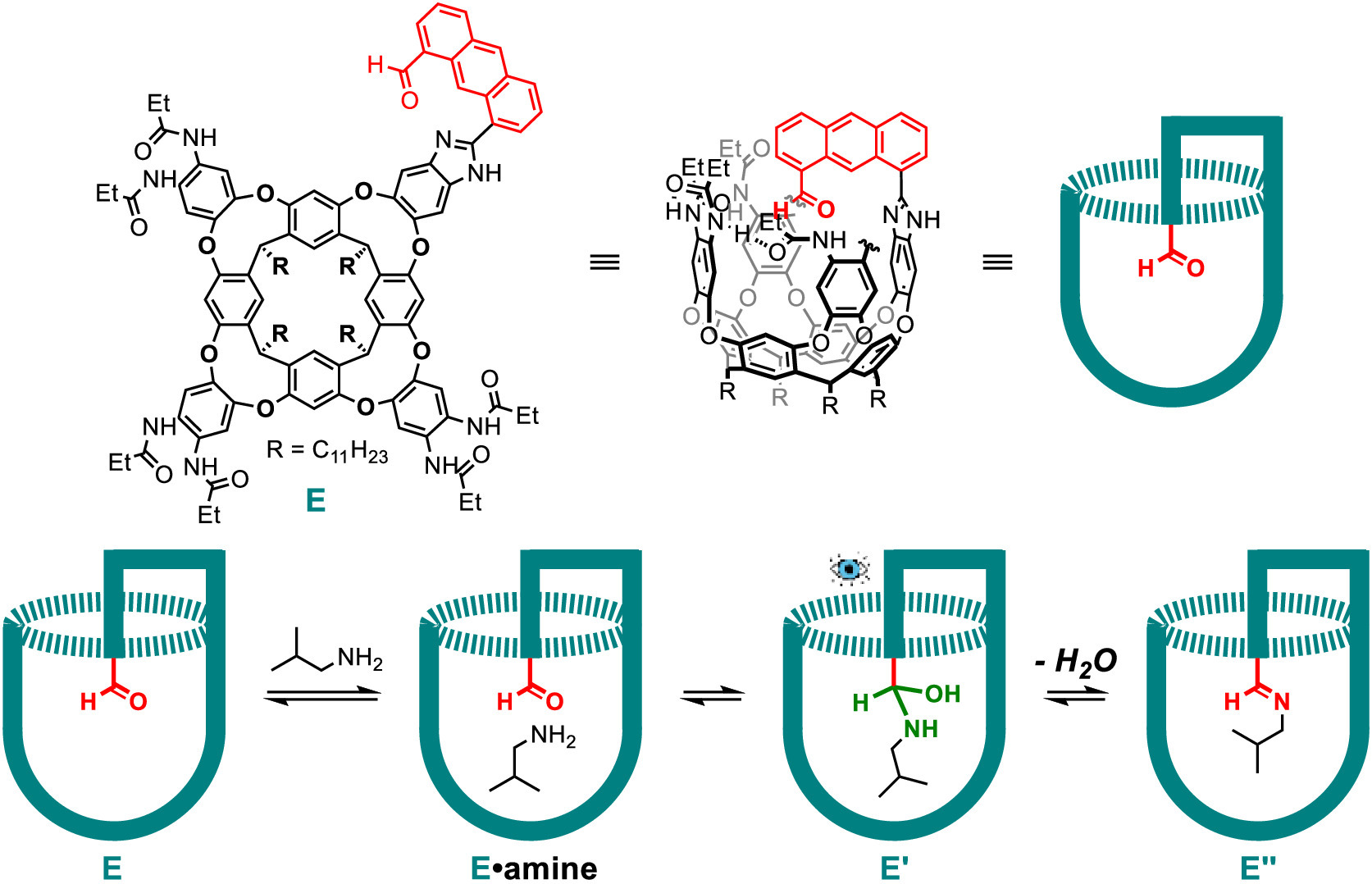
Deep cavitands are vase-shaped macrocycle hosts with one open end that can accomodate various guest molecules [54–60]. They are built up from a resorcinarene core by covalently attaching various walls, and further functionalization of the upper rims endows them with diverse functions for assembling to capsules or participating in catalysis [11–13,15,16,18,61]. Rebek and co-workers have used deep cavitands for stabilization and detection of labile reaction intermediates. The first example in 2007 involved the capture of a tetrahedral hemiaminal intermediate using a functionalized deep cavitand with an "introverted" (inwardly directed) aldehyde group (Fig. 6) [62]. Hemiaminals are intermediates in the addition reaction of primary or secondary amines with carbonyl compounds to afford imines [63]. Hemiaminals are unstable and once formed they either dissociate to starting materials or proceed to the imine product with loss of water, owing to the driving force of forming a π bond. Accordingly, the observation of hemiaminal intermediates was limited to a few special cases [64–66]. The authors designed and synthesized a specific deep cavitand E with an introverted aldehyde functionality. The carbonyl can covalently capture a primary amine, and the container can stabilize the hemiaminal intermediate E' for minutes to hours. This intermediate was observed and characterized through NMR spectroscopy before turning into the final imine product E''. The hemiaminal intermediate is stabilized by three aspects: (1) The (noncovalent) binding of the amine guest into the cavity places it close to the introverted aldehyde group and lowers the barrier for the nucleophilic attack step from the entropic point of view; (2) The secondary amide groups on the upper rim of the host interact with the tetrahedral hemiaminal intermediate through hydrogen bonding, which reduces the enthalpic price of the reaction; (3) The confinement behavior of the host-guest complex prevents the hemiaminal from bases and other amines that accelerate the formation of imine product. More detailed investigation of this stabilization mode and mechanism including kinetic studies and calculation were provided by the same group [67] and Li et al. [68].
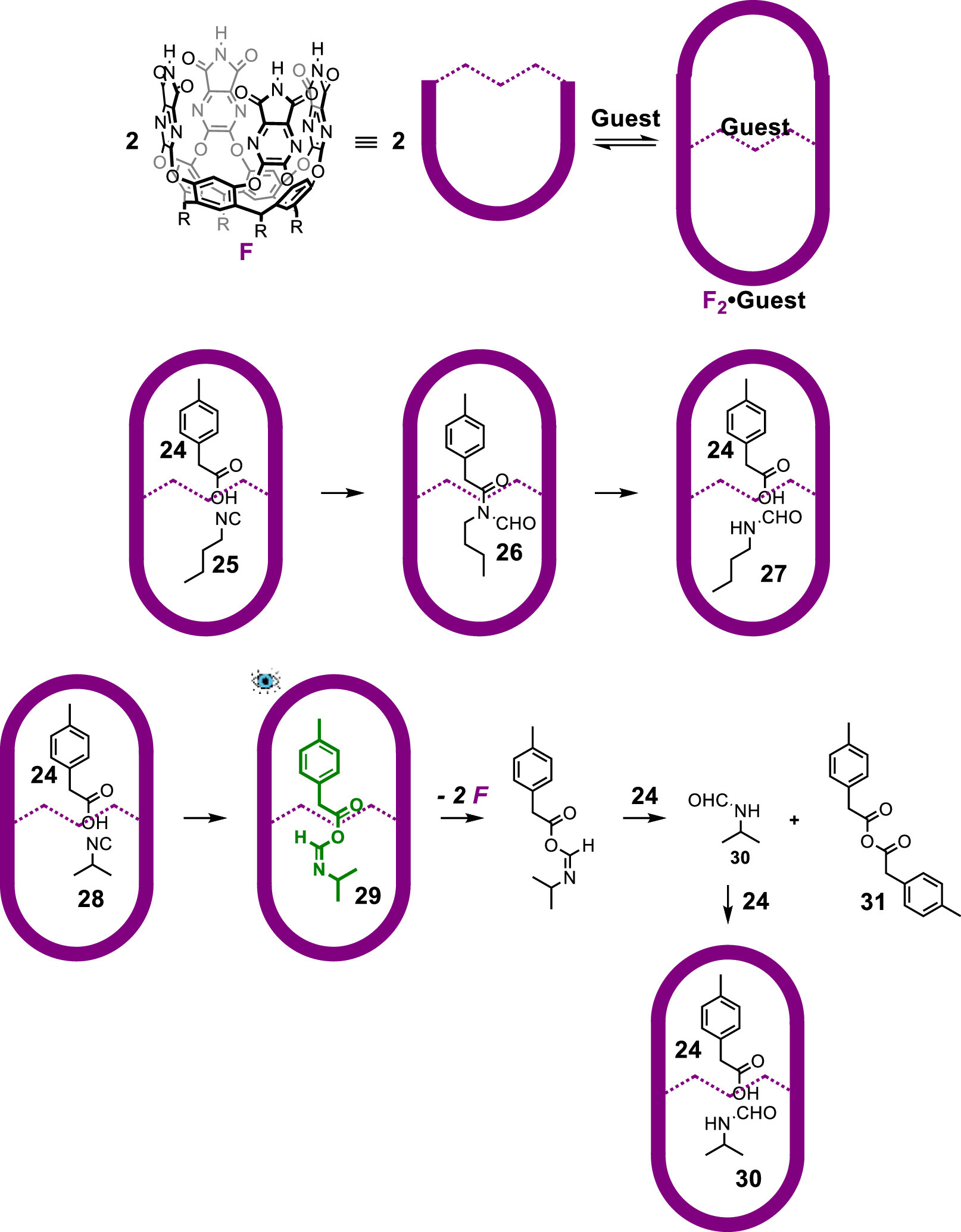
In a following report regarding the reaction of carboxylic acids and isonitriles, the researchers detected another interesting isoamide intermediate inside a self-assembled capsule dimerized from cavitands with benzimidazolone rims [69]. The reaction between carboxylic acids and isonitriles to afford formylated secondary amides had been reported by Danishefsky and co-workers just before this work using microwave heating techniques [70]. The O-acylisoamide (the acyclic version of an azlactone) must be involved in this reaction, followed by the Mumm rearrangement to the formylated secondary amide product. However, the O-acylisoamide intermediate had not been observed owing to its instability to rearrangement. The authors found a way of co-encapsulating the components inside a cylindrical capsule F2 and successfully detected and characterized the intermediate by NMR spectroscopy along the reaction pathway (Fig. 7). Cavitand F2, p-tolylacetic acid (24) and n-butyl isonitrile (25) self-assembled to form a capsular complex on mixing and 1H NMR spectroscopy showed that the acid and isonitrile groups are close to each other in the polar middle of the capsule. The encapsulated starting materials transformed to the formylated secondary amide product 26 even at room temperature. The reaction was completed within 20 h at 40 ℃, during which no trace of O-acylisoamide intermediate was observed in 1H NMR spectrum. A small amount of formamide 27 and acid 24 were also observed co-encapsulated inside the host. However, control experiments showed that no reaction could be observed without the host under the same conditions, and formamide 27 together with anhydride by-product were formed after 2 days at 40 ℃ and 4.0 mol/L concentration. This result indicated that the reaction inside the capsule undergoes a different but accelerated pathway. When a bulkier isonitrile 28 was applied, the O-acylisoamide intermediate 29 was successfully observed inside the capsule in 1H NMR spectrum, but it did not rearrange to the formylated secondary amide product. The four-membered ring intermediate during the acyl rearrangement is highly stained and – relative to the flexible n-butyl group – the branched isopropyl group could not achieve the necessary motions inside to undergo the rearrangement to the formylated secondary amide product. Instead, the O-acylisoamide intermediate 29 escapes the capsule and reacts with another molecule of acid 24 to afford formamide 30 and the symmetrical anhydride 31. The formamide 30 and the acid starting material 24 can be accommodated in the capsule, but the anhydride 31 is too large to fit in the cavity of the host. The successful detection of the O-acylisoamide intermediate in this case was achieved by slowing its rearrangement.
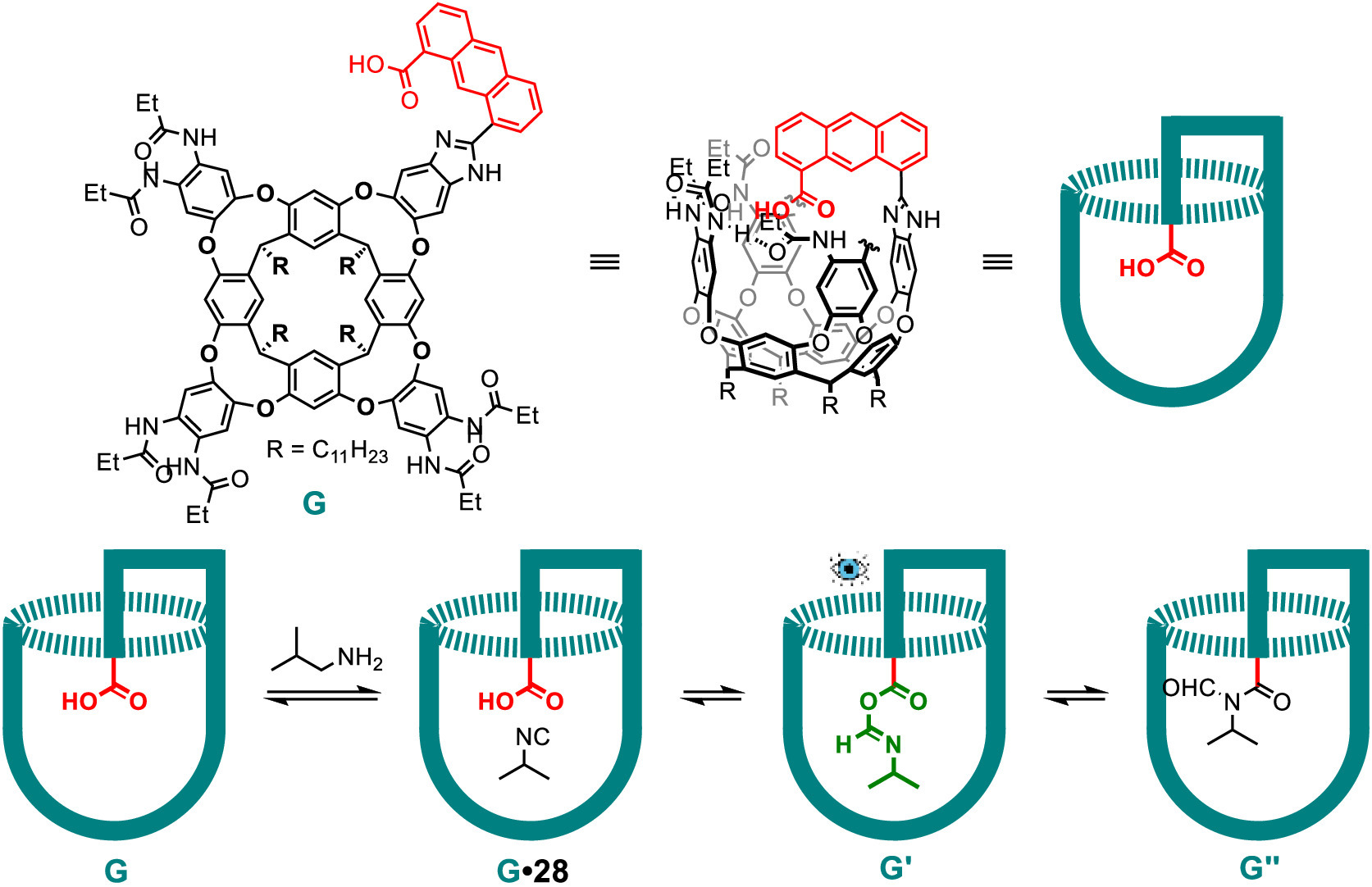
Even though the O-acylisoamide intermediate was detected in the above work, the intermediate underwent a different pathway inside the capsule from the target reaction in solution. Therefore, the authors sought to stabilize the O-acylisoamide intermediate with another mode using a tailored cavitand, which possesses an introverted carboxylic moiety [71]. As shown in Fig. 8, on mixing isopropylisonitrile (28) with cavitand G, both the O-acylisoamide intermediate G' and the formylated secondary amide product G'' were observed in 1H NMR spectrum. The intermediate G' rearranged to the product G'' completely after several hours. In this work, IR spectroscopy was also applied to monitor the reaction process, and a characteristic band at 1771 cm−1 represented the stretching frequency of carbonyl group of the O-acylisoamide intermediate G'. The stabilization of this intermediate is due to three causes: (1) The recognition of the isonitrile guest 28 inside the cavity of the host draws it close to the acid group of the cavitand G and accelerates the formation of the O-acylisoamide intermediate; (2) The aromatic walls of the host and the secondary amide groups on the upper rim of the host can hydrogen bond to the O-acylisoamide intermediate, which offers stabilization; (3) The confinement in the host decelerates the subsequent rearrangement of the O-acylisoamide intermediate through a steric effect. These effects prolong the lifetime of the intermediate for effective detection and characterization. These features demonstrate the advantage of functionalized containers for stabilizing otherwise labile reaction intermediates.
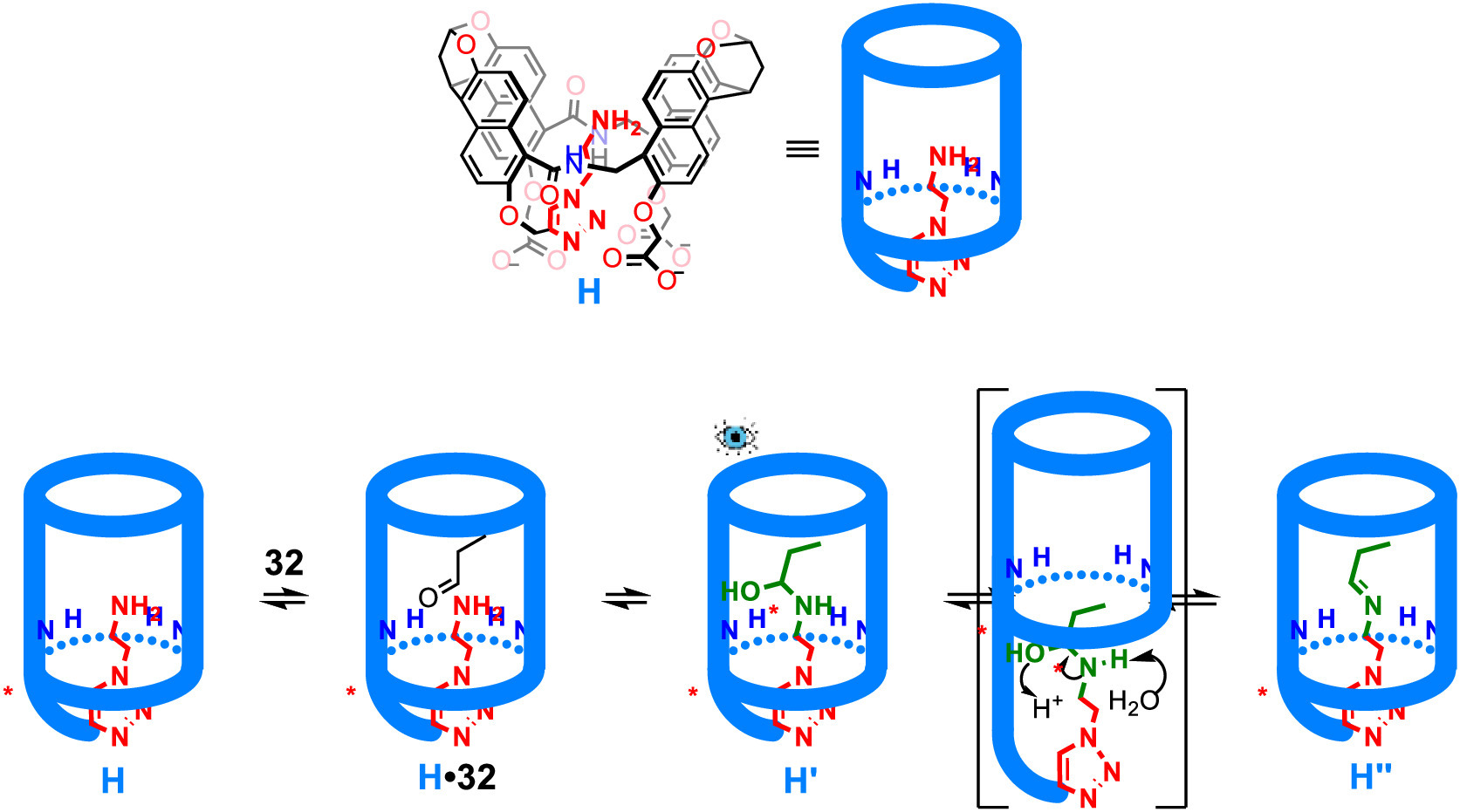
The water-soluble amide naphthotube host was developed by Jiang's group in 2016 [72]. This container proved successful in the recognition of numerous hydrophilic molecules in water through shielded hydrogen bonding [72–75]. The same group reported in 2020 that this naphthotube host could be used to stabilize a closed-ring isomer of spiropyran and applied it to the detection of toxic paraoxon [76] through naked-eye observation. With the further modification of this host, in 2022, Jiang's group reported the stabilization of hemiaminal intermediates (Fig. 9). This involved a tailored, water-soluble amide naphthotube host equipped with an amine functionality (H) [77]. As shown by 1D and 2D NMR spectroscopy, the amine side chain is folded into the cavity of the naphthotube in water and the ethylene arm is rigidified. The amine group was considered to interact with the amide moiety of the host through hydrogen bonding, which was further supported by DFT calculations. In contrast to the functioning mode of the Rebek's work of stabilizing a hemiaminal intermediate, in which an introverted and rigidified aldehyde moiety was introduced to the rim of the cavitand, Jiang and co-workers chose the amine to be attached to the naphthotube in a flexible manner. Once inside the cavity of the host in water, the amine's chain becomes rigidified. Addition of excess propanal (32) to an aqueous solution of the naphthotube host H gives a hemiaminal adduct H' that was detected through NMR and mass spectrometry. Since the host is chiral, two diastereomers were formed in the early stage of the reaction. DFT calculations suggested that one diastereomer (H'-1) formed a single hydrogen bond with the host while the other diastereomer (H'-2) formed two. Indeed, the NMR spectra showed that H'-2 is more stable than H'-1 and the ratio is 0.88 (H'-1/H'-2). Mass spectrometry studies suggested that the hemiaminal intermediate could either release the aldehyde to give back the naphthotube H or go forward to the imine product H'' with the loss of water. Acetaldehyde showed similar behavior to that of propanal but the ratio of the corresponding H'-1/H'-2 is 0.26. However, while monitoring the reaction using larger aldehydes, only imine products were observed without any detection of the hemiaminal intermediates. The NMR spectra of the imine products showed that the ethylene and imine groups were pushed towards the portal of the host, especially for longer aldehydes, where hydrogen bonding interactions between imine and amides were weakened. However, for short aldehydes such as acetaldehyde and propanal, the hemiaminal centers were well shielded and protected inside the cavity of the host and well-located for the hydrogen bonding interactions. It was also showed that the aldehydes were first introduced to the cavity and the reaction occurred inside the host. Both the hydrophobic effect and the hydrogen bonding interaction were responsible for the stabilization of the hemiaminal intermediates. The replacement of longer aldehyde with shorter ones to form new imine products indicated that the reaction is reversible. This work of stabilizing water-sensitive hemiaminal intermediate and imine product in water suggests that water-soluble amine naphthotube could stabilize other reaction intermediates.
To summarize, we have reviewed several cases of stabilizing and characterizing unstable reactive species, with the help of supramolecular containers. First, for intermolecular reactions, recognition of the reagents can draw reactants closer to accelerate the formation of intermediates. The intermediates are then stabilized via the confinement of the supramolecular hosts that isolate and protect them during their reaction to products. Steric effects inside the limited space of the cavity impose specific conformations, which may also slow subsequent transformations. The container walls also protect sensitive intermediates from the outside atmosphere and solvent-borne reagents. These effects cooperate to result in higher steady state concentrations of otherwise reactive intermediates, enough to observe them directly with conventional spectroscopies. The electron-rich π-face of the inner phase and the hydrogen bonding sites of the host can further stabilize the intermediates through pre-organized π bonding and hydrogen bonding. In some cases, the reactive centers are designed to be covalently attached to the frameworks of the hosts as the intermediates are captured inside. Stabilization, detection and characterization of key reactive intermediates on the one hand provide valuable insights for the deciphering and investigating reaction mechanisms, and on the other hand, endow chemists with powerful tool in regulating and controlling reactivity and selectivity by supramolecular containers. We hope this review will encourage researchers to engineer new container molecules for applications in reaction mechanism.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the National Natural Science Foundation of China (Nos. 22071144 and 22101169) and by Shanghai Scientific and Technological Committee (No. 22010500300)
R. Huisgen, Angew. Chem. Int. Ed. 9 (1970) 751–762. doi: 10.1002/anie.197007511
M. Morimoto, S.M. Bierschenk, K.T. Xia, et al., Nat. Catal. 3 (2020) 969–984. doi: 10.1038/s41929-020-00528-3
M. Petroselli, Y.Q. Chen, J. Rebek Jr., Y. Yu, Green Synth. Catal. 2 (2021) 123–130. doi: 10.1016/j.gresc.2021.03.004
C.J. Pedersen, J. Am. Chem. Soc. 89 (1967) 7017–7036. doi: 10.1021/ja01002a035
C.J. Pedersen, Angew. Chem. Int. Ed. 27 (1988) 1021–1027. doi: 10.1002/anie.198810211
J.M. Lehn, E. Sonveaux, A.K. Willard, J. Am. Chem. Soc. 100 (1978) 4914–4916. doi: 10.1021/ja00483a059
J.M. Lehn, S.H. Pine, E.I. Watanabe, A.K. Willard, J. Am. Chem. Soc. 99 (1977) 6766–6768. doi: 10.1021/ja00462a055
D.J. Cram, M.E. Tanner, R. Thomas, Angew. Chem. Int. Ed. 30 (1991) 1024–1027. doi: 10.1002/anie.199110241
W.L. Mock, T.A. Irra, J.P. Wepsiec, T.L. Manimaran, J. Org. Chem. 48 (1983) 3619–3620. doi: 10.1021/jo00168a070
D.C. Rideout, R. Breslow, J. Am. Chem. Soc. 102 (1980) 7816–7817. doi: 10.1021/ja00546a048
A. Galana, P. Ballester, Chem. Soc. Rev. 45 (2016) 1720–1737. doi: 10.1039/C5CS00861A
T.S. Koblenz, J. Wassenaar, J.N.H. Reek, Chem. Soc. Rev. 37 (2008) 247–262. doi: 10.1039/B614961H
R.J. Hooley, J. Rebek Jr., Chem. Biol. 16 (2009) 255–264. doi: 10.1016/j.chembiol.2008.09.015
D.M. Kaphan, F.D. Toste, R.G. Bergman, K.N. Raymond, J. Am. Chem. Soc. 137 (2015) 9202–9205. doi: 10.1021/jacs.5b01261
C.M. Hong, R.G. Bergman, K.N. Raymond, F.D. Toste, Acc. Chem. Res. 51 (2018) 2447–2455. doi: 10.1021/acs.accounts.8b00328
Q. Zhang, L. Catti, K. Tiefenbacher, Acc. Chem. Res. 51 (2018) 2107–2114. doi: 10.1021/acs.accounts.8b00320
G. Olivo, G. Capocasa, D. Del Giudice, O. Lanzalunga, S. Di Stefano, Chem. Soc. Rev. 50 (2021) 7681–7724. doi: 10.1039/D1CS00175B
D.J. Cram, S. Karbach, Y.H. Kim, L. Baczynskyj, G.W. Kalleymeyn, J. Am. Chem. Soc. 107 (1985) 2575–2576. doi: 10.1021/ja00294a076
M.L.C. Quan, D.J. Cram, J. Am. Chem. Soc. 113 (1991) 2754–2755. doi: 10.1021/ja00007a060
E.J. Corey, J. Streith, J. Am. Chem. Soc. 86 (1964) 950–951. doi: 10.1021/ja01059a059
E.J. Corey, W.H. Pirkle, Tetrahedron Lett. 8 (1967) 5255–5256. doi: 10.1016/S0040-4039(01)89655-6
G. Maier, M. Hoppe, K. Lanz, H.P. Reisenauer, Tetrahedron Lett. 25 (1984) 5645–5648. doi: 10.1016/S0040-4039(01)91402-9
J. Kreile, N. Münzel, A. Schweig, H. Specht, Chem. Phys. Lett. 124 (1986) 140–146. doi: 10.1016/0009-2614(86)85133-8
O.L. Chapman, C.L. McIntosh, J. Pacansky, J. Am. Chem. Soc. 95 (1973) 614–617. doi: 10.1021/ja00783a066
C. Y Lin, A. Krantz, J. Chem. Soc., Chem. Commun. (1972) 1111–1112.
G. Maier, S. Pfriem, U. Schäfer, R. Matusch, Angew. Chem. Int. Ed. 17 (1978) 520–521. doi: 10.1002/anie.197805201
G. Maier, A. Alzérreca, Angew. Chem. Int. Ed. 12 (1973) 1015–1016. doi: 10.1002/anie.197310151
S. Masamune, N. Nakarnura, M. Suda, H. Ona, J. Am. Chem. Soc. 95 (1973) 8481–8483. doi: 10.1021/ja00806a064
R. Warmuth, Angew. Chem. Int. Ed. 36 (1997) 1347–1350. doi: 10.1002/anie.199713471
R. Warmuth, Chem. Commun. (1998) 59–60.
N. Münzel, A. Schweig, Chem. Phys. Lett. 147 (1988) 192–194. doi: 10.1016/0009-2614(88)85082-6
J.G. G Simon, N. Münzel, A. Schweig, Chem. Phys. Lett. 170 (1990) 187–192. doi: 10.1016/0009-2614(90)87113-6
J.G. Radziszewski, A. H B Jr., R. Zahradnik, J. Am. Chem. Soc. 114 (1992) 52–57. doi: 10.1021/ja00027a007
A.M. Oerndt, J.C. Facelli, J.G. Radziszewski, et al., J. Am. Chem. Soc. 118 (1996) 846–852. doi: 10.1021/ja953417r
H. Jiao, P. von Ragué Schleyer, B.R. Beno, K.N. Houk, R. Warmuth, Angew. Chem. Int. Ed. 36 (1997) 2761–2764. doi: 10.1002/anie.199727611
R. Warmuth, M.A. Marvel, Angew. Chem. Int. Ed. 39 (2000) 1117–1119. doi: 10.1002/(SICI)1521-3773(20000317)39:6<1117::AID-ANIE1117>3.0.CO;2-E
P.R. West, O.L. Chapman, J.P. LeRoux, J. Am. Chem. Soc. 104 (1982) 1779–1782. doi: 10.1021/ja00370a074
R.J. McMahon, C.J. Abelt, O.L. Chapman, et al., J. Am. Chem. Soc. 109 (1987) 2456–2469. doi: 10.1021/ja00242a034
R. Warmuth, M.A. Marvel, Chem. Eur. J. 7 (2001) 1209–1220. doi: 10.1002/1521-3765(20010316)7:6<1209::AID-CHEM1209>3.0.CO;2-M
R. Warmuth, S. Makowiec, J. Am. Chem. Soc. 127 (2005) 1084–1085. doi: 10.1021/ja044557g
R. Warmuth, S. Makowiec, J. Am. Chem. Soc. 129 (2007) 1233–1241. doi: 10.1021/ja066130a
J. Bredt, H. Thouet, L. Schmitz, Justus Liebigs Ann. Chem. 437 (1924) 1–13. doi: 10.1002/jlac.19244370102
P. Roach, R. Warmuth, Angew. Chem. Int. Ed. 42 (2003) 3039–3042. doi: 10.1002/anie.200351120
R.A. Moss, Carbenic philicity, in: G. Bertrand (Ed. ), Carbene Chemistry: From Fleeting Intermediates to Powerful Reagents, FontisMedia-Marcel Dekker, Lausanne, 2002, pp. 57–101.
R.A. Moss, Acc. Chem. Res. 13 (1980) 58–64. doi: 10.1021/ar50146a005
N.G. Rondan, K.N. Houk, R.A. Moss, J. Am. Chem. Soc. 102 (1980) 1770–1776. doi: 10.1021/ja00526a002
R.A. Moss, Acc. Chem. Res. 22 (1989) 15–21. doi: 10.1021/ar00157a003
W. v. E. Doering, R.G. Buttery, R.G. Laughlin, N. Chaudhuri, J. Am. Chem. Soc. 78 (1956) 3224.
H. Tomioka, E. Iwamoto, H. Itakura, K. Hirai, Nature 412 (2001) 626–628. doi: 10.1038/35088038
S. Goumri-Magnet, T. Kato, H. Gornitzka, A. Baceiredo, G. Bertrand, J. Am. Chem. Soc. 122 (2000) 4464–4470. doi: 10.1021/ja994408b
D. Bourissou, O. Guerret, F.P. Gabbai, G. Bertrand, Chem. Rev. 100 (2000) 39–92. doi: 10.1021/cr940472u
A.J. Arduengo III, R.L. Harlow, M. Kline, J. Am. Chem. Soc. 113 (1991) 361–363. doi: 10.1021/ja00001a054
R.W. Alder, P.R. Allen, M. Murray, A.G. Orpen, Angew. Chem. Int. Ed. 35 (1996) 1121–1123. doi: 10.1002/anie.199611211
J.R. Moran, S. Karbach, D.J. Cram, J. Am. Chem. Soc. 104 (1982) 5826–5828. doi: 10.1021/ja00385a064
J.C. Sherman, D.J. Cram, J. Am. Chem. Soc. 111 (1989) 4527–4528. doi: 10.1021/ja00194a074
E. Dalcanale, P. Soncini, G. Bacchilega, F. Ugozzoli, J. Chem. Soc., Chem. Commun. 8 (1989) 500–502.
J.A. Bryant, C.B. Knobler, D.J. Cram, J. Am. Chem. Soc. 112 (1990) 1254–1255. doi: 10.1021/ja00159a060
J.R. Moran, J.L. Ericson, E. Dalcanale, et al., J. Am. Chem. Soc. 113 (1991) 5707–5714. doi: 10.1021/ja00015a026
P. Roncucci, L. Pirondini, G. Paderni, et al., Chem. Eur. J. 12 (2006) 4775–4784. doi: 10.1002/chem.200600085
Y.Q. Chen, H.W. Guan, K. Kanagaraj, J. Rebek Jr., Y. Yu, Chin. Chem. Lett. 33 (2022) 4908–4911. doi: 10.1016/j.cclet.2022.03.039
P.W.N.M. van Leeuwen, Supramolecular Catalysis, Wiley-VCH, 2008.
T. Iwasawa, R.J. Hooley, J. Rebek Jr., Science 317 (2007) 493–496. doi: 10.1126/science.1143272
E.V. Anslyn, D.A. Dougherty, Modern Physical Organic Chemistry, University Science, Sausalito, CA, 2006.
D.A. Evans, G. Borg, K.A. Scheidt, Angew. Chem. Int. Ed. 41 (2002) 3188–3191. doi: 10.1002/1521-3773(20020902)41:17<3188::AID-ANIE3188>3.0.CO;2-H
L. Floriani, E. Marianucci, P.E. Todesco, J. Chem. Res. 1984 (1984) 126.
J.A. Chudek, R. Foster, D. Young, J. Chem. Soc., Perkin Trans. 2 (1985) 1285–1289.
R.J. Hooley, T. Iwasawa, J. Rebek Jr., J. Am. Chem. Soc. 129 (2007) 15330–15339. doi: 10.1021/ja0759343
L. Xu, S. Hua, S. Li, Chem. Commun. 49 (2013) 1542–1544. doi: 10.1039/c2cc38165f
J.L. Hou, D. Ajami, J. Rebek Jr., J. Am. Chem. Soc. 130 (2008) 7810–7811. doi: 10.1021/ja802288k
X. Li, S.J. Danishefsky, J. Am. Chem. Soc. 130 (2008) 5446–5448. doi: 10.1021/ja800612r
P. Restorp, J. Rebek Jr., J. Am. Chem. Soc. 130 (2008) 11850–11851. doi: 10.1021/ja803854r
G.B. Huang, S.H. Wang, H. Ke, L.P. Yang, W. Jiang, J. Am. Chem. Soc. 138 (2016) 14550–14553. doi: 10.1021/jacs.6b09472
H. Yao, H. Ke, X. Zhang, et al., J. Am. Chem. Soc. 140 (2018) 13466–13477. doi: 10.1021/jacs.8b09157
Y.L. Ma, M. Quan, X.L. Lin, et al., CCS Chem. 3 (2021) 1078–1092. doi: 10.31635/ccschem.020.202000288
L.P. Yang, X. Wang, H. Yao, W. Jiang, Acc. Chem. Res. 53 (2020) 198–208. doi: 10.1021/acs.accounts.9b00415
W.E. Liu, M. Quan, H. Zhou, et al., Chem. Phys. Chem. 21 (2020) 2249–2253. doi: 10.1002/cphc.202000703
M.S. Li, Y.W. Dong, M. Quan, W. Jiang, Angew. Chem. Int. Ed. 61 (2022) e202208508. doi: 10.1002/anie.202208508

Scheme 1 Stabilization and observation of labile reaction intermediates mediated by supramolecular containers.

Figure 4 Stabilization of bicyclo[2.2.2]oct-1-ene and bicyclo[3.2.1]oct-1-ene inside carcerand B.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:







