Citation:
Jun Zhang, Zhiyao Zheng, Can Zhu. Stereochemical editing: Catalytic racemization of secondary alcohols and amines[J]. Chinese Chemical Letters,
2024, 35(5): 109160.
doi:
10.1016/j.cclet.2023.109160
Stereochemical editing: Catalytic racemization of secondary alcohols and amines
English
Stereochemical editing: Catalytic racemization of secondary alcohols and amines
Department of Chemistry, Fudan University, Shanghai 200438, China
b.
Department of Organic Chemistry, Arrhenius Laboratory, Stockholm University, Stockholm 10691, Sweden
* Corresponding author. E-mail address: zhucan@fudan.edu.cn (C. Zhu). 1 These authors contribute equally to this work.
Received Date:
03 June 2023 Accepted Date:
27 September 2023 Revised Date:
20 September 2023 Available Online:
15 May 2024
Abstract:
Chiral alcohols and amines are important structural units widely existing in pharmaceuticals, agrochemicals, and food additives. Dynamic kinetic resolution (DKR) is an efficient strategy to deliver optically active alcohols and amines from their racemates. For the development of DKR method, racemization catalyst plays as a crucial element with the requirement of compatibility with the kinetic resolution (KR) system. In this paper, recent advance in the catalytic racemization of secondary alcohols and amines is summarized based on different types of racemizing intermediates, which are redox racemization via ketone/imine intermediates, racemization via radical intermediates, and racemization via carbocation intermediates. Enzymatic racemization of secondary alcohols and amines is also enclosed.
Optically active alcohols and amines are important substructural units, which have found wide applications in pharmaceuticals, agrochemicals, and food additives [1–3]. Kinetic resolution (KR) is a practical access to optically pure enantiomers from racemic alcohols or amines. However, the theoretical yield of the product (e.g. (R)-product in Scheme 1) in a KR process is limited to 50%, with 50% recovery of the other enantiomer, (S)-substrate, due to the high interconversion energy barrier (ΔGrac) of each substrate enantiomer to the other, which restricts its real applications (Scheme 1) [4]. In view of the above problems, one elegant solution is to utilize dynamic kinetic resolution (DKR) strategy. In combination with the KR process, DKR still relies on stereochemical editing–efficient racemization, in which one enantiomer of the substrate is continuously isomerized to the other by decreasing TSrac energy barrier (from ΔGrac to ΔG'rac). Therefore, DKR delivers the corresponding enantiopure products from racemic forms of secondary alcohols or amines with a theoretical yield of 100% in a one-step manner [5–7].
Scheme 1
Scheme 1.
Kinetic resolution (KR) and dynamic kinetic resolution (DKR). kR is the rate constant for the reaction of (R)-isomer; kS is the rate constant for the reaction of (S)-isomer; krac is the rate constant for the interconversion of the two substrate enantiomers.
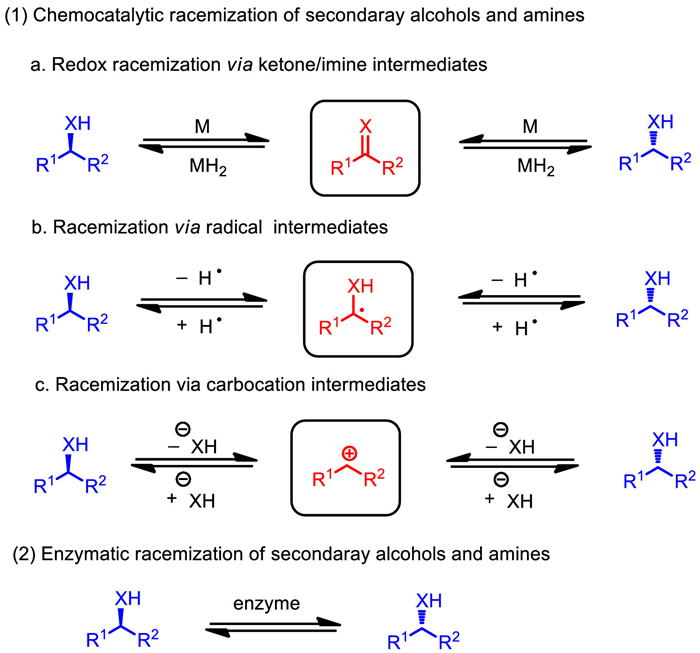
Racemization catalysis plays as a crucial element to ensure the efficient DKR transformations, and there are several requirements: (1) racemization catalysis selectively works on the interconversion of the two enantiomers of the substrate, while inert on the formed chiral product; (2) racemization rate should be competitive, or even faster than the consumption of the reactive enantiomer (k > k > > k); (3) racemization catalysis has to be compatible with the reaction system of KR. Through the summary of relevant literatures, this paper classifies the racemization of secondary alcohols and amines into three chemocatalytic pathways as the first part based on the different types of intermediates, which are redox racemization via ketone/imine intermediates, racemization via radical intermediates, and racemization via carbocation intermediates (Scheme 2). Moreover, enzymatic racemization of secondary alcohols and amines has also been developed as an efficient tool, thus summarized as the second part.
Scheme 2
Scheme 2.
Racemization of secondary alcohols and amines. X = O or NR.
2.
Chemocatalytic racemization of secondary alcohols and amines
2.1
Redox racemization via ketone/imine intermediates
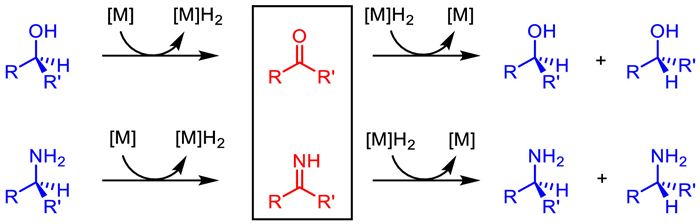
Redox racemization of secondary alcohols and amines under transition-metal catalysis via ketone/imine intermediates is a typical racemization pathway (Scheme 3). In a redox catalytic cycle, as the initial step, a transition metal interacts with a chiral secondary alcohol or amine to deliver ketone/imine intermediate respectively via hydrogen abstraction. Subsequently, the released metal hydride species would reduce the formed ketone/imine intermediate to afford the racemic alcohol or amine, reforming metal catalyst which closes the catalytic cycle. The past few decades have witnessed significant progress in this field. Catalysis based on noble transition metals has been well developed, including rhodium, ruthenium, iridium, and palladium. In recent years, the application of 3d transition metals in the racemization of secondary alcohols and amines, especially iron has drawn increasing attentions, thereby targeting DKR transformations.
Scheme 3
Scheme 3.
Redox racemization of secondary alcohols and amines under transition-metal catalysis via ketone/imine intermediates.
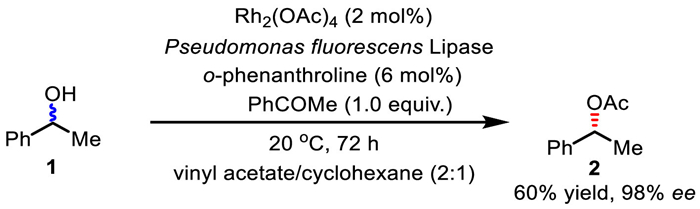
In 1996, Williams and co-workers developed the first chemoenzymatic DKR reaction of secondary alcohols using rhodium acetate as the racemization catalyst (Scheme 4) [8]. With vinyl acetate as the acyl donor, when 1-phenylethanol was employed as the template substrate under dual catalysis of Rh2(OAc)4/o-phenanthroline and PFL (Pseudomonas fluorescens lipase), the reaction delivers single-conformation acetylated product in 60% yield with 98% ee at 20 ℃ in 72 h. Obviously, the enantioselectivity outcome of this reaction is higher than it should be theoretically if proceeding via a simple kinetic resolution (at this conversion, the highest possible enantioselectivity of the ester is 66% ee). The obtained results indicate a simultaneous transformation of each alcohol enantiomer to the other, and Rh2(OAc)4, together with o-phenanthroline are responsible for the interconversion process.
Scheme 4
Scheme 4.
DKR of secondary alcohols under dual catalysis of lipase and rhodium.
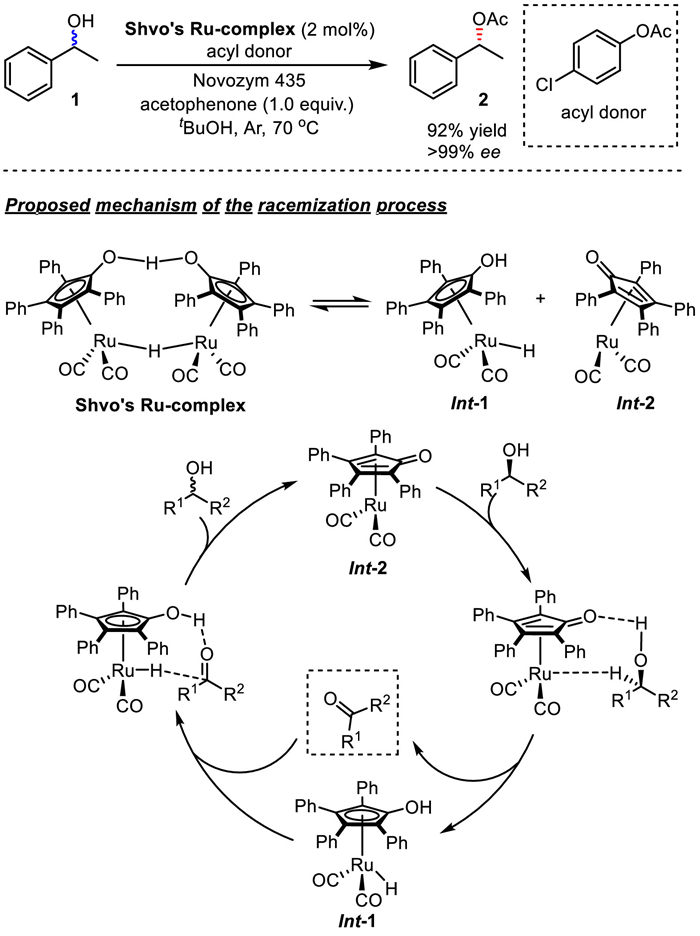
Aiming at the development of DKR of secondary alcohols and amines, homogeneous racemization catalysts based on ruthenium is dominating in this field. Pioneering DKR applications utilizing a homogeneous Ru-based racemization catalyst has been developed by Bäckvall et al. (Scheme 5) [9]. In the reaction system, when Shvo's Ru-complex, {[(η5-Ph4C4CO)]2H}Ru2(CO)4(μ-H) was used as the racemization catalyst, p-chlorophenyl acetate as the acyl donor, the DKR of secondary alcohols was realized with an additive of acetophenone, and chiral product 2 could be obtained from racemic alcohol 1 in 92% yield with > 99% ee. In the catalytic cycle, initially Shvo's Ru-complex could be easily to split into two monomeric species (Int and Int) on heating in solution. The racemization mechanism consists of a combination of outer-sphere dehydrogenation of alcohol substrate by Int, and re-addition of hydrogen to the ketone by similar pathway with Int. Later, the catalytic system developed by the same group and others were also studied to be effective for the DKR of various aliphatic alcohols as well as benzylic alcohols [10–22]. It is worth noting that Bäckvall's group successfully synthesized anti-hypertensive drug propanolol by dynamic kinetic resolution of the racemic azido alcohol in 2001 [19].
Scheme 5
Scheme 5.
Shvo's Ru-complex as the racemization catalyst to realize the DKR of secondary alcohols.
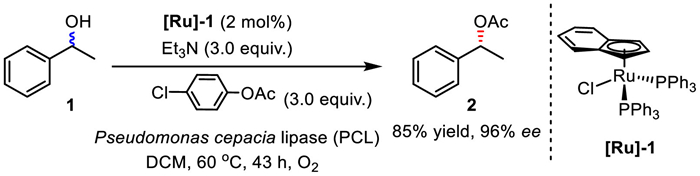
In 1999, Park and co-workers presented a new catalyst, (η5-indenyl)RuCl(PPh3)2 for the racemization of secondary alcohols in the presence of triethylamine and oxygen (Scheme 6) [23]. Unlike previously reported protocols by Bäckvall et al. [9], ketones were not required as hydrogen mediators in this process. This Ru-catalyzed racemization was coupled with enzymatic acetylation for the DKR of secondary alcohols. Notably, stoichiometric amounts of an organic base (Et3N) are necessary in this dual catalytic system.
Scheme 6
Scheme 6.
(η5-Indenyl)Ru complex as the racemization catalyst for the DKR of secondary alcohols.
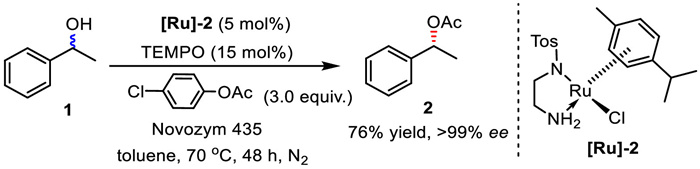
In 2002, Sheldon and co-workers developed a new catalytic system of [TosN(CH2)2NH2]RuCl(p-cymene) ([Ru]−2) and TEMPO to complete the racemization of chiral secondary alcohols (Scheme 7) [24]. Instead of a base, TEMPO was used as the co-catalyst in combination with [Ru]−2, and the racemization system is also compatible with enzyme-catalyzed KR reactions. Finally, enantiomerically pure 1-phenylethyl acetate was obtained in 76% yield with > 99% ee. The alcohol substrate was simultaneously consumed via a side reaction, the oxidation of alcohol by TEMPO to deliver acetophenone as observed during the reaction.
Scheme 7
Scheme 7.
(η6-p-cymene)Ru complex/TEMPO as cooperative racemization catalysts.
Independently, Kim, Park, and co-workers reported a novel ruthenium catalyst (aminocyclopentadienyl ruthenium chloride complex) ([Ru]−3) that can racemize secondary alcohols efficiently at room temperature without the aid of hydrogen mediators (Scheme 8) [25,26]. Furthermore, this racemization conditions are compatible with an enzymatic KR process to develop the DKR transformations.
Scheme 8
Scheme 8.η5-Ru complex ([Ru]−3) as the racemization catalyst for the enzymatic DKR of secondary alcohols.
In 2005, the Bäckvall group achieved a breakthrough in the development of novel and efficient racemization catalysis. When Ru-complex (η5-Ph5C5)Ru(CO)2Cl ([Ru]−4) was activated by potassium t-butoxide, the in-situ generated Int can racemize secondary alcohols efficiently via redox pathway at room temperature (Scheme 9) [27]. The racemization system could be extended in cooperation with lipase for the DKR development, which is investigated to be compatible with a wide range of secondary alcohols. Mechanism studies showed that ligand exchange of Int with a secondary alcohol leading to Int, which underwent β-elimination from an alkoxide complex to produce a hydride ketone complex Int. The reversible migratory insertion subsequently occurs in Int to deliver the racemic alcohol coordinated complex Int. It has to be emphasized that the ketone stays coordinated before the migratory insertion occurs, and does not leave the coordination sphere, supporting inner sphere mechanism [28]. In this way, the formation of ketone, a common side-product produced during redox-racemization, was kept at a low level, thus chiral product could be isolated in high yield. In addition, [Ru]−4 and lipase-cocatalyzed DKR of 1-(6-chloropyridin-3-yl)ethanol, β–chloro alcohol and β-hydroxynitrile as the key step in the synthesis of chiral Me-imidacloprid (a chloronicotinyl insecticide), (R)-bufuralol (a non-selective β-adrenoceptor blocking agent) and duloxetine (an antidepressant drug), respectively [29–31].
Scheme 9
Scheme 9.[Ru]−4 as the racemization catalyst for the DKR of secondary alcohols.
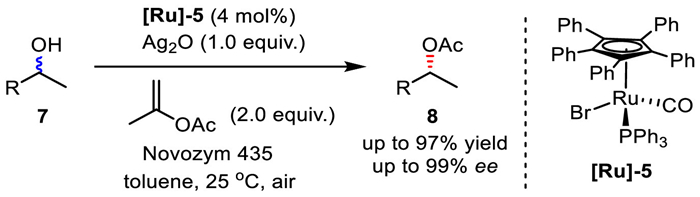
In 2007, Kim and Park's group further modified Bäckvall's ruthenium complex catalyst [Ru]−4 by replacing CO with PPh3, and this new catalyst [Ru]−5 was successfully applied in the DKR of secondary alcohols (Scheme 10) [27,32]. In the racemization operations, the use of strong base is not required, which can improve the compatibility with functional groups, especially those are sensitive under basic conditions. Instead, silver oxide was employed as a weak base to transiently deprotonate the alcohol substrate, thus the analogous alkoxide complex Int would be formed from [Ru]−5via ligand exchange. The following inner sphere mechanism is the same as shown in Scheme 9. Finally, the racemization system was found to be nicely compatible with lipase catalysis, and the DKR of secondary alcohols was consequently realized in ambient atmosphere with isopropenyl acetate as the acylating agent.
In 2012, differing from previous enzymatic DKR processes, Fu et al. developed an unprecedented non-enzymatic DKR of secondary alcohols (Scheme 11) [33]. By using Bäckvall's ruthenium complex [Ru]−4 as the racemization catalyst, Fu's group realized the synthesis of various chiral secondary alcohols via enantioselective acylation with a planar-chiral catalyst of DMAP derivative [(C5Ph5)-DMAP*]. It is noteworthy to mention that this non-enzymatic DKR dramatically extended the substrate scope of secondary alcohols. Besides a methyl group, relatively larger R groups including ethyl, i-Pr, and cyclopentyl can also be tolerated, leading to the corresponding esters in excellent yields with good enantioselectivity. Moreover, the absolute configuration of the produced chiral esters is believed to be accordingly reversed by using an enantiomeric planar-chiral catalyst, which breaks limitation of enzyme catalysis to be able to deliver only one enantiomer. Mechanistic studies indicate that acyl transfer from the catalyst to the alcohol is the rate-determining step of the DKR, and carbonate anion serves as the Brønsted base during the acyl transfer.
Scheme 11
Scheme 11.
Non-enzymatic DKR of secondary alcohols by cooperatively using Bäckvall's ruthenium complex and the planar-chiral catalyst.
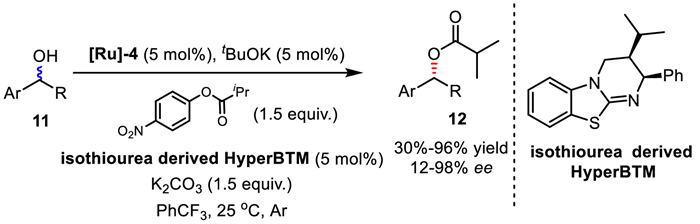
Very recently in 2021, Piotrowski, Suna, and co-workers employed Bäckvall's ruthenium catalyst [Ru]−4 to develop a new non-enzymatic DKR approach of secondary alcohols by using isothiourea-derived HyperBTM as the enantioselective acylation catalyst (Scheme 12) [34]. Compared to Fu's protocols, this method used easy-to-handle crystalline ester as the acylating agent, and DKR takes place at room temperature under an ambient atmosphere. However, only moderate enantioselectivity was obtained in many cases.
Scheme 12
Scheme 12.
Non-enzymatic DKR of secondary alcohols by combining Bäckvall's ruthenium complex with isothiourea-derived HyperBTM catalyst.
In 2006, Zhang and co-workers synthesized a η5-coordination ruthenium complex, (η5-triphenylindenyl)Ru(CO)2Cl ([Ru]−6), performing as an efficient racemization catalyst towards chiral secondary alcohols (Scheme 13). Compared with Bäckvall's ruthenium complex [Ru]−4, a fused benzene ring was introduced to replace two phenyl substituents on the η5-Cp-moiety. This new Ru-complex was easily accessible from 2, 3-diphenylindenone in two steps, and racemization of secondary alcohols could be completed at room temperature in 30 min [35]. Compared with the system using [Ru]−4, a stronger base such as sodium hydride or n-butyllithium was necessary to activate the catalyst to achieve much faster racemization rate than that with KOtBu.
Scheme 13
Scheme 13.
(η5-triphenylindenyl)Ru(CO)2Cl [Ru]−6 as the racemization catalyst.
In Kim and Park's previous work, aminocyclopentadienyl ruthenium chloride complex [Ru]−3 was successfully used in the DKR of secondary alcohols. However, because of its air sensitivity during the transformations, the reaction has to be operated under an inert atmosphere. The development of an air-stable racemization catalyst becomes highly desirable. In 2005, Kim and Park's group synthesized a novel ruthenium complex [Ru]−7 by replacing the amino-substituent in [Ru]−3 with an alkoxyl group. These air-stable ruthenium catalysts could be efficiently employed in the DKR transformations together with Novozym 435 at room temperature in the air (Scheme 14) [36]. Meanwhile, by linking the ruthenium complex to a polymer support, [Ru]−7′ can still maintain its catalytic activity, and was applicable to the DKR of alcohols as a recyclable catalyst.
After that, Kim and Park's group continuously studied the substituent effect on the catalytic activity of [Ru]−7 derivatives in alcohol racemization (Scheme 15) [37]. The authors synthesized a series of derivatives of [{η5-Ar4C4COC(=O)Ar}Ru(CO)2Cl] to investigate the electronic effects of substituents, and concluded that Ru-complexes with an electron-donating substituent exhibited higher activity than those of relative electron-deficiency, e.g., the catalytic performance of [{η5-Ar4C4COC(=O)Ar}Ru(CO)2Cl] (Ar = 4-methoxyphenyl) in the racemization of 1-phenylethanol is much better than that of [{η5-Ph4C4CO(C = O)pH}Ru(CO)2Cl].
Scheme 15
Scheme 15.
Synthesis and reactivity-investigation of [Ru]−7 derivatives in alcohol racemization.
Early in 2000, Kim, Park, and co-workers applied commercially available (p-cymene)-ruthenium(Ⅱ) complex to the DKR of allylic alcohols [38]. This is the first example of non-Cp-coordinated ruthenium complex as the racemization catalyst. The reaction can take place at room temperature and is compatible with a variety of allylic alcohols. Later, the group successfully extended the application scope to general secondary alcohols by using ionic liquid, [BMIM]PF6 ([BMIM] = 1–butyl–3-methylimidazolium) to enhance the racemization activity (Scheme 16) [39].
Scheme 16
Scheme 16.
Employment of non-Cp-coordinated (p-cymene)-ruthenium(Ⅱ) complex.
In 2011, Riant, Aribi-Zouioueche, and co-workers also reported the DKR of various secondary benzylic and aliphatic alcohols by using the same ruthenium(Ⅱ) complex cooperatively with TEMPO, and CALB as the enantioselective acylation catalyst (Scheme 17) [40]. Moreover, they found that the addition of a suitable salenol ligand (L) can significantly improve the racemization efficiency of the system.
Scheme 17
Scheme 17.
Catalytic racemization using (p-cymene)-ruthenium(Ⅱ) complex together with TEMPO and salenol ligand.
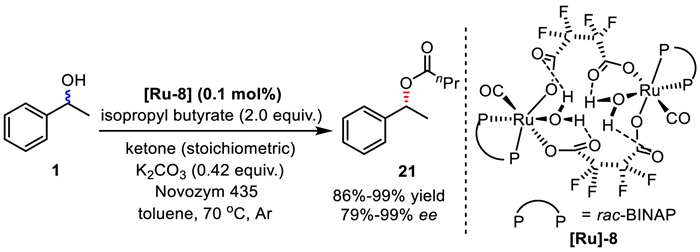
In 2006, Hulshof's group reported a DKR method to access secondary alcohols, where a di-nuclear ruthenium complex was responsible for alcohol racemization, simultaneously bearing tetrafluorosuccinate and rac-BINAP ligands (Scheme 18) [41]. Under dual catalysis of [Ru]−8 (only 0.1 mol%) and Novozym 435, the DKR reaction efficiently produced target product in good yield and excellent ee with isopropyl butyrate as the acyl donor. K2CO3 was the base to activate the Ru catalyst. In addition, the reaction can run smoothly in the absence of ketones as well.
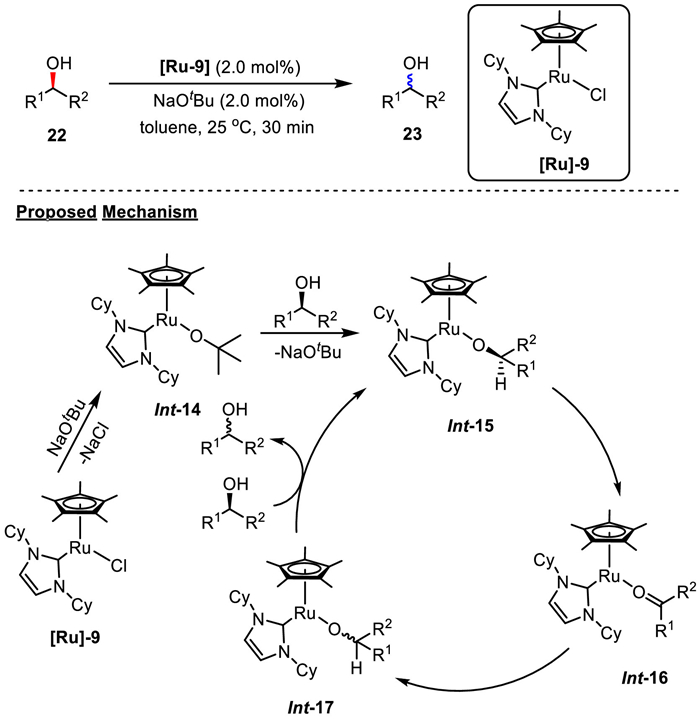
In 2010, a well-defined 16-electron ruthenium complex [Ru]−9 bearing an NHC ligand was reported by Nolan's group, which is applicable to secondary benzylic and aliphatic alcohols (Scheme 19) [42]. Inspired by Shvo's catalyst (Scheme 5), the authors supposed simple well-defined complex adopting the 16-electron configuration might exhibit catalytic activity on alcohol racemization. By using ruthenium catalyst bearing a NHC ligand of N, N'-dicyclohexylimidazol-2-ylidene, alcohol racemization was completed within 30 min at room temperature. The studies showed that steric hindrance of NHC ligand is the key element to ensure the alcohol racemization, which prevented additional coordination of an external ligand to ruthenium, forming a stable 18-electron metal center. The detailed mechanism is the similar as shown in Scheme 9.
Scheme 19
Scheme 19.
Ru-NHC complex [Ru]−9 as the racemization catalyst.
Later in 2011, the same group further developed two well-defined 16-electron ruthenium hydroxide complexes [Ru]−10A and [Ru]−10B, which also bear bulky NHC ligands respectively (Scheme 20) [43]. The difference is the anion of hydroxide in each catalyst, replacing chloride in [Ru]−9. In this way, the use of an external base can be avoided, e.g. KOtBu to activate the inert Ru-hydroxide complex to initiate the racemization process. Both Ru-NHC complexes can be prepared simply by stirring [Cp*Ru(NHC)Cl] with dried CsOH or stirring the precursor [Cp*RuCl]4 with corresponding NHC and CsOH in one-pot. In contrast, [Ru]−10A showed slightly higher efficiency in alcohol racemization, and could be successfully applied to the DKR reactions.
Scheme 20
Scheme 20.
Synthesis and reactivity investigation of Ru-NHC hydroxide complexes in alcohol racemization.
In 2017, by introducing different types of ligands, Joó and co-workers synthetized several other Ru-NHC complexes, i.e. [RuCl2(NHC)(η6-arene)], and Ru-complexes bearing three types of ligands [RuCl(NHC)(η6-arene)(PR3)] [NHC = bmim or emim; arene = benzene or p-cymene; phosphine ligand = PPh3 or 1, 3, 5-triaza-7-phosphaadamantane (pta)] (Scheme 21). These Ru-NHC complexes can also serve as racemization catalysts in the DKR of secondary alcohols [44].
Scheme 21
Scheme 21.
Synthesis of (η6-arene)Ru-complexes bearing different types of ligands.
Aiming to the DKR of amines, in 2002 the Bäckvall group employed Shvo's Ru-complex as the racemization catalyst to the reaction of chiral phenylethanamine (Scheme 22) [45]. By adding 2, 4-dimethyl-3-pentanol as hydrogen donor, the formation of by-products was successfully inhibited. In addition, this catalytic system avoids the use of a strong base and reducing agent so that it has nice tolerance of functional groups.
Scheme 22
Scheme 22.
Shvo's Ru-complex as the racemization catalyst for amines.
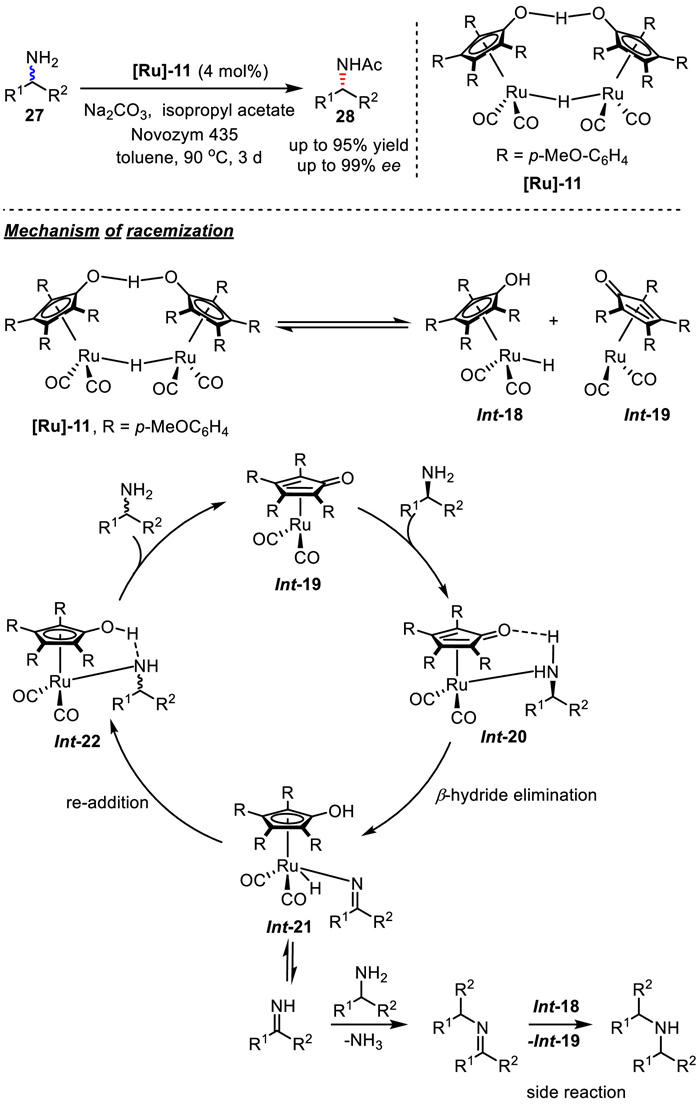
In 2005, the same group modified Shvo's Ru-complex, by replacing all phenyl groups with p-methoxyphenyl substituents on cyclopentadienyl-ligands, the step of re-addition of hydrogen is accelerated, thus reducing the formation of imine as the main side-product (Scheme 23). Finally, by cooperatively using [Ru]−11 and lipase catalysis, target chiral amines were successfully obtained in high yield and enantioselectivity in the presence of Na2CO3 in toluene at 90 ℃ [46]. Later, they studied kinetic isotope effect on the competitive racemization of a 1:1 mixture of deuterated and non-deuterated amine, and the reaction outcomes supported that β-hydride elimination was involved in the rate-determining step. The chemoenzymatic DKR protocol has been applied to the synthesis of norsertraline, an antidepressant of the selective serotonin reuptake inhibitor (SSRI) [47].
Scheme 23
Scheme 23.
Modified [Ru]−11 as the racemization catalyst for the DKR of amines.
In 2019, De Vos and co-workers developed a heterogeneous ruthenium racemization catalyst for the DKR of aliphatic amines [48]. The heterogeneous Ru-catalyst was immobilized on zeolite, and showed well activity on the racemization of amines at 70 ℃ in aprotic polar solvents, but has to be charged with pressurized hydrogen (10 bars). Consequently, a combination of racemization and KR system, DKR delivers aliphatic amines in good yield and enantioselectivity.
2.1.3
Iron catalysts
With the cooperative catalysis of racemization and KR process, DKR have emerged as an effective tool, featuring prominent advantages for the synthesis of chiral secondary alcohols and amines. However, the high cost of precious metals would definitely limit their real applications. Therefore, the use of earth-abundant and cheap metals as racemization catalysts in DKR transformations has attracted considerable attentions. In 2016, Bäckvall's group reported the first example of iron-catalyzed racemization of secondary benzylic alcohols (Scheme 24) [49]. This protocol proceeds under mild reaction conditions, and completely racemic alcohols could be obtained at 50 ℃ within 30 min under [Fe]−1 (2.5 mol%), exhibiting the high efficiency of this octahedral Fe-complex toward alcohol racemization. However, the DKR of secondary alcohols was not realized with [Fe]−1 in this report.
Scheme 24
Scheme 24.
Octahedral complex [Fe]−1 for alcohol racemization.
In the same year, Rueping's group developed the first protocol for the DKR of secondary alcohols by cooperative iron/lipase catalysis (Scheme 25) [50]. Various racemic alcohols can be transformed to the enantioenriched acetates under this dual catalysis, avoiding the use of noble metals. In this reaction, Knölker-type iron complex [Fe]−2 was employed, and a detailed study of the iron complex demonstrated that the iron-catalyst promoted the hydrogen auto-transfer of alcohols under mild reaction conditions, which allows the combination with enzymatic resolution to realize DKR. Choosing p-chlorophenyl acetate as acetylating reagent could reduce the production of the side-product, acetophenone.
Scheme 25
Scheme 25.
Knölker-type iron complex [Fe]−2 for the DKR of alcohols in cooperation with lipase.
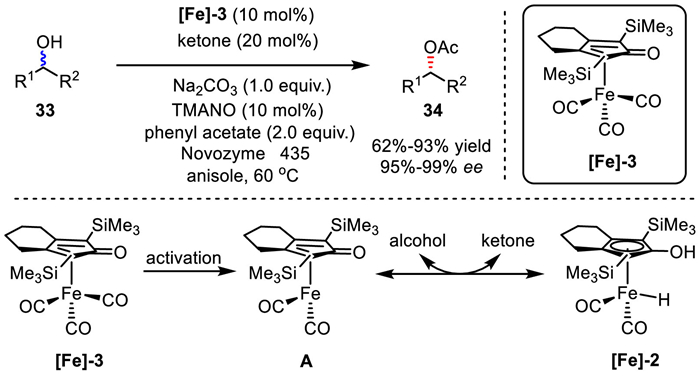
Shortly afterwards in 2017, the Bäckvall group independently reported a metalloenzymatic DKR of secondary alcohols under dual catalysis of iron and lipase (Scheme 26) [51]. The stable iron complex [Fe]−3 is the precursor of active catalytic species A, which was in situ generated via activation with TMANO (trimethylamine N-oxide) from [Fe]−3. Then, species A dehydrogenates the alcohol to deliver the corresponding ketone, and [Fe]−2 hydrogenates ketone back to produce racemic alcohol. Among them, Na2CO3 and catalytic amounts of ketone play important roles in facilitating the re-addition of hydrogen to ketone. This procedure allows various enantiomerically pure benzylic and aliphatic acetates to be prepared directly from racemic alcohols in good yields and with excellent ee.
Scheme 26
Scheme 26.[Fe]−3 as the precursor for alcohol racemization.
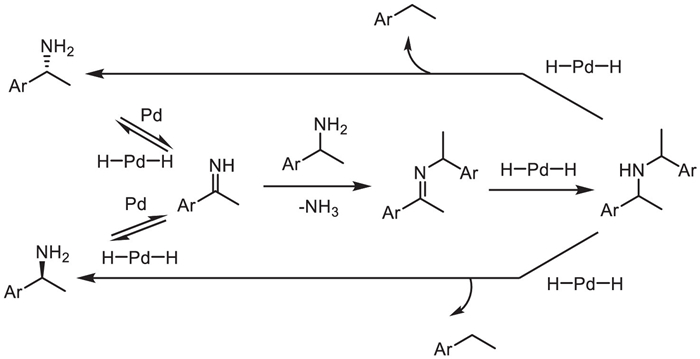
Given the fact that many palladium complexes could catalyze elimination of hydrogen from amine and hydrogenation of imines, palladium is considered as a potential catalyst for amines racemization. Palladium-catalyzed racemization of amines was firstly reported as early as 1996 [52], under dual palladium on charcoal and CALB (Novozym 435) catalysis, the racemization of phenylethylamine compounds was realized by Schimossek's group (Scheme 27). But because of the formation of side-products such as ethylbenzene and bis(1-phenylethyl)amine during racemization, the reaction is low-yielding over long-time running.
Scheme 27
Scheme 27.
Racemization pathway of amine with Pd Catalyst and the possible side reactions.
In the following decade, Jacobs, Park, Kim and co-workers continued to improve the catalytic system. Using heterogeneous catalysts [Pd/BaSO4, metallic palladium nanoparticles (NPs), Pd/AlO(OH)] can inhibit the occurrence of side reaction of condensation between amine and imine intermediates, and improve the catalytic efficiency of palladium [53–59]. In 2013, the Bäckvall group develop efficient multifunctional hybrid catalysts that nanopalladium and enzyme were simultaneously immobilized into the same cavities of the mesocellular foam to achieve the DKR of amines [60].
Metallic palladium nanoparticles (NPs) could efficiently racemize chiral amines, however, their applications in DKR of secondary alcohols was rarely reported. In 2017, Li's group applied a heterogenous catalyst, metallic palladium nanoparticles Pd@SBA-15 into the DKR of secondary alcohols, which bears an aryl substituent (Scheme 28) [61]. Under the cooperative catalysis of Pd@SBA-15 and lipase CALB, one-pot DKR of secondary alcohols was achieved with the assistant of microwave. However, it is noteworthy to mention that the DKR reaction has to be conducted under an atmosphere of 5% H2 to prevent the dehydrogenation of alcohol substrate, forming ketone side-product.
Scheme 28
Scheme 28.
DKR of secondary alcohols with metallic palladium nanoparticles (NPs) as the racemization catalyst.
In 2007, a simple, efficient, iridium-based catalyst system for the racemization of a variety of amines, including tetrahydroisoquinoline (THIQ) was developed by Page's group (Scheme 29) [62,63]. In the reaction, Cp*Ir(Ⅲ) iodide dimer (Cp* = pentamethylcyclopentadienyl) performs as a pre-catalyst, which reacts in situ with amines to form active species for amine racemization, the SCRAM catalyst. This pre-catalyst was air- and water stable, and could be compatible with enzymes in mild conditions.
Scheme 29
Scheme 29.
Pentamethylcyclopentadienyliridium(Ⅲ) iodide dimer for the DKR of amine derivatives.
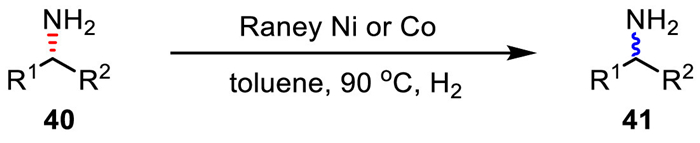
Besides novel transition metals, dynamic racemization catalyzed by non-noble 3d transition metals has attracted even higher attentions. However, amine racemization by 3d transition metals was much less investigated. In 2008, De Vos et al. found that Raney Ni performed high efficiency as a heterogeneous catalyst in the racemization of optically active aliphatic amines (Scheme 30) [64]. Instead, hydrogen pressure was shown as the critical parameter in controlling the chemoselectivity during racemization. Moreover, they also realized amine racemization under the catalysis of Raney Co.
Scheme 30
Scheme 30.
Raney Ni/Co for amine racemization in a hydrogen atmosphere.
Just like palladium, the activity of nickel can be enhanced by working with NPs in racemization reactions. In 2013, the same group continuously utilized a catalyst of nickel nanoparticles (NiNPs) for the racemization of either aliphatic or benzylic primary amines (Scheme 31) [65]. The employed nickel nanoparticles were prepared by reducing nickel(Ⅱ) bromide with sodium hydride in the presence of lithium t-butoxide in toluene, and were stable in the ionic liquid tetrabutylammonium bromide. The catalytic system can complete most racemization processes within a few hours with excellent selectivity.
Scheme 31
Scheme 31.
Raney NiNPs as the racemization catalyst.
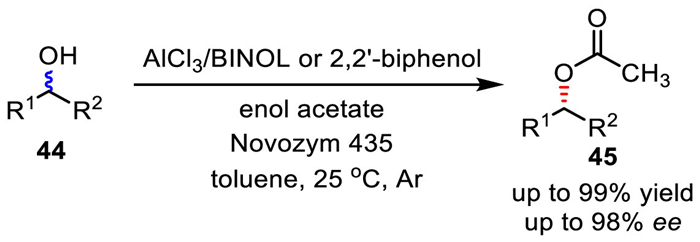
In 2006, Berkessel et al. demonstrated that chemoenzymatic DKR of secondary alcohols is possible in high yields and enantioselectivity through the use of an inexpensive and readily available aluminum catalyst in combination with lipase catalysis (Scheme 32) [66]. Racemization of secondary alcohols by aluminum catalyst proceeds via the Meerwein−Ponndorf−Verley−Oppenauer (MPVO) transformations between the alcohol and the corresponding ketone. The bisphenol-type ligands play a dual role in the DKR. Firstly, they improve the activity of aluminum catalyst by hindering their polymerization. On the other hand, bisphenol aluminum complexes retain alcohol racemic activity in the presence of lipase. In the report, ketone was generated in situ after the acyl group being transferred from enol acetate by lipase. However, enol acetate has to be accordingly used based on the alcohol substrate.
Scheme 32
Scheme 32.
Racemization via the Meerwein-Ponndorf-Verley-Oppenauer (MPVO) reaction mechanism.
Radical mediated reversible hydrogen atom abstraction/transfer (HAA/HAT) from the stereogenic center of compounds is another method of racemization. The transient formation of active radical intermediate Int by a polarity-matched HAT mediator is the key element to promote such alcohol/amine racemization (Scheme 33). In the past few decades, radical-mediated racemization of chiral aromatic amines and aliphatic amines were rapidly developed, especially those based on a thiyl radical. Recently, the combination of radical mediated racemization and enzymatic resolution has become the focus of chemists.
Scheme 33
Scheme 33.
Racemization via active radical intermediate, X = O or NH.
In 2006, Bertrand et al. presented a thiyl-radical-mediated racemization protocol for benzylic amines (Scheme 34) [67]. The authors concluded two key factors to achieve thiyl-radical-mediated racemization of amines: (1) abstractor and substrate must be polarity-matched to each other. The electrophilic thiyl radical and strongly nucleophilic carbon-centered radical of α-position of amine, which were two radical species of opposite polarity, would accelerate the hydrogen atom transfers; (2) bond dissociation energies (BDEs) of the thiol S–H bond and the C–H bond of the substrate, has to match with each other. In the racemization system of benzylic amines, the stoichiometric amounts of thiol were added as racemization mediator, with 2, 2′-azobis(2-methylpropionitrile (AIBN) as radical initiator. Among them, α-C–H BDE is calculated to be 340–360 kJ/mol for benzylic amines, while the S-H BDE of p-methylthiophenol is 340–350 kJ/mol, indicating the matched BDEs of the involved bond during racemization. It is noteworthy to mention that competitive oxidation of the intermediate α-amino radical makes the method more efficient for secondary and tertiary amines compared to primary ones.
Scheme 34
Scheme 34.
Thiyl-radical-mediated racemization of amines.
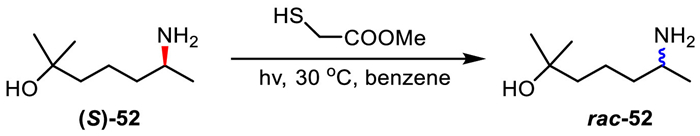
Later, Bertrand's group extended this protocol to non-activated aliphatic amines, and further realized thiyl-radical-mediated racemization of more challenging amines in the presence of AIBN (Scheme 35a) [68]. Because of higher BDEs of α-C-H bonds in aliphatic amines than those in benzylic amines, thiols having stronger S-H bonds were chosen for aliphatic amines. Guided by the calculated α-C−H BDEs and S-H BDE, the racemization of non-activated aliphatic amines was successfully achieved with alkanethiols and methyl thioglycolate (The S-H BDEs of alkanethiols and methyl thioglycolate are 360–370 kJ/mol). Authors discovered that the knowledge of the reaction enthalpy is critical to select the appropriate thiol.
Scheme 35
Scheme 35.
Thiyl-radical-mediated DKR of aliphatic amines in cooperation with Novozym 435.
In a previous study, Bertrand found the α-C−H BDE of amine is stronger after acylation (approximately increased by 17 kJ/mol) [68], implying that the absolute configuration of the chiral acylated product can be maintained in the presence of a suitable thiyl-radical mediator. Based on it, later in 2007, they successfully developed the DKR of aliphatic amines by combining thiyl-radical-mediated racemization with lipase-catalyzed enzymatic resolution (Scheme 35b) [69]. In this reaction, thiol 3 was used as racemization catalyst, and Novozym 435 was used as co-catalyst to react at 80 ℃ for 2 h, delivering optically pure amides with high enantioselectivities.
Bertrand's group continued to extend the application scope of the radical-mediated racemization of amines. In 2008, Bertrand et al. employed UV irradiation to trigger the formation of active thiyl-radical species, thus racemizing non-activated aliphatic amines under mild reaction conditions (Scheme 36). The process could realize efficient racemization of aliphatic amines at 30 ℃ [70]. The racemization efficiency depends on the complex interplay of several factors such as α-C-H BDE, amine ionization potential, amine basicity, and thiol acidity. The relatively low-temperature range creates conditions for subsequent research on the DKR of aliphatic amines cooperatively with thermolabile enzymes.
Scheme 36
Scheme 36.
UV-irradiation-assisted radical-mediated racemization of aliphatic amines.
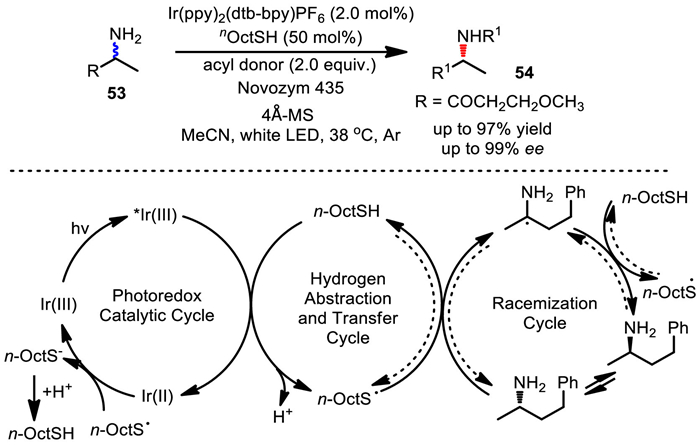
Based on the concept of racemization via radical intermediates, in 2018, Zhou's group developed a photoenzymatic DKR of amines by coupled visible-light photoredox and enzyme catalysis (Scheme 37) [71]. The iridium-photocatalyst was irradiated by a white LED lamp (32 W) to produce a photoexcited complex, which was reduced by nOctSH to generate the active thiyl radical, triggering the HAT process and consequently racemizing amine substrates. The DKR protocol was successfully achieved under mild conditions (38 ℃), thus allows a variety of primary amines to be converted into chiral products in high yield and with excellent enantioselectivity. Impressively, this method can also be extended to deliver chiral 1, 4-diamine derivatives with high diastereo- and enantioselectivities.
Scheme 37
Scheme 37.
DKR of amines by visible-light photoredox and enzyme catalysis.
In 2023, Collins' group reported a remarkable chemoenzymatic DKR of secondary alcohols and amines proceeding a racemization pathway via radical intermediates. They employed a heteroleptic copper complex and a commercially available lipase (Candida antarctica lipase B) (Scheme 38) [72]. Racemization secondary alcohols and amines were achieved by combining copper-based photocatalysis with thiyl-radical-promoted hydrogen atom transfer process. The radical-mediated racemization is compatible with the enzyme-catalyzed KR system, and optically secondary alcohols and amines were obtained in 60%−97% yield with 76%−99% ee. It is noteworthy to mention that this is the first DKR example of secondary alcohols achieved through a radical-racemization pathway.
Scheme 38
Scheme 38.
DKR of alcohols enabled by photoredox and enzyme catalysis.
Chiral secondary alcohols could be racemized under the catalysis of acid through a sequential and reversible protonation and water loss to form a transient sp2-carbenium ion (Scheme 39). In this way, a Lewis acid or Brønsted acid can promote the racemization of secondary alcohols in principle. However, strong acids are generally incompatible in DKR process due to the general formation of corresponding alkenes as the main side-product. Akai and Jacobs developed the DKR of secondary alcohols by using oxovanadium catalyst, a Lewis acid. Ruggeri and House independently employed heterogeneous acids AmberlystI and DeloxanI acid resins as the catalyst to realize racemization of benzylic alcohols [73,74]. These methods greatly enriched the acid-catalyzed racemization of secondary alcohols.
Scheme 39
Scheme 39.
Racemization of secondary alcohols by forming a transient sp2-carbenium ion; LA = Lewis acid.
In 2006, Akai and co-workers developed a novel DKR process of allylic alcohols by using oxovanadium compound [VO(OSiPh3)3] as the racemization catalyst (Scheme 40) [75]. The reaction could take place under mild conditions to afford chiral products with excellent enantiomer resolution and chemical yields. The racemization process enabled by [VO(OSiPh3)3] proceeds via a transient π-allyl carbocation. In this way, dynamic equilibrium was set up between different isomers of allylic alcohol, and the reaction undergoes chemo- and enantioselective esterification by lipase with the unique selectivity of less-hindered side. Therefore, [VO(OSiPh3)3] does not only perform as the racemization catalyst of allylic alcohols, but also accounts for 1, 3-transposition of the hydroxy group during the overall transformations. Later, the same group used polymer-bound vanadyl phosphate and mesoporous-silica-immobilized oxovanadium (V-MPS) as heterogenous racemization catalyst, and successively achieved efficient DKR extension to other types of secondary alcohols, and asymmetric synthesis of (R)-imperanene [76,77].
Scheme 40
Scheme 40.
Racemization of allyl alcohols under the catalysis of oxovanadium.
In 2007, the Jacobs group further reported the racemization of benzylic alcohols with inorganic oxovanadium compounds, vanadyl sulfate (VOSO4) or vanadium oxide (V2O4) (Scheme 41) [78]. This V-catalyzed racemization was also compatible to combine with a lipase-catalyzed KR, leading to a chemoenzymatic DKR of benzylic alcohols. The DKR was operated in a one-pot manner, and does not need any additives or specifically designed acyl donors. Under optimized conditions, the DKR process delivered the corresponding esters in good yield with high optical purity. Unfortunately, the system failed to be applicable to general aliphatic alcohols (R1 = alkyl group).
Scheme 41
Scheme 41.
Vanadyl sulfate as the racemization catalyst.
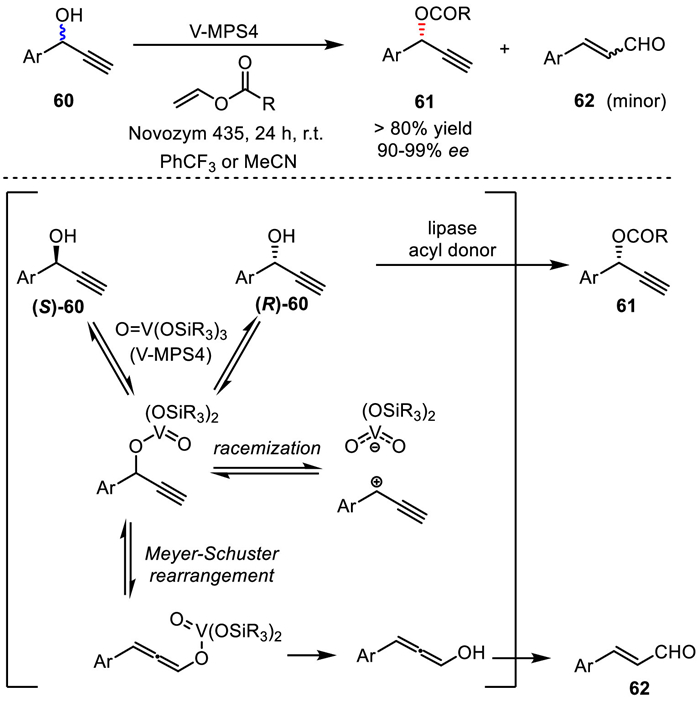
In 2019, Akai and co-workers continuously applied the developed lipase/oxovanadium co-catalytic system in the DKR of propargylic alcohols (Scheme 42) [79]. In this report, V-MPS4, with oxovanadium moieties being covalently bound to the inner surface of mesoporous silica, not only catalyzes the racemization of propargylic alcohols, but also can promote a side reaction to form the corresponding conjugate enals 62 through Meyer−Schuster rearrangement of propargylic alcohols. By screening the reaction conditions, the authors found that a magic solvent (trifluoromethylbenzene) could suppress the rearrangement of propargyl alcohols while effectively accelerating the racemization. Finally, the DKR process could produce the corresponding optically active esters in good yields with excellent ee.
Scheme 42
Scheme 42.
Lipase/oxovanadium co-catalyzed DKR of propargylic alcohols.
In 2003, Jacobs and co-workers firstly realized biphasic DKR of a series of benzylic alcohols by using heterogeneous acid zeolites (Scheme 43) [80,81]. Biphasic DKR of racemic 1-phenylethanol could be achieved under dual catalysis of H-β zeolite and Novozym 435 at 60 ℃ in two phases. The overall DKR transformations consist of H-β zeolite-catalyzed racemization in water layer, and the lipase-catalyzed KR in organic layer. Unfortunately, the protocol is incompatible with electron-rich benzylic alcohols substrates, because of the formation of stable ether in the zeolite pores. Later in 2014, Bäckvall, Deska, and co-worker extended the biphasic strategy to the DKR of allylic alcohols under cooperative catalysis of Novozym 435 and Dowex 50Wx4 [82].
Scheme 43
Scheme 43.
Biphasic DKR of secondary alcohols with acid zeolites.
In 2007, Jaenicke's group developed the DKR of secondary alcohols in a single nonaqueous liquid phase by combining zeolite-catalyzed racemization and enzyme-catalyzed transesterification (Scheme 44) [83]. The authors achieved DKR transformations in a single-phase by doping β zeolites at a low level with other metals (e.g. Sn, Zr, Al, Ti). Among them, hydrophobic Zr-β and Al-β were investigated to give the best performances. During racemization, styrene was formed as the major side product. Along with the racemization of alcohol materials, zeolite catalyst also caused a loss of the enantiomeric purity by racemizing the product esters. However, this could be significantly suppressed by using larger acyl-transfer reagents, such as vinyl octanoate. The resulting bulky ester is not allowed to enter the active sites, avoiding subsequent racemization in the pore channels of zeolite catalyst. In this way, (R)-esters were produced in good yield with excellent enantioselectivity.
Scheme 44
Scheme 44.
DKR of benzylic alcohols using Zr-β or Al-β zeolites.
In 2010, the group of Yang realized highly efficient DKR of secondary benzylic alcohols with acid resin, the racemization catalyst (Scheme 45A) [84]. In this way, chiral ester was efficiently afforded under dual catalysis of acid resins (CD8604) and Novozym 435. Notably, styrene was not detected as the side-product in this catalytic system, which could be generally formed during alcohol racemization via a carbocation intermediate. Later in 2012, Zhang's group realized a similar DKR by using H-β nanozeolite microsphere (β-ZMS) to racemize alcohol substrate (Scheme 45B) [85]. Compared to commercially available zeolite β (C-β), β-ZMS was equipped with more externally accessible acid sites and short microporous channel, which endows the catalyst with higher selectivity for small substrates by reducing diffusion time of substrate/product in the three-dimensional framework. Meanwhile, the authors also found that β-ZMSs showed higher racemization efficiency for the large sec–alcohol molecules, such as racemic 1-(1-naphthyl)-ethanol. In previous work on the DKR for secondary alcohols [84], Yang et al. found that the acid resins (CD8604) were able to gradually dissolved in the solvent, which would reduce the enzyme activity because of the solved acid group-SO3H. In 2013, they continued to develop a new DKR protocol by using a low-cost solid super acid TiO2/SO42− to racemize alcohol substrates (Scheme 45C) [86]. TiO2/SO42− catalyst is sufficiently stable and compatible with enzyme-catalyzed KR system. In 2014, the same group used a more cost-efficient sulfonated sepiolite for the DKR of secondary benzylic alcohols at room temperature (Scheme 45D) [87]. Sepiolite is one of the cheapest minerals with large specific surface areas. Owing to these characters, sulfonated sepiolite displayed higher efficiency of catalytic activity in racemizing 1-phenylethanol, compared to previously reported acid resins (CD-550, CD-8604), and solid super acid TiO2/SO42−.
Scheme 45
Scheme 45.
Acid catalysts for alcohol racemization via carbocation intermediates.
Recently in 2017, Bäckvall and co-workers reported a heterogeneous acid-catalyzed racemization of tertiary alcohols (Scheme 46) [88]. By examining the activity of several acidic resins with different acidities, Dowex 50WX8 was investigated as the optimal resin to exhibit the best performance. In this way, chiral tertiary alcohols could complete the racemization within 5–8 h at room temperature. It is noteworthy to mention that racemization of tertiary alcohols is conceptually impossible through other pathways, e.g. redox racemization or racemization via radical intermediates. Although the DKR of tertiary alcohols has not been realized yet, due to the relatively poor KR performance of lipase towards bulky alcohols [89], this method could be considered as optional racemization tool for the development of DKR transformations in the future.
Scheme 46
Scheme 46.
Dowex 50WX8 as the racemization catalyst for tertiary alcohols.
3.
Enzymatic racemization of secondary alcohols and amines
Enzymatic racemization of optical activity secondary alcohols and amines are cost‐effective. Harsh reaction conditions are generally nonessential, endowing the catalytic process with high compatibility with the KR process. Because of mild reaction conditions and green environmental protection in enzymatic racemization, it has attracted considerable attentions to explore the DKR of secondary alcohols and amines by utilizing an enzyme-catalyzed racemization. However, due to the specificity of biosynthetic pathways, most of active molecules in living organisms are chiral isomers, which would selectively interact with one enantiomer of the substrate, limiting the nature to proceed racemization pathways. Exploring application potentials of racemases in the DKR reaction has been continuously pursued by chemists [5].
In 1999, Faber and co-worker reported a stepwise deracemization reaction of (±)-mandelic acid by using a two-enzyme system, i.e. lipase and mandelate racemase (Scheme 47) [90]. Firstly, Pseudomonas sp. lipase catalyzed enantioselective O-acylation of (±)-mandelic acid; subsequently, mandelate racemase acts as a racemization catalyst to convert the configuration of unreacted (R)-mandelic acid in aqueous buffer. After four cycles of KR and racemization, (S)‐O‐acetylmandelic acid was obtained in good yield with excellent enantioselectivity. Later, the same group extended the applicability of this stepwise deracemization protocol to other α-hydroxycarboxylic acids bearing α-aliphatic and α-aromatic substituents respectively by using whole resting cells of Lactobacillus paracasei DSM 20207 [91,92]. In 2006, they realized racemization of α–hydroxy ketones at physiological conditions by using Lactobacillus paracasei DSM 20207 (Scheme 48) [93].
Scheme 47
Scheme 47.
Dual enzymatic DKR of α-hydroxycarboxylic acids.
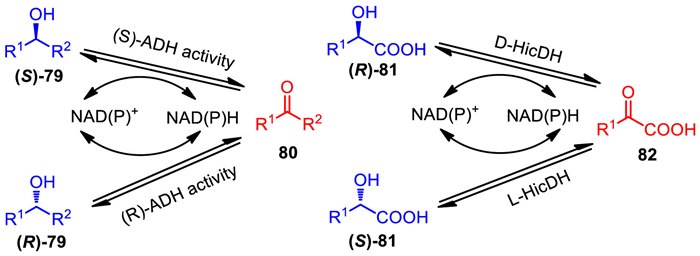
In 2007, Kroutil's group developed a racemization strategy that is based on the reversible interconversion of substrate enantiomers by using a pair of stereo-complementary biocatalysts. To prove the effectiveness of this strategy, the authors successfully achieved racemization of various sec–alcohols under the catalysis of a pair of stereo-complementary biocatalysts ADHs (ADH-"A" from Rhodococcus ruber, and LK-ADH, ADH from Lactobacillus kefir). Interconversion of alcohol enantiomers was realized via the intermediate of their corresponding ketones (Scheme 49, left) [94]. In 2009, Faber's group extend dual enzymatic system to α-hydroxycarboxylic acids [95]. By using a pair of stereo-complementary α-hydroxycarboxylic acid dehydrogenases (D-HicDH from Lactobacillus confusus DSM 20196, and l-HicDH from Lactobacillus paracasei DSM 20008) as coupled active catalysts, efficient racemization of various α-hydroxycarboxylic acids was completed via a reversible oxidation–reduction sequence (Scheme 49, right). These racemization strategies are expected to be further combined with a lipase mediated kinetic resolution.
Scheme 49
Scheme 49.
Dual enzymatic racemization system by using a pair of stereo-complementary biocatalysts.
In 2013, Musa and co-workers achieved racemization of secondary alcohols by using alcohol dehydrogenase, from Thermoanaerobacter ethanolicus (W110A TeSADH) (Scheme 50) [96]. The enzymatic racemization catalysis of W110A TeSADH exhibits good thermal stability and high tolerance with organic solvents, allowing the racemization process taking place in media containing up to 50% (v/v) of organic solvents. Subsequently, the authors further improved the efficiency of the enzymatic system by using Trp-110 mutants of TeSADH in the presence of oxidized and reduced forms of nicotinamide–adenine dinucleotide. The authors also found that the W110G mutation and triple mutant W110A/I86A/C295ATeSADH gave a noticeable enhancement in racemization performance than the previously reported mutant W110A TeSADH [97].
Scheme 50
Scheme 50.
Alcohol dehydrogenase W110A TeSADH as the racemization catalyst.
In 2016, Musa and co-workers described the preliminary results of dual enzymatic DKR of secondary alcohols (Scheme 51) [98]. Combining xerogel-immobilized W110A racemization catalysis with Candida antarctica lipase B (CALB)-catalyzed KR, the authors developed the dual-enzymatic DKR transformation in a one-pot operation. The enzymatic racemization could take place in organic solvents, which provides conditions for subsequent CALB-catalyzed KR. However, the efficiency of dual enzymatic DKR is still at a relatively low level, thus needs to be further improved in the future.
Scheme 51
Scheme 51.
Dual enzymatic DKR of secondary alcohols.
In 2018, Kroutil and co-workers described a biocatalytic racemization by employing the triple mutant W110A/I86A/C295A TeSADH (alcohol dehydrogenase from Thermoanaerobacter pseudoethanolicus) (Scheme 52) [99]. By mutating specific sites of the wild-type enzyme, not only can the substrate range be broadened, but the original selectivity is also preserved. TeSADH WIC was employed as a non-stereoselective ADH to racemize secondary alcohols, and in this way the dual-enzymatic DKR of 1‐phenyl‐2‐propanol and 2‐hexanol were realized by combining racemization in aqueous phase with KR in organic phase. In the system, the authors added catalytic amounts of ketone to increase the racemization rate, and used a polyvinylidene difluoride (PVDF) membrane in a Tea-Bag construction for compartmentalization of the two reaction systems, impelling the dual and biphasic enzymatic DKR of secondary alcohols.
Scheme 52
Scheme 52.
Dual enzymatic DKR of secondary alcohols.
Due to the necessity for harsh conditions to enable racemization, amine racemization generally faces to higher challenge than alcohol racemization, bringing disadvantages regarding the issue of compatibility with KR processing [100]. In 2005, Asano's group reported an efficient DKR of amino acid amide in the presence of α-amino-ε-caprolactam (ACL) racemase from Achromobacter obao and d-aminopeptidase (Scheme 53) [101]. The system could produce (R)-amino acid with excellent yield and ee from l-amino acid amide by a combination of ACL racemase (EC 5.1.1.15) and d-stereospecific hydrolase.
Scheme 53
Scheme 53.
ACL racemase as the racemization catalyst for amines.
In 2011, the same group developed the first DKR access to chiral α-amino acids from α-aminonitriles (Scheme 54) [102]. As the initial step, racemic α-aminonitrile was hydrolyzed by non-stereoselective nitrile hydratase (NHase) from Rhodococcus opacus 71D. Subsequently, the resulting racemic α-amino acid amide was hydrolyzed by stereoselective amino acid amide hydrolase d-aminopeptidase from Ochrobactrum anthropi C1–38 to deliver chiral α-amino acids, and meanwhile the remaining α-amino acid amide was racemized by ACL racemase.
Scheme 54
Scheme 54.
DKR transformations to chiral α-amino acids from α-aminonitriles.
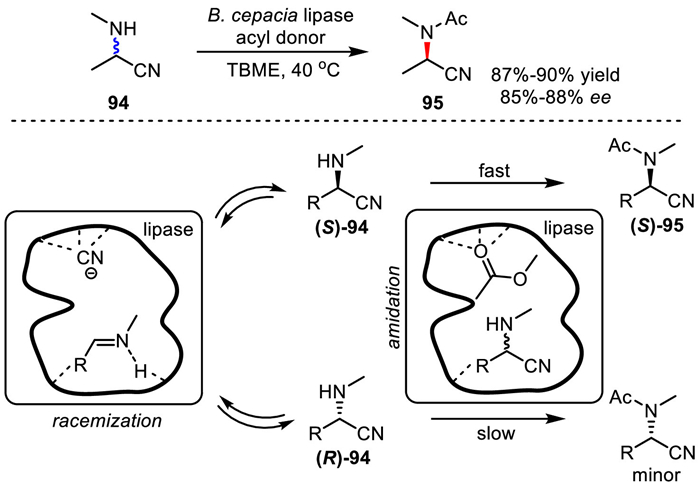
In 2011, Ramström's group reported a dual-function DKR of α-aminonitriles by a single catalyst of B. cepacia lipase (Scheme 55) [103]. The enzyme has both racemase activity and normal lipase transesterification activity in the system. The reactions were performed with phenyl acetate as acyl donor under the catalysis of B. cepacia lipase at 40 ℃ in tbutyl methyl ether (TBME). In this way, chiral amide products were obtained in good yields with high ee values. The racemization mechanism is proposed to proceed via continuous retro‐Strecker C–C bond breaking and Strecker bond reformation reactions.
Scheme 55
Scheme 55.
Single lipase-catalyzed DKR of α-aminonitriles.
In 2011, Faber and co-workers realized enzymatic racemization of α-chiral primary amines by a reversible deamination/amination sequence catalyzed by commercially available stereo-complementary ω-transaminases (Scheme 56) [104]. In the system, employing a pair of ketone/amine co-additives as amino-group shuttles, various primary amines could be efficiently racemized under mild reaction conditions.
Scheme 56
Scheme 56.
Stereo-complementary ω-transaminases as the coupled racemization catalysts.
Recently in 2018, Berglund and co-workers reported a novel biocatalytic method for enzymatic racemization of primary amines by using stereo-complementary amine transaminases (Aspergillus oryzae (R)-ATA [Ao-(R)-ATA] and the Cv-(S)-ATA) (Scheme 57) [105]. This one-pot enzymatic racemization of 1-methyl-3-phenylpropylamine was achieved by using enantiopure d- or l-alanine as amino donor. It is necessary to emphasize that, compared with previous reports, this process shows reduced complexity and better atom economy.
Scheme 57
Scheme 57.
Stereo-complementary amine transaminases for amine racemization.
The advances of racemization of secondary alcohols and amines through different catalytic pathways during the past three decades have been concluded in this review, i.e. redox racemization via ketone/imine intermediates, racemization via radical intermediates, racemization via carbocation intermediates, and enzymatic racemization. These stereochemical editing strategies [106,107] would be expected to be compatible with KR catalysis, to apply in the DKR to provide new methods for the synthesis of enantiopure alcohols and amines. The redox racemization of secondary alcohols and amines is mainly achieved by employing noble transition metals complexes, based on Ru, Ir, Rh, Pd, etc. In addition, the use of earth abundant and more readily available metals, such as Al and Fe is the development trend of this approach. Radical mediated racemization of amines was initially developed by Bertrand's group, which can be compatible with enzyme-catalyzed KR to deliver optical active products. With the efforts of Bertrand and Zhou et al., radical-mediated racemization has been continuously improved, especially by introducing photochemical initiation. In addition, the DKR of secondary alcohols with racemization via radical intermediates has been realized by Collins' group. Racemization via carbocation intermediates has been realized based by forming a transient sp2-carbenium ion. In the presence of a Lewis acid or Brønsted acid, racemization of secondary alcohols was able to occur under either homogeneous catalysis such as oxovanadium catalyst, or heterogeneous catalysis such as acid resins and acid zeolites. Notably, this racemization strategy is still limited to alcohol substrates currently. Enzymatic racemization could be operated under mild reaction conditions. This protocol is generally environmentally benign, exhibiting application potentials in bis‐enzymatic DKR in a one-pot manner. However, the reported bis‐enzymatic DKR examples were still limited, and need to be further developed.
In the future, chemists will pay more attentions to the following aspects when developing catalytic racemization systems of alcohols and amines: (1) More efficient and environmentally benign catalysis will be focused on, especially those based on 3d transition-metals [108–113] (2) The catalytic systems should have higher compatibility with KR processes, and tolerate various functional groups with broader substrate scope; (3) Easy to handle and salable catalysis would be followed with interests, which can be applied to industrial processes.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We acknowledge the National Natural Science Foundation of China (No. 22271054), the "1000-Youth Talents Plan", and Fudan University (start-up grant) for financial support.
[1]
A. Bartoszewicz, N. Ahlsten, B. Martin-Matute, Chem. Eur. J. 19 (2013) 7274–7302. doi: 10.1002/chem.201202836
J.H. Choi, Y.K. Choi, Y.H. Kim, et al., J. Org. Chem. 69 (2004) 1972–1977. doi: 10.1021/jo0355799
[27]
B. Martín-Matute, M. Edin, K. Bogár, F.B. Kaynak, J.E. Bäckvall, J. Am. Chem. Soc. 127 (2005) 8817–8825. doi: 10.1021/ja051576x
[28]
B. Martín-Matute, J.B. Aberg, M. Edin, J.E. Bäckvall, Chem. Eur. J. 13 (2007) 6063–6072. doi: 10.1002/chem.200700373
[29]
P. Krumlinde, K. Bogár, J.E. Bäckvall, J. Org. Chem. 74 (2009) 7407–7410. doi: 10.1021/jo9014276
[30]
E.V. Johnston, K. Bogár, J.E. Bäckvall, J. Org. Chem. 75 (2010) 4596–4599. doi: 10.1021/jo100936f
[31]
A. Träff, R. Lihammar, J.E. Bäckvall, J. Org. Chem. 76 (2011) 3917–3921. doi: 10.1021/jo2003665
[32]
S.B. Ko, B. Baburaj, M.J. Kim, J. Park, J. Org. Chem. 72 (2007) 6860–6864. doi: 10.1021/jo071065o
[33]
S.Y. Lee, J.M. Murphy, A. Ukai, G.C. Fu, J. Am. Chem. Soc. 134 (2012) 15149–15153. doi: 10.1021/ja307425g
[34]
A. Kinens, S. Balkaitis, O.K. Ahmad, D.W. Piotrowski, E. Suna, J. Org. Chem. 86 (2021) 7189–7202. doi: 10.1021/acs.joc.1c00545
[35]
Q. Xi, W. Zhang, X. Zhang, Synlett (2006) 945–947.
[36]
N. Kim, S.B. Ko, M.S. Kwon, M.J. Kim, J. Park, Org. Lett. 7 (2005) 4523–4526. doi: 10.1021/ol051889x
[37]
J.H. Lee, N. Kim, M.J. Kim, J. Park, ChemCatChem 3 (2011) 354–359. doi: 10.1002/cctc.201000304
[38]
D. Lee, E.A. Huh, M.J. Kim, et al., Org. Lett. 2 (2000) 2377–2379. doi: 10.1021/ol006159y
[39]
M.J. Kim, H.M. Kim, D. Kim, Y. Ahn, J. Park, Green Chem. 6 (2004) 471–474. doi: 10.1039/B405651E
[40]
M. Merabet-Khelassi, N. Vriamont, L. Aribi-Zouioueche, O. Riant, Tetrahedron Asymmetry 22 (2011) 1790–1796. doi: 10.1016/j.tetasy.2011.10.007
[41]
S.F.G.M. v. Nispen, J. v. Buijtenen, J.A.J.M. Vekemans, J. Meuldijk, L.A. Hulshof, Tetrahedron Asymmetry 17 (2006) 2299–2305. doi: 10.1016/j.tetasy.2006.08.003
[42]
J. Bosson, S.P. Nolan, J. Org. Chem. 75 (2010) 2039–2043. doi: 10.1021/jo1001005
X.Y. Liu, H.B. Zhu, Y.J. Shen, J. Jiang, T. Tu, Chin. Chem. Lett. 28 (2017) 350–353. doi: 10.1016/j.cclet.2016.09.006
Scheme 1
Kinetic resolution (KR) and dynamic kinetic resolution (DKR). kR is the rate constant for the reaction of (R)-isomer; kS is the rate constant for the reaction of (S)-isomer; krac is the rate constant for the interconversion of the two substrate enantiomers.


 DownLoad:
DownLoad:





































 下载:
下载:


























































