School of Environmental and Chemical Engineering, International Joint Laboratory of Catalytic Chemistry, College of Sciences, Shanghai University, Shanghai 200444, China
b.
BASF Environmental Catalyst and Metal Solutions, Iselin, NJ 08830, United States
Received Date:
01 May 2023 Accepted Date:
19 September 2023 Revised Date:
28 August 2023 Available Online:
15 June 2024
Abstract:
Development of hydrothermally stable, low-temperature catalysts for controlling nitrogen oxides emissions from mobile sources remains an urgent challenge. We have prepared a metal oxide-zeolite composite catalyst by depositing Mn active species on a mixture support of CeO2/Al2O3 and ZSM-5. This composite catalyst is hydrothermally stable and shows improved low-temperature SCR activity and significantly reduced N2O formation than the corresponding metal oxide catalyst. Comparing with a Cu-CHA catalyst, the composite catalyst has a faster response to NH3 injection and less NH3 slip. Our characterization results reveal that such an oxide-zeolite composite catalyst contains more acidic sites and Mn3+ species as a result of oxide-zeolite interaction, and this interaction leads to the generation of more NH4+ species bound to the Brønsted acid sites and more reactive NOx species absorbed on the Mn sites. Herein, we report our mechanistic understanding of the oxide-zeolite composite catalyst and its molecular pathway for improving the low-temperature activity and N2 selectivity for NH3-SCR reaction. Practically, this work may provide an alternative methodology for low-temperature NOx control from diesel vehicles.
Nitrogen oxides (NOx) discharged from mobile and stationary sources are the main culprit of atmospheric pollution such as photochemical smog and acid rain [1-3]. To comply with the increasingly stringent regulations worldwide, selective catalytic reduction of NOx (SCR) with NH3 or urea has been broadly used for NOx emission controls from diesel vehicles [4,5]. The development of advanced diesel engine technologies has increased fuel combustion efficiency and at the same time resulted in reduced exhaust gas temperatures [6]. Typically, a commercial SCR catalyst is quite efficient to remove NOx at temperatures above 250 ℃, but leaving most of the NOx emissions unconverted below 200 ℃ such as during engine cold-start [7].
Commercially, there are two major types of catalysts for diesel NOx controls, Cu-CHA and V2O5/WO3-TiO2, each with its own performance and durability characteristics [8]. In general, Cu-CHA is more active than V2O5/WO3-TiO2 at low temperatures. However, even Cu-CHA does not have sufficient activity below 200 ℃. In addition, due to its large NH3 adsorption capacity, Cu-CHA cannot be immediately active for SCR upon NH3 (or urea) dosing; its activity reaches its maximum only after the catalyst has been filled with NH3. The slow response could result in low NOx efficiency during engine cold-start or serious NH3 slip during abrupt vehicle acceleration [9]. In this regard, metal oxides catalysts with relatively small NH3 adsorption capacities could be considered as potential candidates to replace Cu-CHA in diesel vehicles for some applications or as add-on technologies to broaden the effective temperature window. Although the V2O5/WO3-TiO2 based catalysts show some advantages over Cu-CHA in sulfur tolerance and cost, they have some serious drawbacks, including low low-temperature activity, low hydrothermal stability, and potential escape of toxic materials (V2O5) to the environment [10,11]. On the durability side, a SCR catalyst used for heavy-duty diesel vehicles needs to be stable not only in the harshest engine exhaust conditions (500–550 ℃ with > 10% water vapor content) but also during the diesel particulate filter (DPF) regeneration, with the filter downstream temperature exceeding 600 ℃ [12]. Due to the complicated running conditions of diesel vehicles, there have been extensive efforts devoted to exploiting environmentally friendly SCR catalysts with better low-temperature catalytic activity, hydrothermal stability and fast response to NH3 reactants.
Among various non-vanadium metal oxide catalysts, Mn-based catalysts display superior low-temperature SCR performance due to their variable valence states that are conducive to enhanced redox properties [13,14]. However, Mn-based catalysts typically show inferior N2 selectivity, especially at high temperatures, due to the non-selective NH3 oxidation. There have been significant efforts by many researchers to improve the N2 selectivity of Mn-based catalysts, and these efforts may be grouped into two main strategies. One strategy is the modification of Mn active species by transition metal elements, such as Ce [15], Sm [16], Mo [17], Nb [18], to enhance the electron transport between the active sites and metal additives and to increase the number of acidic sites. Another strategy is the use of carriers with special features, such as microporous TiO2 [19], carbon nanotubes [20], various zeolites [21], to indirectly enhance the catalytic performance by their special catalyst structures. These metal additives and supports have been shown to improve the N2 selectivity to a certain extent. However, low N2 selectivity at high temperatures and poor thermal stability remain to be great challenges for Mn-based catalysts, limiting their application in mobile sources.
Al2O3 support is widely used in three-way conversion (TWC) catalysts due to its low cost, high specific surface area, moderate acidity and high temperature resistance [22]. Therefore, Al2O3 is a promising candidate of support material for the development of highly efficient SCR catalysts with excellent hydrothermal stability. In addition, Jeong et al. reported a TWC catalyst with 100% dispersion of active metals (Pt, Pd and Rh) obtained by impregnating the metal precursor on the pre-reduced activated γ-alumina with nanoceria particles anchored on the Alpenta3+ sites, and this catalyst showed excellent low-temperature activity and hydrothermal stability for TWC reaction [23]. Inspired by this literature, a commercially available CeO2 modified Al2O3 (CeO2/Al2O3) material was considered as a more appropriate support to disperse Mn oxides, which was expected to improve the electron transfer on the supported Mn oxide catalysts and thus the SCR performance. However, the acidic sites on the CeO2/Al2O3 materials that affect the adsorption and activation of the reactants are still limited and needed to be enhanced.
Moreover, zeolites are also widely used as supports for SCR catalysts because of their special pore structures, large specific surface area and abundance of acidic sites [24-26]. Chen et al. reported that Mn-Ce/X catalysts synthesized by impregnating Mn and Ce on zeolite X had more Brønsted acid sites and higher fractions of Mn4+ and Ce3+ species. These catalysts were shown to have better redox properties than those of Mn-Ce/ZSM-5, Mn-Ce/Beta and Mn-Ce/Y catalysts [27]. Shao et al. investigated a set of MnOx/ZSM-5 catalysts prepared by ethanol dispersion and found that these catalysts, due to their high specific surface areas and abundant acidic sites, to be highly selective for NH3-SCR (> 90% N2 selectivity at 120–240 ℃) [28]. However, the role of zeolite in promoting SCR activity and improving N2 selectivity remains little understood.
In this work, we attempt to improve the SCR activity and N2 selectivity of a MnOx-based catalyst from two fronts. A commercial CeO2/Al2O3 (20 wt% CeO2) material (ALCE20) was selected as the support for MnOx to better disperse the Mn species and generate a right mix of oxidation states through Mn-Ce interaction. A zeolite component was incorporated into the Mn-based catalyst to increase acidity. Herein, we report improved SCR activity and greatly enhanced N2 selectivity on the composite catalysts that have gone through a hydrothermal aging process simulating heavy-duty truck operations. Through extensive catalyst characterizations, we present mechanistic understanding of catalysts and reveal the molecular pathway for reduced N2O formation.
Deposited MnOx catalysts were prepared by impregnation using the incipient wetness technique. The required amount of ALCE20 and NH4+-ZSM-5 (provided by BASF Corporation) were weighed and mixed by grinding to obtain a mixture support. A calculated quantity of Mn(NO3)2·4H2O (provided by Sinopharm Chemical Reagent Co., Ltd. (China)) was dissolved in absolute ethanol under ultrasound, denoted as active component solution. The active component solution was added to the mixture support in a crucible under stirring to reach incipient wetness state, which was then dried at 80 ℃ in an oven for 2 h. The resulting material was finally calcined by ramping temperature to 500 ℃ at a rate of 2 ℃/min and kept there for 3 h. The obtained sample is considered as fresh sample and contains 7 wt% MnO2. The catalyst compositions are designated as Mn/(ALCE20+xZSM-5)-F (x = 20, 40, 60, 80), where x (in wt%) is the content of zeolite in the mixture support. Moreover, for comparison purpose, the Cu-CHA-F and V2O5-WO3/TiO2-F (denoted as VWTi-F) with 3 wt% V2O5 and 8 wt% WO3 were prepared. More details about the catalyst synthesis are supplied in Supporting information.
The hydrothermal aging treatment of Mn/(ALCE20+xZSM-5)-F, Mn/(ALCE20+20Beta)-F, Mn/(ALCE20+20Ferrierite)-F, Mn/(ALCE20+20Mordenite)-F, Mn/ALCE20-F, Cu-CHA-F and VWTi-F catalysts was carried out at 650 ℃ for 50 h in a self-made unit made up with a muffle furnace, a thermostat water bath and an air generator with a feed gas containing 10 vol% H2O in air (0.2 L/min). The aged samples are marked as Mn/(ALCE20+xZSM-5)-HA, Mn/(ALCE20+20Beta)-HA, Mn/(ALCE20+20Ferrierite)-HA, Mn/(ALCE20+20Mordenite)-HA, Mn/ALCE20-HA, Cu-CHA-HA and VWTi-HA, respectively.
The NH3-SCR performance test was carried out in a quartz tube fixed bed reactor with an inner diameter of 4 mm. Prior to activity test, the as-prepared catalysts were sieved into 40−60 mesh fraction and weighed (0.15 g) for testing. The feed gas consisted of 500 ppm NO, 500 ppm NH3, 10 vol% O2, 5 vol% H2O, 5 vol% CO2 and balanced N2. The total flow rate of the feed was 500 mL/min, which corresponds to a mass-based space velocity of 200, 000 mL g−1 h−1. The outlet concentrations of NH3, NO, NO2 and N2O were measured using a FTIR spectrometer (Protea atmosFIR-AFS-B2T-C-1911). The NOx conversion was calculated as follows (Eq. 1):
(1)
where NOx is the sum of NO and NO2. [NOx]in and [NOx]out denote the corresponding inlet and outlet gas concentrations, respectively. More details on the characterization of these catalysts are supplied in Supporting information.
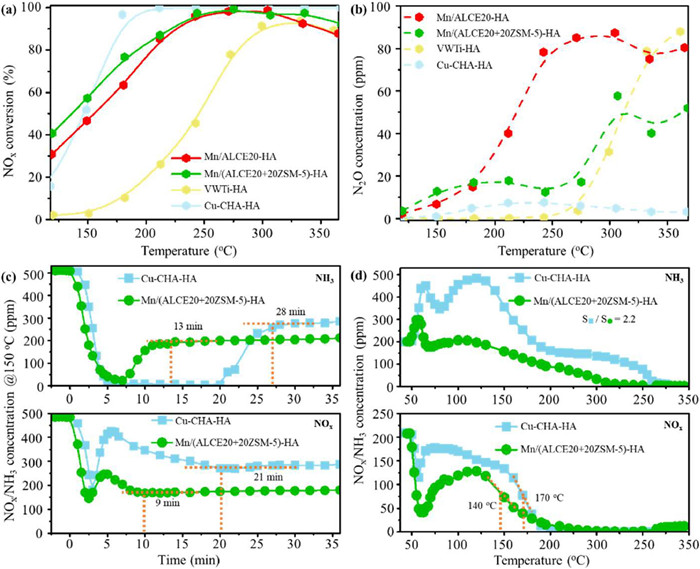
To obtain an optimal catalyst composition, an initial catalyst screening was conducted by adjusting the oxide to zeolite ratio. The best catalyst is MnOx supported on a mixture containing 20% ZSM-5 and 80% ALCE20, which shows the optimal SCR performance after hydrothermal aging (Fig. S1 in Supporting information). Moreover, Mn/(ALCE20+20ZSM-5)-F shows obviously higher SCR activity than the other reference catalysts including Mn/ALCE20-F, VWTi-F, Cu-CHA-F and Mn/Al2O3-F in the whole temperature range, reaching more than 90% NOx conversion at 150 ℃ (Fig. S2 in Supporting information). As shown in Fig. 1a, comparing with Mn/ALCE20-HA, Mn/(ALCE20+20ZSM-5)-HA shows an appreciable increase in low-temperature (< 225 ℃) activity, reaching about 80% NOx conversion at 180 ℃. Surprisingly, as shown in Fig. 1b, N2O formation is significantly reduced on the composite catalyst between 180 ℃ and 360 ℃. At 240 ℃, the N2O concentration is 80 ppm on the zeolite-free reference but < 15 ppm on the zeolite-containing catalyst, which is considerably lower than most Mn-based catalysts used for SCR reaction. The promoting effect is also found on catalysts containing other types of zeolites (Fig. S3 in Supporting information). For comparison, the VWTi-HA catalyst shows poor catalytic activity after hydrothermal aging, not reaching 90% NOx conversion until 300 ℃. Cu-CHA-HA, on the other hand, is quite active above 180 ℃ but is inferior to Mn/(ALCE20+20ZSM-5)-HA below 150 ℃. Thus, the composite catalyst with a certain oxide to zeolite ratio is shown to be advantageous to the commercial VWTi oxide and Cu-CHA zeolite catalysts for low-temperature SCR activity.
Figure 1
Figure 1.
NOx conversion (a) and N2O formation (b) as a function of temperature over Mn/ALCE20-HA, Mn/(ALCE20+20ZSM-5)-HA, VWTi-HA and Cu-CHA-HA. Response profiles (c) of NH3 and NOx on Cu-CHA-HA and Mn/(ALCE20+20ZSM-5)-HA during a transient SCR reaction at 150 ℃; Transient SCR performance (d) as a function of temperature on Cu-CHA-HA and Mn/(ALCE20+20ZSM-5)-HA. Prior to test (c) and (d), each catalyst was pretreated in air at 300 ℃ for 1 h. For test (a) and (b): [NO] = [NH3] = 500 ppm, [O2] = 10 vol%, [H2O] = 5 vol%, [CO2] = 5 vol%, N2 as balance, and WHSV = 200, 000 mL g−1 h−1. For test (c), upon the catalyst was cooled to 150 ℃ after the air pretreatment, a flow of 500 ppm NO + 500 ppm NH3 + 10 vol% O2 was introduced. For test (d), upon the catalyst was cooled to 50 ℃ after the air pretreatment, NH3 (500 ppm NH3, 10 vol% O2, N2 as balance) was injected for 1 h until saturation. The NH3-saturated catalyst was then heated to 350 ℃ at 5 ℃/min in a flow of 200 ppm NO + 200 ppm NH3 + 10 vol% O2.
Transient response to urea (NH3) injection is a relevant performance measurement related to the application of SCR catalyst in vehicles, especially during engine cold-start. Fig. 1c shows NH3 and NOx concentration profiles as a function of time upon contacting catalysts (Cu-CHA-HA and Mn/(ALCE20+20ZSM-5)-HA) with a SCR feed at 150 ℃. On both catalysts, upon contacting the catalyst with the SCR feed, the NH3 concentration quickly decreased to near zero and then gradually increased to a steady-state level. These two catalysts differ in the time required to reach the steady-state level and in the stabilized NH3 concentration. It takes about 28 min for Cu-CHA-HA to reach its stabilized NH3 concentration (around 280 ppm) but only 13 min for Mn/ALCE20+20ZSM-5-HA (around 200 ppm). The transient NH3 consumption is likely the result of NH3 adsorption, SCR reaction and parasitic NH3 oxidation. Simultaneously, the NOx concentration drops precipitously upon contacting the SCR feed, quickly increases, then gradually decreases again and eventually reaches a steady-state level. Similar to the NH3 profiles, it took twice the time for Cu-CHA-HA to reach its stabilized NOx concentration (around 280 ppm) compared to Mn/(ALCE20+20ZSM-5)-HA (around 180 ppm). Thus, it takes much shorter time for the composite catalyst to reach its maximum NOx than the Cu-CHA-HA reference. To simulate the acceleration of diesel vehicle after a cold start, pre-saturated Cu-CHA-HA and Mn/(ALCE20+20ZSM-5)-HA catalysts were evaluated using temperature programmed SCR reaction (from 50 ℃ to 350 ℃, 5 ℃/min). As shown in Fig. 1d, at the start of the temperature ramp, there is an initial increase in NH3 concentration, which is believed to be due to flushing out the residual NH3 used to saturate the catalysts from the gas line. The subsequent increase is due to NH3 desorption from the catalysts. The amount of NH3 desorbed from Cu-CHA-HA (S▪) is around 2.2 times than that of Mn/(ALCE20+20ZSM-5)-HA (S•), further confirming that a high NH3 adsorption capacity on Cu-CHA could delay its activation at low temperature. Meanwhile, on both catalysts, the NOx concentration decreases rapidly at first, then increases slowly, and finally reduces tardily to zero with increasing temperature. Mn/(ALCE20+20ZSM-5)-HA reaches the maximum consumption rate at a lower temperature, indicating its higher NOx reduction activity at low temperature. The transient experiments shown in Figs. 1c and d imply that on Mn/(ALCE20+20ZSM-5)-HA we could achieve a much quicker response on urea injection and much less NH3 slip upon a sudden vehicle acceleration.
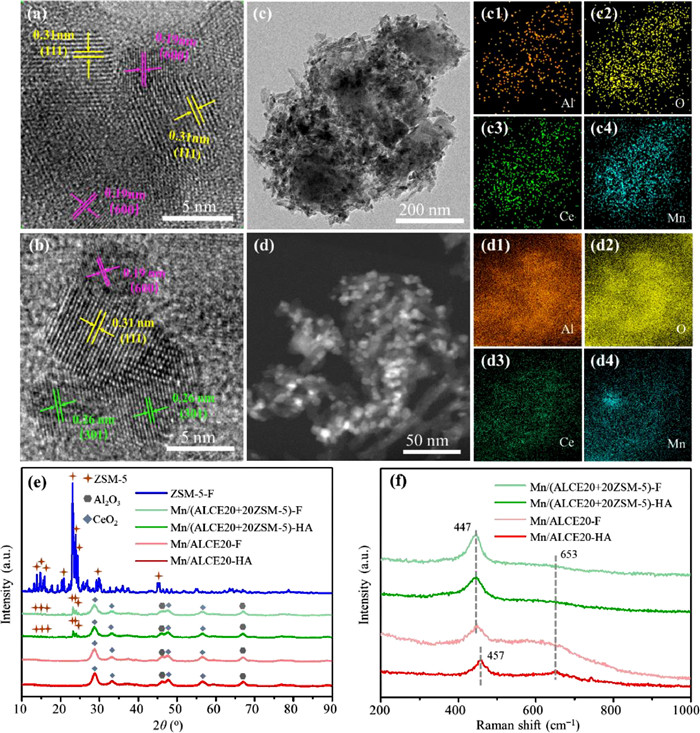
N2 adsorption-desorption measurements were performed on Mn/AlCE20 and Mn/(AlCE20+20ZSM-5) catalysts, and the results were summarized in Table S1 (Supporting information). Relative to the Mn/ALCE20-F reference, incorporating 20% zeolite significantly increases the specific surface area and the total pore volume of the Mn/(ALCE20+20ZSM-5)-F catalyst and at the same time substantially decreases the average pore diameter. These changes are the result of mathematical average between an oxide component and the zeolite component (large micro-pore surface area with high total pore volume). After hydrothermal aging, compared to Mn/ALCE20-HA catalyst (108.0 m2/g), the surface area of Mn/(ALCE20+20ZSM-5)-HA catalyst (142.7 m2/g) increases significantly because the ALCE20+20ZSM-5 support is prepared by partially replacing ALCE20 with ZSM-5 zeolite with high specific surface area (341.0 m2/g). This high surface area due to the addition of zeolites can facilitate the dispersal of Mn species by providing more dispersion sites, which is consistent with the results reported by Lu et al. [29]. The microstructures of the catalysts were examined by high resolution transmission electron microscopy (HR-TEM). As shown in Fig. 2, different lattice fringes are clearly observable on Mn/(ALCE20+20ZSM-5)-HA (Fig. 2a) and Mn/ALCE20-HA (Fig. 2b). On Mn/(ALCE20+20ZSM-5)-HA (Fig. 2a), lattice spacing of 0.31 nm belongs to CeO2 (111) crystalline planes, and lattice spacing of 0.19 nm corresponds to the Al2O3 (400) surface. No crystalline planes are associated with Mn species. On the low-magnification image (Fig. 2c), ZSM-5 particles with smooth surface are surrounded by ALCE20. EDS element mappings show that Mn distribution is uniform throughout the sample. The Si mapping (Fig. S4 in Supporting information) indicates that ZSM-5 is uniformly distributed in the whole area. On Mn/ALCE20-HA (Fig. 2b), the CeO2 (111) crystal plane, the crystal plane of Al2O3 and the MnO2 (301) plane (0.26 nm) lattice spacing are observed. The corresponding EDS mapping (Fig. 2d) further indicates that some Mn species aggregate on Mn/ALCE20-HA. These results show that Mn species have a better dispersion on Mn/(ALCE20+20ZSM-5)-HA.
Figure 2
Figure 2.
HR-TEM images of Mn/(ALCE20+20ZSM-5)-HA (a) and Mn/ALCE20-HA (b). TEM images and the corresponding EDS element mappings of Al (c1, d1), O (c2, d2), Ce (c3, d3) and Mn (c4, d4) for Mn/(ALCE20+20ZSM-5)-HA (c) and Mn/ALCE20-HA (d). XRD patterns (e) and Raman spectra (f) of Mn/ALCE20-HA, Mn/ALCE20-F, Mn/(ALCE20+20ZSM-5)-HA and Mn/(ALCE20+20ZSM-5)-F.
X-ray diffraction (XRD) was performed on Mn/ALCE20 and Mn/(ALCE20+20ZSM-5) catalysts. As illustrated in Fig. 2e, Al2O3 (PDF #04–0877) and CeO2 (PDF #34–0394) are the main crystal phases on all catalysts. The crystal pattern corresponding to ZSM-5 (PDF #47–0638) is also observed on the Mn/(ALCE20+20ZSM-5) catalysts with main diffraction peaks at 23.2°, 24.0° and 24.5°, indicating that the structure of ZSM-5 has been preserved on Mn/(ALCE20+20ZSM-5) catalysts, even after hydrothermal aging. Conversely, the presence of ZSM-5 does not seem to have any influence on the crystal structure of Al2O3 and CeO2. The similarity between fresh and aged catalysts in XRD pattern indicates that the CeO2/Al2O3 support is very stable. Interestingly, no peaks related to Mn oxides are observed on any samples, implying that Mn species are fully dispersed.
The Raman spectra was used to further examine the local structures of these catalysts. As illustrated in Fig. 2f, the band within 447–457 cm−1 corresponds to the structural vibration of CeO2 [30,31]. Comparing with Mn/ALCE20-F, the CeO2 band has a red shift (from 447 cm−1 to 457 cm−1) on Mn/ALCE20-HA, indicating that the hydrothermal aging treatment increases the bond strength of Ce-O, making it harder to activate the Ce-O bond and weaking the interaction between Mn and Ce species [32]. For the composite catalysts, the CeO2 changes little after hydrothermal aging, indicating that the CeO2 is quite stable on the mixture support. In addition, the band within 600–700 cm−1 can be observed on all catalysts, which may be ascribed to the Mn species [33]. This band is more intense on the oxide reference (Mn/ALCE20) than the composite catalyst, and the contrast is more obvious on aged catalysts. This indicates a low lattice symmetry for Mn species on the Mn/(ALCE20+20ZSM-5) catalyst and thus a higher Mn dispersion [34].
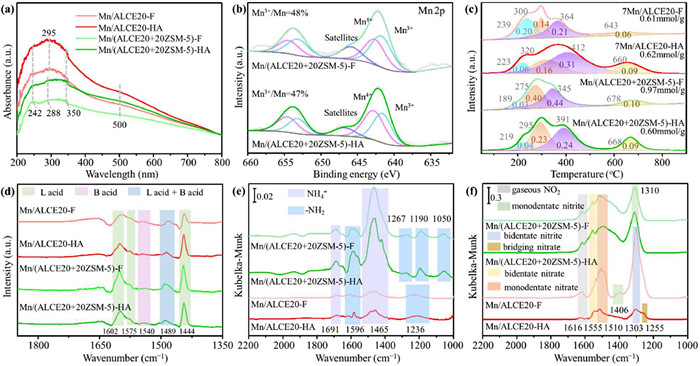
Diffuse reflectance UV–vis spectroscopy was used to identify the oxidation states and coordination state of metal complexes on the composite and metal oxide reference catalysts, and the results are shown in Fig. 3a. The absorption bands around 242 and 350 nm on all catalysts are related to the charge transfer process between Mn2+ and O2−, and between Mn3+ and O2−, respectively [35-37]. Based on the UV–vis spectra collected on the ALCE20 support (Fig. S5 in Supporting information), the absorption band at 295 nm on Mn/(ALCE20+20ZSM-5)-F and Mn/(ALCE20+20ZSM-5)-HA is assigned as the dispersed CeO2 nanoparticles. However, the CeO2 band is down shifted to 288 nm on Mn/(ALCE20+20ZSM-5)-F and Mn/(ALCE20+20ZSM-5)-HA. This red shift is caused by the chemical interaction between CeO2 particles and the surrounding manganese oxides, which reduces the electron density of CeO2 particles [38]. The enhancement of absorption bands around 242 and 350 nm on Mn/(ALCE20+20ZSM-5) catalyst also confirms the existence of stronger chemical interaction between Mn and Ce species. In addition, unlike Mn/ALCE20, the absorption band around 500 nm, attributed to the charge transfer transition of Mn4+ [39], is not evident on Mn/(ALCE20+20ZSM-5), possibly also due to the influence by CeO2 nanoparticles. Note, the intensity of the UV–vis spectra of Mn/(ALCE20+20ZSM-5) catalysts are noticeably weaker than those of Mn/ALCE20 catalysts. This could be a result of a dilution effect in the composite catalyst due to incorporating ZSM-5. These results indicate the existence of some kind of interaction between cerium oxide and Mn species on the surface of Mn/(ALCE20+20ZSM-5) catalysts, which results in more stable surface Mn species with lower valence states even after hydrothermal aging.
Figure 3
Figure 3.
UV–vis spectra (a) of Mn/ALCE20-HA, Mn/ALCE20-F, Mn/(ALCE20+20ZSM-5)-HA, and Mn/(ALCE20+20ZSM-5)-F. Mn 2p XPS (b) of Mn/(ALCE20+20ZSM-5)-HA and Mn/(ALCE20+20ZSM-5)-F. H2-TPR profiles (c) and FTIR spectra with pyridine adsorption (d) for Mn/ALCE20-HA, Mn/ALCE20-F, Mn/(ALCE20+20ZSM-5)-HA and Mn/(ALCE20+20ZSM-5)-F. For the FTIR experiment, pyridine adsorption was carried out at 50 ℃ for 1 h. (e) In situ DRIFTS of NH3 desorption. NH3 adsorption was carried out at 50 ℃ with 1000 ppm NH3 for 1 h. (f) In situ DRIFTS of NO + O2 desorption. NO + O2 adsorption was performed at 50 ℃ with 1000 ppm NO and 5 vol% O2 for 1 h. The desorption spectra of (e) and (f) were collected at 100 ℃ over Mn/ALCE20-HA, Mn/ALCE20-F, Mn/(ALCE20+20ZSM-5)-HA and Mn/(ALCE20+20ZSM-5)-F.
The surface elemental composition and chemical states of these catalysts were further determined by X-ray photoelectron spectroscopy (XPS) and deconvoluted using Gaussian functions. As shown in Fig. 3b and Fig. S6 (Supporting information), the Mn 2p XPS of both Mn/(ALCE20+20ZSM-5) and Mn/ALCE20 shows two main peaks around 642 eV and 654 eV, attributed to Mn 2p3/2 and Mn 2p1/2, respectively. The Mn 2p3/2 peak could be divided into three sub-peaks at 641.4–641.6 eV, 642.4–642.9 eV and 645.6–647.0 eV, corresponding to the Mn3+, Mn4+ and satellite peaks, respectively [13,40-42]. The relative atomic contents of Mn species are determined by computing the areas of the corresponding peaks. The Mn3+/Mn ratio, where Mn is the total Mn species, is higher on Mn/(ALCE20+20ZSM-5)-F (48%) than that on Mn/ALCE20-F (38%). After hydrothermal aging, the proportion of Mn3+ on Mn/(ALCE20+20ZSM-5)-HA (47%) decreases slightly but is still higher than that on Mn/ALCE20-HA (46%). It has been reported that low valence Mn species exhibits higher activity [42,43], which is why composite catalyst is more active at low temperature. Ce 3d XPS of Mn/(ALCE20+20ZSM-5) and Mn/ALCE20 were obtained (Figs. S7 and S8 in Supporting information) and deconvoluted into 8 peaks by Gaussian-Lorentz fitting. The peaks marked as v', v'', v''', u', u'', and u''' are related to Ce4+, and the peaks marked as v and u are attributed to Ce3+ [29,44]. The relative atomic content of Ce3+ in Mn/(ALCE20+20ZSM-5)-F (13%) is lower than that in Mn/ALCE20-F (16%), and the trend is same on the aged catalysts. These findings further indicate that there should exist an electron transfer process between the surface Ce species and the Mn species on the catalysts, i.e., Mn4+ + Ce3+ → Mn3+ + Ce4+, which can be regarded as electronic evidence for the interaction between Mn species and the Ce-containing support. This electron transfer process remains viable even after hydrothermal aging. The stable existence of this electron transfer can facilitate the chemical redox cycles and is beneficial to the improvement of low-temperature catalytic activity [45]. In addition, the change in Mn3+ and Ce3+ contents due to aging is smaller on Mn/(ALCE20+20ZSM-5) than that on Mn/ALCE20, indicating that the electron transfer between Mn and Ce species on Mn/(ALCE20+20ZSM-5) catalyst is stable. Mn species with low oxidation states are expected to be less active for the unselective NH3 oxidation reaction, which mainly produces N2O [46,47].
Redox property is an important performance indicator for Mn-based catalysts. H2-TPR was performed to investigate the reducibility of fresh and aged Mn/ALCE20 and Mn/(ALCE20+20ZSM-5) catalysts. On a Mn-based catalyst, normally multiple reduction peaks can be observed between 200 ℃ and 600 ℃, due to the consecutive reduction process: MnO2 → Mn2O3 → Mn3O4 → MnO [27,48]. As shown in Fig. 3c, four reduction peaks (around 260, 330, 360 and 630 ℃) can be deconvoluted from each TPR profile, which could be attributed to the following four stepwise reduction steps: MnO2 → Mn2O3, Mn2O3 → Mn3O4, Mn3O4 → MnO/CeO2 → CeOx, and CeO2 → CeOx [49,50], with the corresponding H2 consumption amount (in mmol/g) labeled in each deconvoluted peak. As shown in Fig. S9 (Supporting information), the reduction peaks around 411 and 622 ℃ appear on the AlCE20-F support, both of which belong to the reduction of CeO2 species, and the proportion of Ce species reduced at high temperature (67%) is larger. After loading Mn oxides, the proportion of Ce species reduced at around 600 ℃ decreases significantly in Fig. 3c, and the proportion of Ce species reduced at around 400 ℃ increases significantly, accompanied by the emergence of Mn species reduction peaks. This indicates that the electron transfer between Mn and Ce species occurs on Mn/ALCE20 catalyst, which increases the Ce species reduced at low temperature and improves the redox ability of Ce species. After incorporating ZSM-5, compared with Mn/ALCE20 catalyst, although the reduction peak of Ce species at about 600 ℃ on Mn/(ALCE20+20ZSM-5) catalyst does not change significantly, the reduction peaks of Mn species not only shift to lower temperature, which indicates that Mn/(ALCE20+20ZSM-5) catalyst has the enhanced redox ability, but also dominate by the reduction of low-valent Mn species (Mn2O3 → Mn3O4 and Mn3O4 → MnO), further revealing that compounding ZSM-5 zeolite enhances the electron transfer for Mn and Ce species and promotes the formation of low-valent Mn species. Although the H2 consumption of Mn/(ALCE20+20ZSM-5) catalyst decreases significantly (from 0.97 mmol/g to 0.62 mmol/g) after hydrothermal aging, it is still comparable to that of Mn/ALCE20 catalyst. Moreover, different from the obvious change of Mn species distribution of Mn/ALCE20 catalyst before and after hydrothermal aging, Mn/(ALCE20+20ZSM-5) catalyst maintains a similar Mn species distribution, and the proportion of lower valence Mn species maintains at a high level, which further indicates that the Mn4+ species prone to deeply reduce are greatly decreased, thereby relieving the excessive oxidation of NH3 and improving N2 selectivity. In addition, consistent with the results of Mn 2p XPS, these low valence Mn species are expected to be less active for the unselective NH3 oxidation reaction, which is an important factor for N2 selectivity improvement.
Surface acidity is another important factor for deNOx performance. To identify the types of acidity on these catalysts, FTIR spectra with pyridine adsorption were collected (Fig. 3d) and peaks quantified by integrating the areas (Fig. S10 in Supporting information). The bands at 1602 cm−1, 1575 cm−1 and 1444 cm−1 are the contributions from Lewis acid sites. The bands at 1540 cm−1 can be ascribed to Brønsted acid sites, while the bands at 1489 cm−1 correspond to both Lewis and Brønsted acid sites [51,52]. Compared to Mn/ALCE20-F, the overall intensity of the bands ascribed to the Lewis acid sites on Mn/(ALCE20+20ZSM-5)-F is much higher, and this trend remains even after hydrothermal aging, albeit at a reduced level. As shown in Fig. S10, the number of Lewis acid sites has significantly increased by combining ALCE20 and 20ZSM-5 as fresh catalyst and after aging, and this would result in more lower valence Mn sites and better NH3 adsorption. This finding confirms the H2-TPR results that Mn/(ALCE20+20ZSM-5) catalysts contain more lower valence Mn species.
NH3-TPD-MS experiment (using mass spectrometer to measure NH3 desorption) was conducted to further determine the acidic properties of catalysts. As shown in Fig. S11 (Supporting information), all catalysts show three distinct desorption peaks at 215–250 ℃, 293–399 ℃ and 416–572 ℃, designated as weak, medium and strong acid sites, respectively [30]. The corresponding NH3 desorption is quantified (in µmol/g) and labeled in each deconvoluted peak. As illustrated in Fig. S11a, Mn/ALCE20-F and Mn/ALCE20-HA show similar desorption peaks, NH3 desorption quantity and proportion of each desorption peak, indicating similar NH3 adsorption behavior and capacity over these two catalysts, which is also similar to the ALCE20-F support. On fresh and aged Mn/(ALCE20+20ZSM-5) catalysts, a new medium-temperature NH3 desorption peak appears around 300 ℃ (306 ℃ and 293 ℃ for fresh and aged, respectively) (Fig. S11b in Supporting information). This peak is related to the increased fraction of lower valence Mn species facilitated by the presence of ZSM-5 or due to the unsaturated Al sites in the zeolite itself. Further, the low-temperature peak on the composite catalyst is somewhat lower than that of the oxide reference (by 35 ℃ and 7 ℃ for fresh and aged catalysts, respectively). This downshift in peak temperature is evidently caused by the interaction between the oxide component and ZSM-5. In addition, Mn/(ALCE20+20ZSM-5)-F (1.23 µmol/g) has 40% more total NH3 desorption than Mn/ALCE20-F (0.87 µmol/g) due to the contribution from ZSM-5, but the total NH3 desorption quantities are comparable for Mn/ALCE20-HA and Mn/(ALCE20+20ZSM-5)-HA (0.82 µmol/g and 0.88 µmol/g, respectively). These results suggest that the most consequential acid site for the improved SCR activity and N2 selectivity on the composite catalyst is the medium-strength acid site created by the oxide-zeolite interaction (NH3 desorption peak around 300 ℃). This acid site is believed to facilitate NH3 activation for SCR catalysis at lower temperatures with minimized NH3 oxidation.
In order to understand how NH3 and NO interact with the catalysts in depth, in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) was used at elevated temperatures with either pre-adsorbed NH3 or NO + O2. Fig. S12a (Supporting information) shows the desorption spectra of all catalysts collected at six temperatures between 50 ℃ and 300 ℃ after a pre-adsorption of NH3 at 50 ℃, and Fig. 3e compares the spectra collected at 100 ℃ as a function of catalyst composition/condition. As shown in Fig. 3e, several bands remain on all four catalysts at 100 ℃, which belong to the coordinated ammonia (-NH2) bound to Lewis acid sites (at around 1596, 1267, 1236, 1050 and 1190 cm−1) and ionic NH4+ bound to Brønsted acid sites (at around 1691 and 1465 cm−1), respectively [53,54]. Comparing to Mn/ALCE20-F, significantly higher amounts of reactive -NH2 and NH4+ are found on Mn/(ALCE20+20ZSM-5)-F. The increased NH4+ bound to Brønsted acid sites due to the ZSM-5 component in the support, while the increase in -NH2 bound to the Lewis acid sites may be due to improved dispersion of the Mn species and/or increased portion of lower valence Mn species facilitated by the presence of ZSM-5 or due to the unsaturated Al sites in the zeolite itself. Interestingly, after hydrothermal aging the intensities of the -NH2 groups bound to Lewis acid sites and the ionic NH4+ groups bound to Brønsted acid sites remain the same on the zeolite-containing catalyst, demonstrating its high hydrothermal stability for NH3 adsorption and activation.
Fig. 3f shows the desorption spectra collected on these catalysts at 100 ℃ after pre-adsorption of NO + O2 at 50 ℃. Several obvious adsorption bands can be observed, corresponding to the gaseous NO2 (at around 1616 cm−1), bidentate nitrate (at around 1555 cm−1), monodentate nitrate (at around 1510 cm−1), monodentate nitrite (at around 1406 and 1310 cm−1), bidentate nitrite (1303 cm−1) and bridging nitrate (1255 cm−1) [55,56]. It is generally reported and believed that the adsorption and activation of NOx species always happen at the surface-labile O species bound to metal active sites among SCR catalysts [57]. On Mn/(ALCE20+20ZSM-5)-F higher intensity of monodentate nitrite (1310 cm−1), nitrates (1555 and 1510 cm−1) and gaseous NO2 (1616 cm−1) is observed relative to Mn/ALCE20-F. Moreover, though more bidentate nitrites (1303 cm−1) adsorption is detected on Mn/ALCE20-F, these bidentate nitrite species decrease significantly on Mn/ALCE20-HA. More nitrates, gaseous NO2 and more stable monodentate nitrites on Mn/(ALCE20+20ZSM-5) catalysts are likely responsible for its high NOx reduction activity and excellent hydrothermal stability. As shown in Fig. S12b (Supporting information), with raising temperature, the adsorption intensity of monodentate nitrate and monodentate nitrite groups decreases rapidly on Mn/(ALCE20+20ZSM-5) catalysts, while the adsorption intensity of NOx species on the Mn/(ALCE20+20ZSM-5)-HA is also stronger than that of Mn/ALCE20-HA. Therefore, these results show that more active Mn species are generated on the Mn/(ALCE20+20ZSM-5) catalyst, which results in more active adsorbed NOx species. After hydrothermal aging, these adsorbed species remain well on the Mn/(ALCE20+20ZSM-5)-HA catalyst, which demonstrates that active Mn species can be maintained after the hydrothermal aging process, thus promoting the continuous adsorption and activation of NOx.
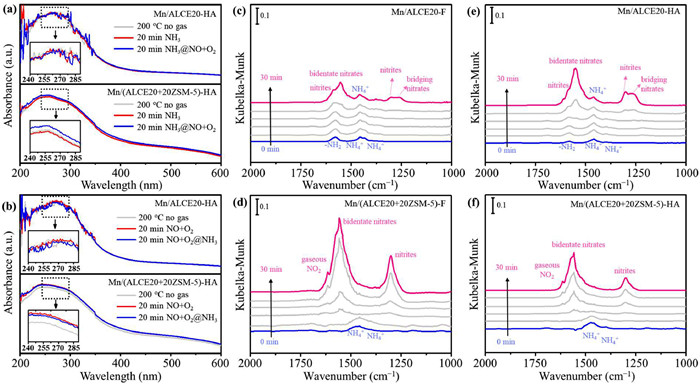
In order to study the structural changes of these catalysts during SCR reaction, we collected in situ UV–vis spectra after exposing the catalysts with various reacting gasses at 200 ℃. Different active Mn sites in the absence of any reactive gas were first measured by UV–vis spectra, the changes in peak position and intensity of spectra after the addition of the reactants would be the result of adsorption and activation of NH3 or NO + O2 at active Mn sites. As illustrated in Fig. 4a, after Mn/ALCE20-HA and Mn/(ALCE20+20ZSM-5)-HA are pre-adsorbed with 1000 ppm NH3 for 20 min at 200 ℃, no peak shift is detected in the spectra, suggesting that NH3 is primarily adsorbed on Brønsted acid sites not on active Mn sites. After injection of NO + O2, a band shift is observed on Mn/(ALCE20+20ZSM-5)-HA. This shift could be caused by two possible reactions with the Mn sites: One is related to NO + O2 titrating the adsorbed NH3 species thus restoring the initial Mn sites, and the other may be due to newly adsorbed NOx species on the active Mn sites. In contrast, the band shift is not obvious on Mn/ALCE20-HA, indicating its relatively low reaction rate. In addition, as shown in Fig. 4b that a band offset is observed on Mn/(ALCE20+20ZSM-5)-HA after pre-adsorption of NO + O2, suggesting that NO is adsorbed and activated on the catalyst. After introducing NH3, a slight band shift is observed, which indicates that NH3 quickly adsorbs on the acid sites and reacts directly with both the adsorbed NOx species and gaseous NO. For Mn/ALCE20-HA, the band shift caused by pre-adsorption of NO + O2 and subsequent NH3 injection is not obvious, illustrating the limited ability of Mn/ALCE20-HA in adsorption and activation of reactants. Combining Figs. 4a and b, it may be concluded that Mn/(ALCE20+20ZSM-5)-HA is benefited from the composite support that provides more acid sites and results in highly dispersed Mn species.
Figure 4
Figure 4.
UV–vis spectra of the transient reaction between pre-adsorbed 1000 ppm NH3 and 1000 ppm NO + 5 vol% O2 at 200 ℃ (a) and between pre-adsorbed 1000 ppm NO + 5 vol% O2 and 1000 ppm NH3 at 200 ℃ (b) over Mn/ALCE20-HA and Mn/(ALCE20+20ZSM-5)-HA catalysts. Inset showed the enlarged labeled section. In situ DRIFTS of the transient reaction between pre-adsorbed NH3 (with 1000 ppm NH3 for 30 min) and 1000 ppm NO + 5 vol% O2 at 180 ℃ as a function of time over (c) Mn/ALCE20-F, (d) Mn/(ALCE20+20ZSM-5)-F, (e) Mn/ALCE20-HA and (f) Mn/(ALCE20+20ZSM-5)-HA.
The types of adsorbed species, active intermediate species and reaction mechanism in SCR reaction were further probed by in situ DRIFTS. As illustrated in Figs. 4c–f, after NH3 adsorption (with 1000 ppm NH3 for 30 min) at 180 ℃, the vibration peaks of NH4+ (1457 and 1433 cm−1) and -NH2 (1578 cm−1) are observed on Mn/ALCE20-F and Mn/ALCE20-HA [58]. On Mn/(ALCE20+20ZSM-5)-F and Mn/(ALCE20+20ZSM-5)-HA, the NH4+ bands at 1457 and 1433 cm−1 are quickly enhanced right after the NH3 adsorption, indicating that more Brønsted acid sites are introduced to the composite catalyst by incorporating ZSM-5. With the introduction of NO + O2, the peaks assigned to the adsorbed NH3 species is impaired on both catalysts with variable degrees. At the same time, nitrites (1588 cm−1 and 1301 cm−1), bidentate nitrates (1550 cm−1), bridge nitrates (1253 cm−1) and gaseous NO2 (1617 cm−1) appeared [59]. The rate of accumulation of the adsorbed NO species is obviously faster on Mn/(ALCE20+20ZSM-5)-F than on Mn/ALCE20-F. In contrast to Mn/(ALCE20+20ZSM-5)-F, the adsorbed NH3 species on Mn/ALCE20-F begin to consume only after about 30 min, with a large part of NH4+ species (1457 cm−1) adsorbed on Brønsted acid sites still visible. Interestingly, gaseous NO2 band is observed on Mn/(ALCE20+20ZSM-5)-F in the later part of the experiment, which could be a product of NH4NO3 decomposition or NO oxidation. The adsorbed nitrates can combine with the NH4+ bound acid sites to form NH4NO3 intermediates, which can then react with gaseous NO to produce NO2 and NH4NO2; the latter product is unstable and can readily decompose to N2 and H2O. On the other side, NO oxidation to NO2 is a known pathway, especially when the adsorbed NH3 species is depleting from the surface. In any case, the presence of gaseous NO2 most definitely would accelerate the NOx reduction rate due to the fast SCR reaction and had been demonstrated (Fig. 1). The "Fast SCR" reaction, NH4NO3 + NO → NH4NO2 + NO2, was also reported to occur on other catalysts with zeolites. For example, Chen et al. found that zeolite X supported Mn-Ce catalyst could provide active sites for the reaction between NH4NO3 species and NO species to generate N2 and H2O following the L-H mechanism [45]; Zhu et al. observed that the NOx conversion of Cu-SSZ-39 was promoted under the fast SCR conditions, via the "NH4NO2 path" (NO forms nitrite species and then reacts with NH4+ species bound to Bronsted acid sites to form N2 and H2O) and "NH4NO3 path" (NO reacts with NH4NO3 to form NO2 and NH4NO2) [60]. In addition, more adsorbed NH3 species on the composite catalyst is able to quickly react with the nitrites species produced by the highly dispersed MnOx, resulting in increased N2 selectivity. This conclusion is in agreement with the results of in situ UV–vis spectra. The in situ DRIFTS tests of Mn/(ALCE20+20ZSM-5)-HA show the similar reaction phenomenon as Mn/(ALCE20+20ZSM-5)-F, indicating that the reaction mechanism remain the same after hydrothermal aging.
The transient reaction between pre-adsorbed NO + O2 and the gaseous NH3 stream was investigated by using the in situ DRIFTS on these catalysts as well. As shown in Fig. S13 (Supporting information), after pre-adsorption of NO + O2 at 180 ℃ for 30 min, gaseous NO2 (1617 cm−1), bidentate nitrates (1550 cm−1) and nitrites (1301 cm−1) are detected on Mn/(ALCE20+20ZSM-5)-F, and bidentate nitrates (1550 cm−1), nitrites (1590 cm−1) and bridge nitrates (1253 cm−1) are observed on Mn/ALCE20-F. After the injection of NH3, the -NH2 species (1542, 1510, 1280 and 1302 cm−1) and NH4+ species (1457 cm−1) are found to progressively increase with time on Mn/(ALCE20+20ZSM-5)-F and Mn/(ALCE20+20ZSM-5)-HA with a simultaneous decrease for nitrate species [61]. The consumption rate of nitrate species is noticeably faster on Mn/(ALCE20+20ZSM-5) than on Mn/ALCE20 regardless of aging condition. On the Mn/(ALCE20+20ZSM-5) catalysts, the NH4+ and -NH2 species grow to be the dominate species by 30 min compared to those on the Mn/ALCE20 catalysts. These results indicate that on the composite catalyst the pre-adsorbed nitrate species are more reactive (higher rate of disappearance) upon titration by gaseous NH3 at 180 ℃. After the nitrate species is completely consumed, the surface of the catalyst was dominated by adsorbed ammonia species (NH4+ and -NH2). The same process also takes place on the Mn/ALCE20 catalyst but at a slower rate. Evidently, the difference between these two catalyst compositions in the rate of titration (in situ SCR) is the result of different states of Mn species on these catalysts. An additional advantage of the composite catalyst seems to be its ability to store more NH3, resulting in a kinetic advantage for the SCR reaction.
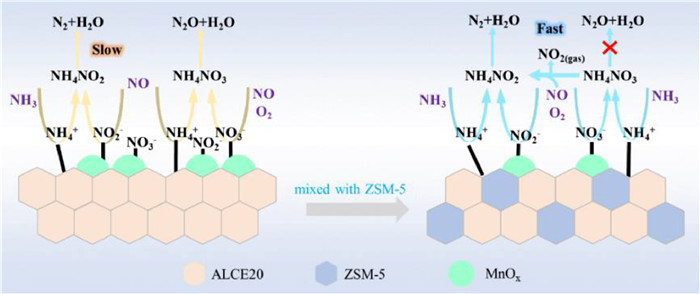
Our mechanistic understanding of the composite catalyst is schematically illustrated by Fig. 5. Due to the incorporation of zeolite, the composite catalyst contains more acidic sites, which offers a kinetic advantage to the oxide-alone catalyst. Benefiting from the oxide-zeolite interaction, the composite catalyst contains more lower valence Mn species, which leads to the formation of more active nitrates and nitrites. These absorbed nitrogen oxides species can quickly react with the adsorbed surface NH3 to form NH4NO3 and NH4NO2 intermediates. On the composite catalyst, the NH4NO3 intermediates are thought to preferentially react with gaseous NO to form NO2 and NH4NO2; the former enables the alleged "Fast SCR" reaction (NO + NO2 + 2NH3 → 2N2 + 3H2O) and the latter can readily decompose to N2 and H2O. This pathway reduces the probability for the decomposition of NH4NO3 to N2O and therefore improves the N2 selectivity. Although both NH4NO3 and NH4NO2 intermediates were found on Mn/ALCE20 catalysts (in situ DRIFTS results), no obvious reaction path leading its conversion to N2 could be found at 180 ℃. Its decomposition to mostly N2O either thermally or catalytically at higher temperatures is expected and has been confirmed (Fig. 1b). The oxide-zeolite composite catalyst appears to inhibit the excessive dehydrogenation of adsorbed NH3 species and thereby improves N2 selectivity. It is precisely because of these favorable factors that the composite catalyst exhibits good low-temperature activity and hydrothermal stability as well as higher N2 selectivity.
Figure 5
Figure 5.
Schematic representation of molecular pathways for enhanced N2 selectivity over Mn/(ALCE20+20ZSM-5).
In this work, we report a novel SCR catalyst that has a good low-temperature activity and N2 selectivity, excellent hydrothermal stability as well as faster response to NH3 injection and less NH3 slip. This catalyst was prepared by depositing MnOx on the mixture of CeO2/Al2O3 and ZSM-5. The oxide-zeolite composite along with the oxide reference were extensively characterized with various techniques both as fresh and aged catalysts. The NH3-TPD-MS and in situ DRIFTS results show that the composite catalyst contains more acidic sites. Due to the oxide-zeolite interaction, the MnOx species is found to be better dispersed on the composite catalyst and lower valence. In situ DRIFTS experiments indicate that more active nitrates and nitrites species are formed on the composite catalyst. These adsorbed nitrogen oxides could combine with NH4+ species bound to Brønsted acid sites to form NH4NO3 and the key NH4NO2 intermediates that lead to N2 and H2O formation. Importantly, the formation and intensity of these reactive intermediates on the composite catalyst do not show appreciable change after hydrothermal aging, which is key to its good hydrothermal stability. Therefore, this study provides a new method for applying manganese oxide based catalysts to low-temperature deNOx from diesel vehicle exhaust.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was in part supported by BASF Environmental Catalyst and Metal Solutions. The authors acknowledge the support of the National Natural Science Foundation of China (Nos. 21976117, 22125604 and 22276119), and the sponsor by "Chenguang Program" supported by Shanghai Education Development Foundation and Shanghai Municipal Education Commission (No. 21CGA48).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.109132.
Figure 1
NOx conversion (a) and N2O formation (b) as a function of temperature over Mn/ALCE20-HA, Mn/(ALCE20+20ZSM-5)-HA, VWTi-HA and Cu-CHA-HA. Response profiles (c) of NH3 and NOx on Cu-CHA-HA and Mn/(ALCE20+20ZSM-5)-HA during a transient SCR reaction at 150 ℃; Transient SCR performance (d) as a function of temperature on Cu-CHA-HA and Mn/(ALCE20+20ZSM-5)-HA. Prior to test (c) and (d), each catalyst was pretreated in air at 300 ℃ for 1 h. For test (a) and (b): [NO] = [NH3] = 500 ppm, [O2] = 10 vol%, [H2O] = 5 vol%, [CO2] = 5 vol%, N2 as balance, and WHSV = 200, 000 mL g−1 h−1. For test (c), upon the catalyst was cooled to 150 ℃ after the air pretreatment, a flow of 500 ppm NO + 500 ppm NH3 + 10 vol% O2 was introduced. For test (d), upon the catalyst was cooled to 50 ℃ after the air pretreatment, NH3 (500 ppm NH3, 10 vol% O2, N2 as balance) was injected for 1 h until saturation. The NH3-saturated catalyst was then heated to 350 ℃ at 5 ℃/min in a flow of 200 ppm NO + 200 ppm NH3 + 10 vol% O2.
Figure 2
HR-TEM images of Mn/(ALCE20+20ZSM-5)-HA (a) and Mn/ALCE20-HA (b). TEM images and the corresponding EDS element mappings of Al (c1, d1), O (c2, d2), Ce (c3, d3) and Mn (c4, d4) for Mn/(ALCE20+20ZSM-5)-HA (c) and Mn/ALCE20-HA (d). XRD patterns (e) and Raman spectra (f) of Mn/ALCE20-HA, Mn/ALCE20-F, Mn/(ALCE20+20ZSM-5)-HA and Mn/(ALCE20+20ZSM-5)-F.
Figure 3
UV–vis spectra (a) of Mn/ALCE20-HA, Mn/ALCE20-F, Mn/(ALCE20+20ZSM-5)-HA, and Mn/(ALCE20+20ZSM-5)-F. Mn 2p XPS (b) of Mn/(ALCE20+20ZSM-5)-HA and Mn/(ALCE20+20ZSM-5)-F. H2-TPR profiles (c) and FTIR spectra with pyridine adsorption (d) for Mn/ALCE20-HA, Mn/ALCE20-F, Mn/(ALCE20+20ZSM-5)-HA and Mn/(ALCE20+20ZSM-5)-F. For the FTIR experiment, pyridine adsorption was carried out at 50 ℃ for 1 h. (e) In situ DRIFTS of NH3 desorption. NH3 adsorption was carried out at 50 ℃ with 1000 ppm NH3 for 1 h. (f) In situ DRIFTS of NO + O2 desorption. NO + O2 adsorption was performed at 50 ℃ with 1000 ppm NO and 5 vol% O2 for 1 h. The desorption spectra of (e) and (f) were collected at 100 ℃ over Mn/ALCE20-HA, Mn/ALCE20-F, Mn/(ALCE20+20ZSM-5)-HA and Mn/(ALCE20+20ZSM-5)-F.
Figure 4
UV–vis spectra of the transient reaction between pre-adsorbed 1000 ppm NH3 and 1000 ppm NO + 5 vol% O2 at 200 ℃ (a) and between pre-adsorbed 1000 ppm NO + 5 vol% O2 and 1000 ppm NH3 at 200 ℃ (b) over Mn/ALCE20-HA and Mn/(ALCE20+20ZSM-5)-HA catalysts. Inset showed the enlarged labeled section. In situ DRIFTS of the transient reaction between pre-adsorbed NH3 (with 1000 ppm NH3 for 30 min) and 1000 ppm NO + 5 vol% O2 at 180 ℃ as a function of time over (c) Mn/ALCE20-F, (d) Mn/(ALCE20+20ZSM-5)-F, (e) Mn/ALCE20-HA and (f) Mn/(ALCE20+20ZSM-5)-HA.

 DownLoad:
DownLoad:

 下载:
下载:






