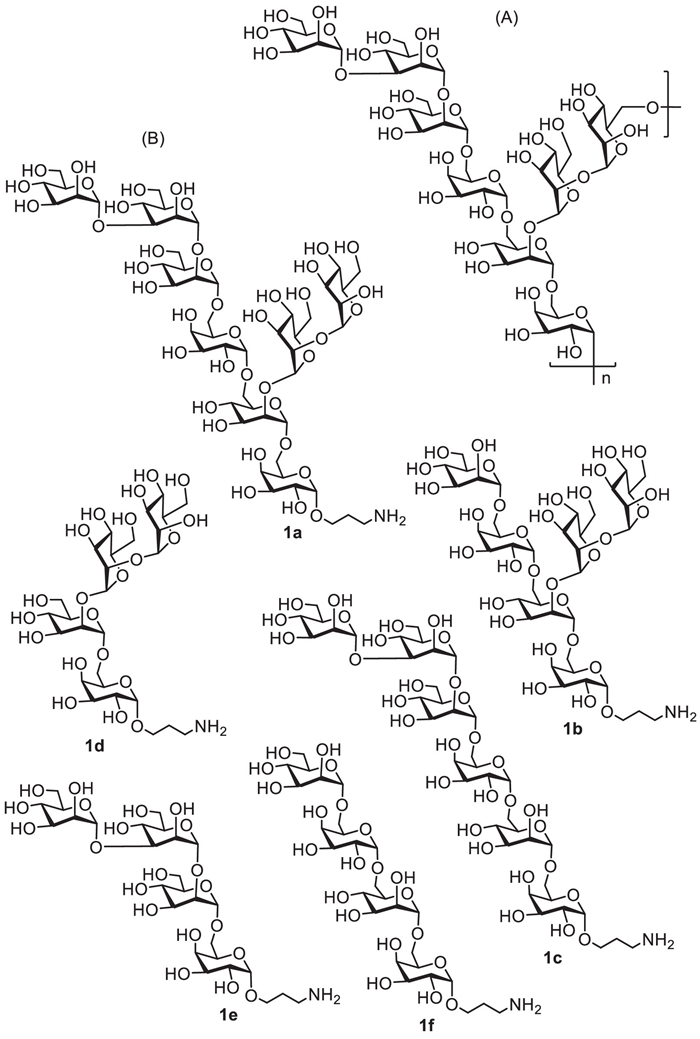
Figure 1.
The chemical structures of the octasaccharide repeating unit (A) of AC galactomannan and the target oligosaccharide derivatives 1a–1f (B) in this study.
Convergent synthesis and immunological study of oligosaccharide derivatives related to galactomannan from Antrodia cinnamomea
Shuying Li , Weiwei ZhuGe , Xuan Sun , Chongzhen Sun , Zhaojun Liu , Chenghe Xiong , Min Xiao , Guofeng Gu

Medicinal mushrooms have long been widely concerned because of their multiple pharmaceutical activities, including antibacterial, antiviral, antitumor, antioxidant, and immunomodulatory activities [1-3]. For example, Antrodia cinnamomea (AC), known as a medicinal fungus that is uniquely distributed in Taiwan region, has been attracted much attention owing to its extremely broad medicinal applications in treatment of various diseases, such as abdominal pain, drug poisoning, skin itching, hypertension, cancer and liver ailment, etc. [4, 5]. Polysaccharides in AC have been identified as one of the main pharmacologically active components and showed great potential as immunostimulants or adjuvants in immunotherapy and vaccination development [6]. In 2017, Wu and co-workers reported the isolation and purification of a cold-water soluble galactomannan from AC, and disclosed that it exhibited significant immunostimulatory ability on the phagocytosis and bactericidal activity of J774A.1 macrophages [5]. Further studies revealed that the isolated AC galactomannan could extremely elicit tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) secretion in J774A.1 macrophages and human dendritic cells speculatively through activating protein kinase C-α (PKC-α) and mitogen activated protein kinases (MAPK) signaling pathways after binding to the Toll-like receptor 4 (TLR4) on cell surface [6]. However, the underlying mechanism of its immunomodulation still remain poorly understood; therefore, the screening bioactive oligosaccharide epitope of AC galactomannan is worthwhile for exploration.
Structurally, as depicted in Fig. 1A, the AC galactomannan has been chemically elucidated as the following structure: {→6)-α-D-Manp-(1→2)-α-D-Manp-(1→2)-[α-D-Manp-(1→3)-α-D-Manp-(1→2)-α-D-Manp-(1→6)-α-D-Galp-(1→6)]-α-D-Manp-(1→6)-α-D-Galp-(1→}n [5, 6]. Its repeating unit is an octasaccharide that composed of a tetrasaccharide backbone chain and a tetrasaccharide side chain, in whose structure D-mannose and D-galactose residues were interconnected via α−1,2, α−1,3 and α−1,6 glycosidic bonds, respectively. Thus far, there are no reports on the synthesis and biological activity of the oligosaccharide fragments related to this AC galactomannan. Accordingly, we reported herein the first chemical synthesis of intact repeating unit, octasaccharide 1a, of AC galactomannan and its substructures, including two hexasaccharide fragments 1b and 1c, and three tetrasaccharide fragments 1d–1f, for in-depth structure-activity relationship immunological study (Fig. 1B). With the structurally defined AC oligosaccharides, we also preliminarily investigated their immunostimulatory activity on viability, phagocytosis and cytokines secretion in Raw264.7 cell line in vitro.
Considering that the target molecules 1a–1f were all even number in chain length, we planned to assembly them by the convergent [2 + 2], [2 + 2 + 2] or [2 + 2 + 2 + 2] glycosylation strategy and employ the acyl-mediated neighboring group participation effect (NGPE) [7] and the ether-assisted solvent effect [8] to efficiently control the dominating formation of α-mannosyl and α-galactosyl bonds. Accordingly, as shown in Scheme 1, retrosynthetic disconnection of 1a–1f issued in five disaccharides 2–6 as the key synthetic intermediates. Among them, thioglycosides 2, 3, and 4, which were utilized as glycosyl donors in subsequent oligosaccharide assembly, could be easily prepared from mannosyl imidate donor 7 [9], mannosyl acceptors 9 [10, 11], and 10 [12], and galactosyl acceptor 11 [13, 14], whereas disaccharides 5 and 6 could be readily assembled from mannosyl imidate donors 7 and 8 with galactosyl acceptors 11 and 12, and were then served as glycosyl acceptors later after selective removal of the acetyl (Ac) group at O-2 position or tert-butyldimethylsilyl (TBS) group at O-6 position of D-mannose residue. Additionally, selective deacetylation of disaccharide 4 would afford latent glycosyl acceptor used in construction of linear hexasaccharide 1c in our design. The detailed synthetic procedures for monosaccharide intermediates 7–12 were outlined Schemes S1 and S2 (Supporting information).
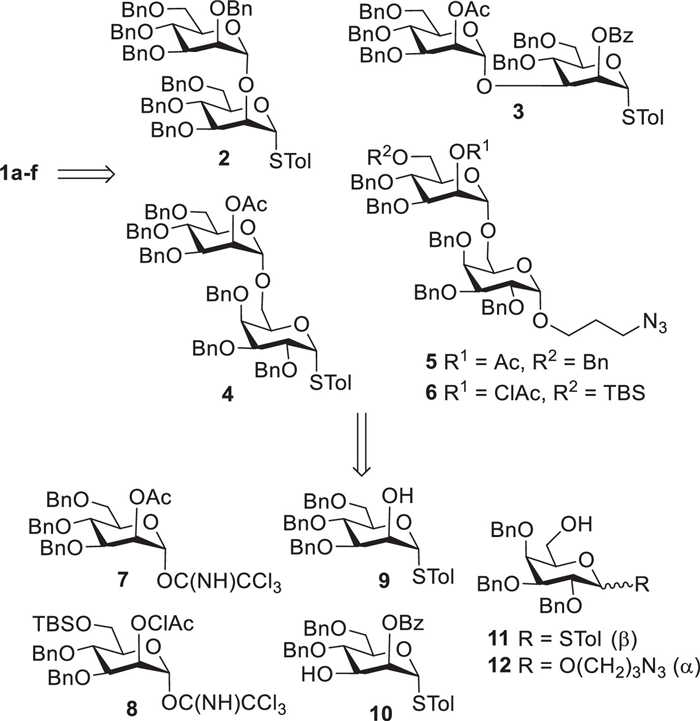
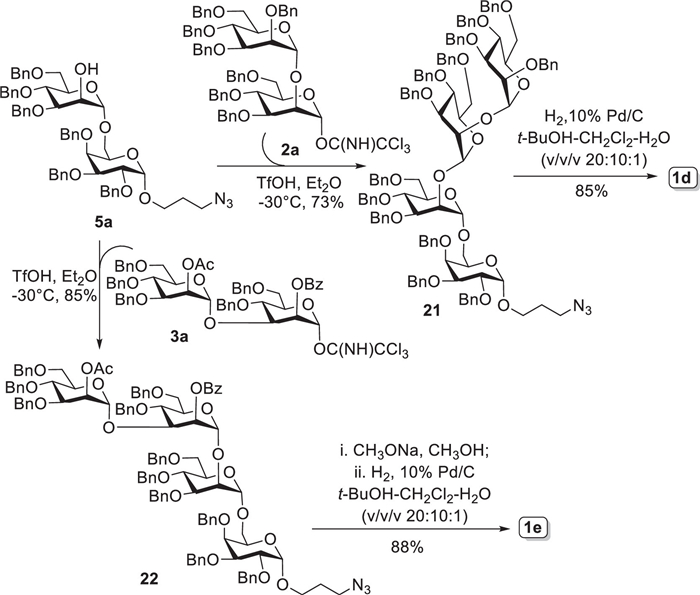
In our design, as outlined in Scheme S3 (Supporting information), mannosyl trichloroacetimidate 7 acted as a universal glycosyl donor to react with different glycosyl acceptors, i.e., 9–12, to produce the key disaccharide intermediates 2–5. For example, the coupling reaction between imidate 7 (1.1 equiv.) and 9 by the catalytic amount of trimethylsilyl triflate (TMSOTf) smoothly generated the desired disaccharide product followed by the conversion of protecting group (Ac→Bn) at mannosyl 2-O position, affording disaccharide 2 in overall 73% yield (3 steps). Thiomannoside 10 was glycosylated with 7 under the promotion of TMSOTf (0.1 equiv.) to yield disaccharide 3 in 87% yield. Likewise, condensation of thiogalactoside acceptor 11 and 7 with TMSOTf as catalyst reposefully furnished disaccharide 4 in excellent yield (92%). Galactoside acceptor 12 was reacted with 7 in activation of TMSOTf to produce disaccharide 5 in high yield of 87%. With the similar protocol established above, disaccharide 6 was smoothly prepared in a moderate yield of 65% from the glycosylation reaction of 12 with 2,6-orthogonally protected imidate donor 8. The new α-mannosyl bonds formed in disaccharides 2–6 were well guaranteed due to the acyl (Ac or ClAc) group-mediated NGPE. Moreover, disaccharyl thioglycosides 2–4 were further converted into more active trichloroacetimidate donors 2a–4a for purpose of efficient construction of large oligosaccharide chain later. This transformation reaction was achieved through the following two steps: (ⅰ) hydrolysis of the reducing thioglycoside with NIS-AgOTf co-catalysis in wet solvent [15], and (ⅱ) subsequent activation of the resultant hemiacetal with trichloroacetonitrile (Cl3CCN) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) [16]. Furthermore, deacetylation of 5 under Zempén condition gave disaccharide acceptor 5a in 98% yield, which was expected to subsequently assembly target tetrasaccharides 1d and 1e. Alternatively, selective cleavage of TBS group in 6 was carried out with BF3·Et2O in chloroform [17], successfully providing disaccharide acceptor 6a in excellent yield, which would be used for construction of target molecules 1a-1c and 1f in following study.
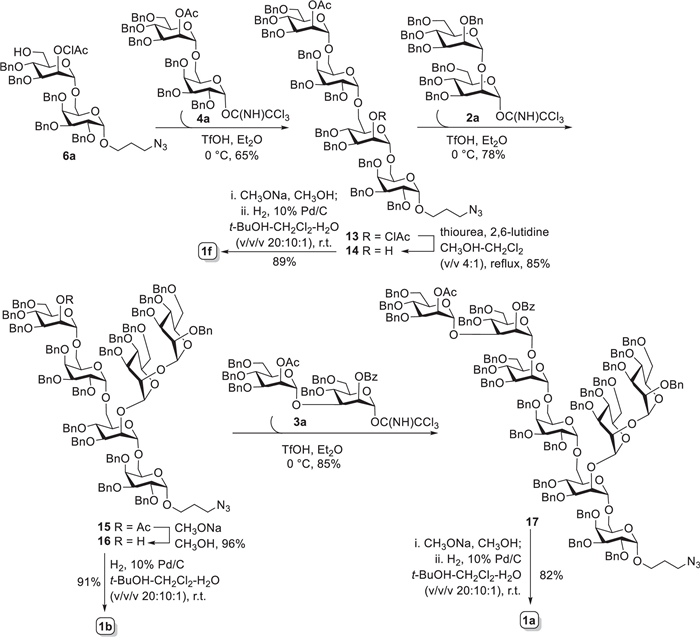
The octasaccharide 1a, as shown in Scheme 2, was designedly assembled via [2 + 2 + 2 + 2] strategy that required disaccharide building blocks 2a, 3a, 4a, and 6a involved. Given that the construction of α-galactosyl bond would be the most difficult task in the synthesis, we commenced with the synthesis of tetrasaccharide 13 first (Scheme 2). Unfortunately, direct glycosylation of 6a and thioglycoside 4 under different catalysts and reaction conditions (Table S1 in Supporting information) proceeded inefficiently, which merely generated the desired 13 in 10%−20% yields. However, when changing to disaccharyl trichloroacetimidate 4a as glycosyl donor, the above glycosylation reaction produced 13 with a satisfying yield. Up to 65% yield of 13 could be achieved with triflic acid (TfOH) as the promotor in Et2O solvent. The newly formed α-galactosyl bond in 13 was readily judged from the small coupling constant (3J1, 2 = 3.0 Hz) of H-1Gal signal at δ 5.10 ppm in 1H NMR spectrum. It should be noted here that β-isomeric product was also observed in above [2 + 2] reaction but inseparable from 13. Thereafter, thiourea-promoted cleavage of chloroacetyl group (ClAc) [18] on the mannosyl 2-O-postion furnished tetrasaccharide acceptor 14 in 85% yield, which was then glycosylated with imidate donor 2a (1.2 equiv.) in presence of catalytic amount of TfOH in Et2O successfully afforded hexasaccharide 15 in 78% yield. All α-glycosidic bonds in 15 were undoubtedly confirmed from the 1JC-1, H-1 coupling constants (> 169 Hz) between C-1s and H-1s in its 1H-coupled HSQC spectrum [19, 20]. Similarly, trace amount of inseparable β-isomer generated during the [4 + 2] glycosidation reaction. Next, treatment of 15 with CH3ONa in CH3OH smoothly provided 2-OH hexasaccharide acceptor 16 in excellent yield. The more active disaccharide imidate 3a was preferentially chosen here for further glycosylation. As expected, condensation of 16 and 3a (1.2 equiv.) under the promotion of TfOH accomplished perfectly the synthesis of fully protected octasaccharide 17 (85% yield). Again, the 1JC-1, H-1 coupling constants observed from 1H-coupled HSQC spectrum were all over 169 Hz, indicating the formation of α-glycosidic bonds in 17. Finally, deacylation of 17 with CH3ONa in CH3OH, followed by Pd-catalyzed hydrogenolytic debenzylation and azide reduction in t-BuOH−CH2Cl2−H2O co-solvents (v/v/v, 20:10:1) furnished target octasaccharide 1a in 82% yield, after purification by size-exclusion chromatography on Sephedex G-10 column. Furthermore, global deprotection of 16 and 14 by Zempén condition and/or hydrogenolysis with 10% Pd/C as the catalyst generated the desired hexasaccharide 1b (91%) and tetrasaccharide 1f (89%), respectively, which were further purified on Sephedex G-10 column.
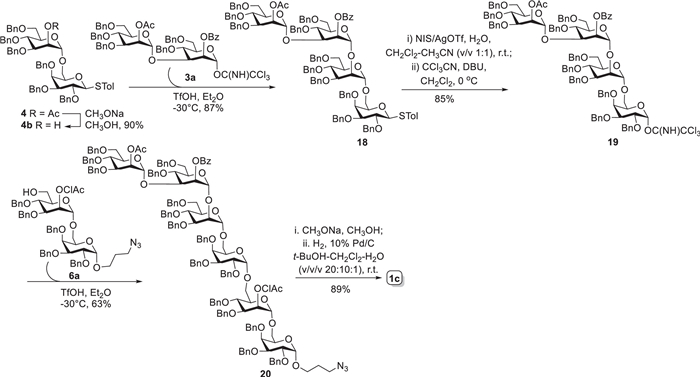
As outlined in Scheme 3, the linear hexasaccharide 1c was assembled via a convergent [2 + 2 + 2] strategy with disaccharide building blocks 3a, 4b and 6a. Glycosylation reaction of acceptor 4b, generated from Zempén deacetylation of thioglycoside 4, and imidate 3a using TfOH (0.1 equiv.) in Et2O gave tetrasaccharide 18 in high yield (87%). The transformation of tetrasaccharide thioglycoside 18 into trichloroacetimidate form 19 (85%) went smoothly in two steps, as described for preparation of disaccharide imidates 2a–4a. Then, the active imidate donor 19 reacted with acceptor 6a under the activation of TfOH to provide the fully protected hexasaccharide 20 in 63% yield. This [4 + 2] reaction was not well stereoselectively controlled by solvent effect of Et2O with production of inseparable β-isomer (ca. 30%). Likewise, the small coupling constant (3J1, 2 = 3.6 Hz) of H-1Gal signal at δ 5.09 ppm in 1H NMR confirmed the α-configuration of the newly formed galactoside. Eventually, the target hexasaccharide 1c was obtained by global deprotection in two steps, as described above for 1a, in overall 89% yield.
The synthesis of tetrasaccharides 1d and 1e using disaccharide 2a, 3a, and 5a was shown in Scheme 4. Acceptor 5a was coupled with imidate 2a to afford predominantly tetrasaccharide 21 (α/β = 10:1) in good yield of 73%, which was subjected to hydrogenolytic debenzylation and purification as described above to give desired tetrasaccharide 1d in 85% yield. The 1JC-1, H-1 values (> 169 Hz) between C-1s and H-1s calculated from 1H-coupled HSQC spectrum guaranteed the α-glycosidic bonds in 21. Alternatively, the reaction of 5a with imidate 3a promoted by TfOH in dry Et2O produced tetrasaccharide 22 in 85% yield. Ultimately, global deprotection of 22 using the aforementioned protocols for 1a and 1c afforded target tetrasaccharide 1e in 88% yield. Collectively, the target AC oligosaccharides 1a-1f together with all synthetic intermediates involved above were completely characterized by 1D- and 2D-NMR and MS spectra.
With enough amount of the synthesized AC oligosaccharides 1a–1f in hand, we thereby explored preliminarily their immunostimulatory activity toward Raw264.7 cells. First, the viability of Raw264.7 cells incubated with different concentration of 1a–1f was examined using CCK-8 assay [21]. As shown in Fig. 2A, except for tetrasaccharide 1f that showed none proliferative effect against macrophage cells, all other five oligosaccharides 1a–1e exhibited strong proliferation effect. The cell viability was significantly promoted with oligosaccharide concentrations under 25 µg/mL, whereas its growths were decreased markedly as oligosaccharide concentration exceeding this dose. Among tested AC oligosaccharides, tetrasaccharide 1d, hexasaccharide 1b, and octasaccharide 1a, which all contained the tetrasaccharide backbone structure, exhibited much stronger immunostimulatory activity towards Raw264.7 cell viability than those having the tetrasaccharide sidechain structure, i.e. tetrasaccharide 1e and hexasaccharide 1c. Most notably, tetrasaccharide 1d exerted the best proliferation ability at concentration of 25 µg/mL, indicating its great immunostimulatory capacity for further investigation.
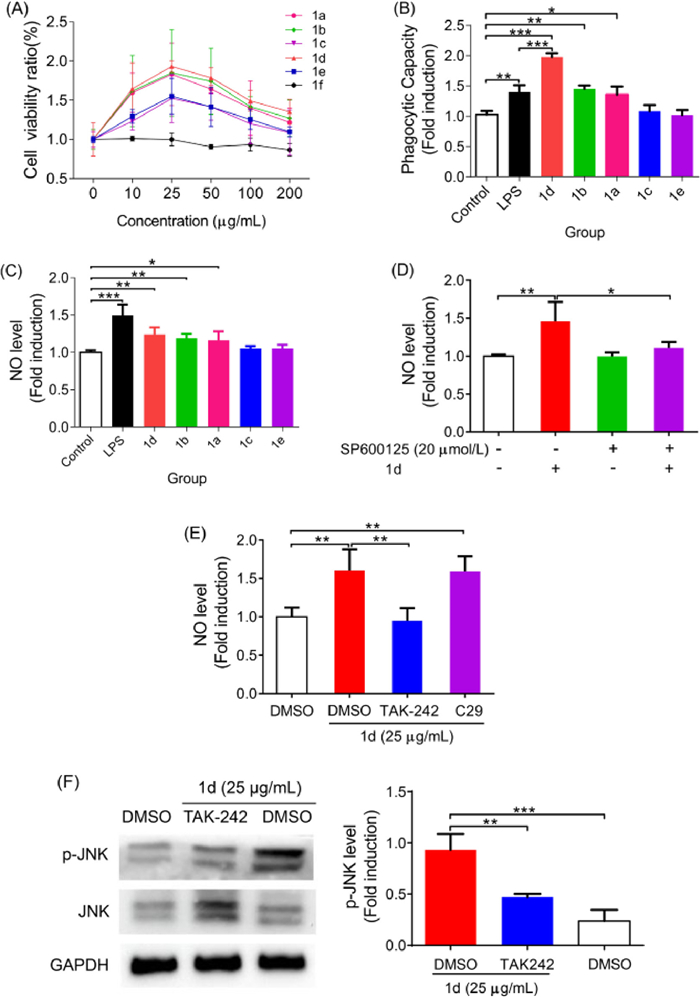
Next, the effect of 1a–1e on the phagocytic activity of Raw264.7 cells were characterized via measuring neutral red uptake assay [22]. These studies were carried out at the optimal concentration of 25 µg/mL of 1a–1e using lipopolysaccharide (LPS, 1.0 µg/mL), a well characterized macrophage stimulator, as the positive control. As depicted in Fig. 2B, compared with the blank group, the Raw264.7 cells treated with octasaccharide 1a, hexasaccharide 1b, or tetrasaccharide 1d exhibited significantly phagocytic ability to phagocytize neutral red, whereas those cells treated with hexasaccharide 1c or tetrasaccharide 1e did not show phagocytosis at all. Likewise, tetrasaccharide 1d exerted the best ability on influencing macrophage phagocytosis towards intake of neutral red. Additionally, tetrasaccharide 1d was also more effective than LPS in enhancing the phagocytic activity of macrophages, further revealing the excellent immunomodulatory role of 1d on Raw264.7 macrophages.
The effect of 1a–1e (25 µg/mL) on production of pro-inflammatory cytokines, including NO, TNF-α, IL-6, and IL-1β from Raw264.7 macrophages was further evaluated using LPS (1.0 µg/mL) as a positive control. Clearly, oligosaccharides 1a, 1b and 1d significantly stimulated the secretion of NO, TNF-α, and IL-6 in different degree at the tested concentration (Fig. 2C and Figs. S1A-C in Supporting information). Moreover, 1b and 1d could promote Raw264.7 macrophages to accumulate IL-1β production as compared with the blank control. However, 1c and 1e did not exhibit the stimulatory activity on the induction of NO, TNF-α, IL-6, and IL-1β levels (except for 1e on TNF-α). These findings, together with the above proliferation and phagocytosis results, have again demonstrated that AC oligosaccharides containing tetrasaccharide backbone structure, especially for 1d, have exerted the excellent immune-enhancing activity on Raw264.7 cells and could be used as a potential immunostimulatory agent.
As well known, macrophage activation is usually associated with mitogen-activated protein kinase (MAPK, including JNK, p38, and ERK1/2), nuclear factor-kappa B (NF-κB), and/or phosphatidylinositide 3-kinase (PI3K)/AKT signaling pathways [23]. Based on the observed significant immunostimulatory activity of tetrasaccharide 1d on macrophages, the potential signaling pathways activated by 1d were further probed. To this end, Raw264.7 cells were pretreated with SP6000125 (JNK inhibitor), SB203580 (p38 inhibitor), PD98059 (ERK1/2 inhibitor), BAY11–7082 (IKK inhibitor) or LY294002 (PI3K inhibitor) for 2 h and then co-treated with 1d (25 µg/mL) for 24 h. The secretion level of NO in Raw264.7 cells was determined by Griess assay [24]. As shown in Fig. 2D, the NO production induced by 1d was significantly decreased in macrophages that pretreated with SP600125 inhibitor. However, SB203580 (p38 inhibitor) and BAY11–7082 (IKK inhibitor) attenuated slightly the NO production on 1d-mediated macrophages (Figs. S2A and B in Supporting information). Furthermore, neither inhibition of ERK1/2 signaling pathway by PD98059 nor blockade of PI3K/AKT signaling pathway by LY294002 affected 1d-induced NO production (Figs. S2C and D in Supporting information). Our findings suggested that tetrasaccharide 1d might stimulate the NO secretion mainly through the activation of JNK signaling pathway. This speculation was further demonstrated by the results that the inhibition of JNK signaling pathway with SP600125 could dramatically suppressed the expression of other immune cytokines (IL-1β, IL-6, and TNF-α) promoted by 1d (Figs. S3A-C in Supporting information). To further confirm the effect of 1d on the activation of JNK signaling pathway, the phosphorylation level of JNK protein was measured by western blot analysis [25] after treating Raw264.7 cells with 10 or 25 µg/mL of 1d. As shown in Fig. S3D (Supporting information), the phosphorylation level of JNK protein in 1d-treated Raw264.7 cells were significantly enhanced as compared with the blank control. These results indicated that tetrasaccharide 1d might activate the JNK signaling pathway.
It has been well documented that functional glycan ligands could bind to Toll-like receptors (TLRs), such as TLR2 and TLR4, to further activate MAPK signaling pathway, leading to the secretion of various immune factors [26]. Accordingly, we explored the potential role of TLR2 and TLR4 in the activation of immune factors by tetrasaccharide 1d in Raw264.7 cells. As compared to the cells treated with 1d alone, the inhibition of TLR2 with C29 inhibitor (100 µmol/L) had no influence on the NO production triggered by 1d in cells, whereas the inhibition of TLR4 by TAK242 inhibitor (5 µmol/L) significantly reduced 1d-stimulated NO production in cells (Fig. 2E). Furthermore, the significant down-regulated expression of TNF-α, IL-6 and IL-1β by 1d has been observed in Raw264.7 cells that pretreated with TAK242 (Figs. S4A-C in Supporting information). In addition, the inhibition of TLR4 with TAK242 could suppress the phosphorylation of JNK protein induced by 1d as comparison to that treated with 1d alone (Fig. 2F). These results indicated that TLR4 might be the main receptor for tetrasaccharide 1d binding to activate macrophages via the JNK signaling pathway.
In summary, we described here the efficient synthesis of homogenous and structurally well-defined AC galactomannan tetra-, hexa- and octa-saccharides 1a–1f via highly convergent [2 + 2], [2 + 2 + 2] and [2 + 2 + 2 + 2] glycosylation strategy. In these syntheses, disaccharide trichloroacetimidates 2a–4a were proved to be more active glycosyl donors, as compared with their corresponding thioglycosides 2–4, to efficiently complete the synthesis of target molecules. Also, all α-glycosidic bonds in 1a–1f were stereoselectively controlled via the neighboring group participation effect (Ac, ClAc, or Bz group) and the solvent effect (Et2O solvent) and successfully achieved in satisfying yields. The preliminary immunostimulant effect of the synthesized oligosaccharides 1a–1f on Raw264.7 macrophages have disclosed that oligosaccharides carried with tetrasaccharide backbone structure of AC galactomannan polysaccharide, such as octasaccharide 1a, hexasaccharide 1b, and tetrasaccharide 1d, could significantly promote the proliferation, phagocytosis, and cytokine production of NO, TNF-α, IL-6 and IL-1β in macrophages. Most particularly, tetrasaccharide 1d exerted the best immunomodulatory activity towards Raw264.7 cells. Furthermore, the inhibition of TLR4 and JNK signaling pathway with TAK-242 and SP6000125 inhibitors, respectively, extremely reduced the 1d-induced secretion of pro-inflammation cytokines and, as well, the JNK phosphorylation. All these findings indicated that TLR4 receptor on the surface of macrophages might be the putative receptor for AC oligosaccharide binding first, which in turn activates intracellular JNK signaling pathway and thus induces cytokine production. Collectively, based on the fine structure and excellent immunomodulatory activity observed in this study, the synthetic tetrasaccharide 1d has been identified as a potential immunomodulator candidate to enhance immunity. Its immunostimulatory potential on nonspecific immune responses is undergoing and will be communicated in due course.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by grants from the National Key Research and Development Program of China (No. 2018YFA0902000) and the National Natural Science Foundation of China (No. 21877074).
Supplementary material associated with this article can be found, in the online version, at doi:
A. Bhambri, M. Srivastava, V.G. Mahale, et al., Front. Microbiol. 13 (2022) 837266. doi: 10.3389/fmicb.2022.837266
V. Bell, C.R.P.G. Silva, J. Guina, T.H. Fernandes, Front. Nutr. 9 (2022) 1050099. doi: 10.3389/fnut.2022.1050099
J. Xu, R. Shen, Z. Jiao, et al., Nutrients 14 (2022) 2622. doi: 10.3390/nu14132622
S.H. Tu, C.H. Wu, L.C. Chen, et al., Agric. Food Chem. 60 (2012) 3612–3618. doi: 10.1021/jf300221g
N. Perera, F.L. Yang, C.M. Chang, et al., Org. Lett. 19 (2017) 3486–3489. doi: 10.1021/acs.orglett.7b01468
N. Perera, F.L. Yang, Y.T. Lu, et al., Int. J. Biol. Sci. 14 (2018) 1378–1388. doi: 10.7150/ijbs.24564
C.S. Chao, C.Y. Lin, S. Mulani, W.C. Hung, K.K.T. Mong, Chem. Eur. J. 17 (2011) 12193–12202. doi: 10.1002/chem.201100732
A. Kafle, J. Liu, L. Cui, Can. J. Chem. 94 (2016) 894–901. doi: 10.1139/cjc-2016-0417
F. Yamazaki, S. Sato, T. Nukada, Y. Ito, T. Ogawa, Carbohydr. Res. 201 (1990) 31–50. doi: 10.1016/0008-6215(90)84223-H
K. Chayajarus, D.J. Chambers, M.J. Chughtai, A.J. Fairbanks, Org. Lett. 6 (2004) 3797–3800. doi: 10.1021/ol048427o
D. Wang, D.C. Xiong, X.S. Ye, Chin. Chem. Lett. 29 (2018) 1340–1342. doi: 10.1016/j.cclet.2017.12.014
K.K.T. Mong, K.S. Shiau, Y.H. Lin, K.C. Cheng, C.H. Lin, Org. Biomol. Chem. 13 (2015) 11550–11560. doi: 10.1039/C5OB01786F
C. Li, Y. Sun, J. Zhang, et al., Carbohydr. Res. 376 (2013) 15–23. doi: 10.1016/j.carres.2013.02.008
C.H. Wang, S.T. Li, T.L. Lin, et al., Angew. Chem. Int. Ed. 52 (2013) 9157–9161. doi: 10.1002/anie.201302540
D. Wang, W. Zhuge, Z. Guo, G. Gu, Carbohydr. Res. 442 (2017) 41–51. doi: 10.1016/j.carres.2017.03.004
R.R. Schmidt, J. Michel, Angew. Chem. Int. Ed. 19 (1980) 731–732. doi: 10.1002/anie.198007311
K. Ruda, J. Lindberg, P.J. Garegg, S. Oscarson, P. Konradsson, J. Am. Chem. Soc. 122 (2000) 11067–11072. doi: 10.1021/ja001515t
M. Bertolini, C.P.J. Glaudemans, Carbohydr. Res. 15 (1970) 263–270. doi: 10.1016/S0008-6215(00)88010-6
K. Bock, C. Pedersen, J. Chem. Soc., Perkin Trans. 2 (1974) 293–297.
J. Duus, C.H. Gotfredsen, K. Bock, Chem. Rev. 100 (2000) 4589–4614. doi: 10.1021/cr990302n
Y. Li, M. Liu, K. Yang, J. Tian, Chin. Herb. Med. 14 (2022) 254–262.
Q.M. Liu, S.S. Xu, L. Li, et al., Carbohydr. Polym. 165 (2017) 189–196. doi: 10.1016/j.carbpol.2017.02.032
Z. Liu, Z. Liu, L. Li, et al., Food Sci. Nutr. 10 (2022) 1093–1102. doi: 10.1002/fsn3.2735
H. Sun, J. Zhang, F. Chen, et al., Carbohydr. Polym. 121 (2015) 388–402. doi: 10.1016/j.carbpol.2014.12.023
Y. Chen, P. Li, Y. Peng, et al., Free Radic. Biol. Med. 172 (2021) 590–603. doi: 10.1016/j.freeradbiomed.2021.07.005
N.G. Geum, H.J. Eo, H.J. Kim, et al., J. Funct. Foods 73 (2020) 104139. doi: 10.1016/j.jff.2020.104139

Figure 1 The chemical structures of the octasaccharide repeating unit (A) of AC galactomannan and the target oligosaccharide derivatives 1a–1f (B) in this study.

Scheme 2 Synthesis of the octasaccharide 1a of the galactomannan intact repeating unit and its hexa- and tetra-saccharide fragments, 1b and 1f.

Figure 2 The effect of AC oligosaccharides 1a–1f on proliferation (A), phagocytosis (B) and NO secretion (C) of Raw264.7 macrophages. (D) The effect of JNK signaling pathway on 1d-mediated NO production in Raw264.7 cell lines. The effect of TLR4 on NO production (E) and JNK signaling pathway activation (F) in Raw264.7 cell line. Results are expressed as the means ± SD obtained from triplicate experiments. *P < 0.05, **P < 0.01, ***P < 0.001.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:


