yqlan@m.scnu.edu.cn (Y.-Q. Lan). 1 These authors contributed equally to this work.
Received Date:
24 April 2023 Accepted Date:
12 July 2023 Revised Date:
30 May 2023 Available Online:
15 May 2024
Abstract:
The fascinating chemical structure and broad application prospect of Keggin-type polyoxometalates (POMs) have attracted many chemists to explore and discover continuously. Unlike the traditional Keggin, larger metal atomic radius, higher metal coordinated numbers, lower metal valence states and other features allow the group IVB metal-based Keggin (IVB-Keggin) more space and unknown in terms of structure and performance. Herein, density functional theory (DFT) calculations were performed to explore the influences including cores, shells, caps, and terminal ligands, et al. on IVB-Keggin, and analyze the possibility of novel structure synthesis. From the perspective of multi-layer onion-like clusters, molecular energy level, host-guest interaction energy, surface charge and covalent bond polarity can be further adjusted to achieve the oriented design of functional IVB-Keggin. These insights are expected to provide theoretical support for experimental synthesis, opening a new perspective to understand the growth of Keggin.
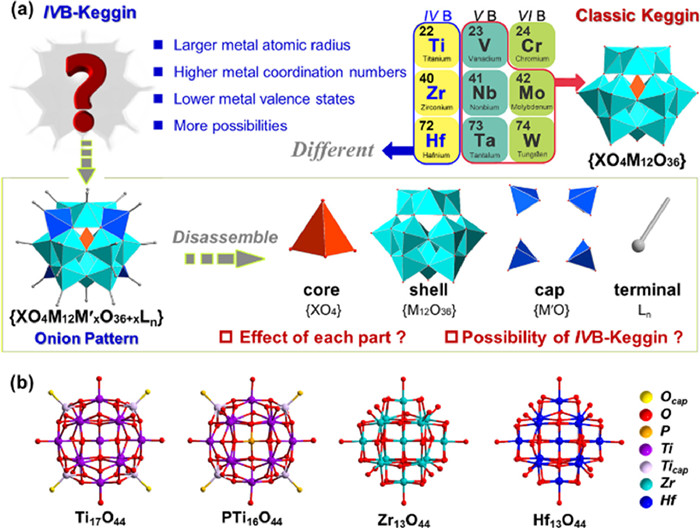
Keggin-type polyoxometalates (POMs) has been highlighted great attention due to the unique structure and remarkable properties, which have been widely reported to exhibit good application properties in materials, catalysts, and pharmaceuticals [1–7]. The archetypical Keggin structures were most constructed of highest oxidation state ions of the late transition metals (Fig. 1a), bridged by oxide ligands to generated the clusters, which the structural motif is the (XM12O40) (X = P, Si, Ge…, M = MoⅥ, WⅥ…) [8–11]. The classic Keggin POMs can be regarded as multi-layer onion-like structures. Such a Td symmetric anion cluster contains an inner layer of a central tetrahedron core {XO4} and a second {M12O36} shell connected by twelve octahedron {MO6} groups [12,13]. In contrast to these classic MoⅥ- and WⅥ-based Keggin architectures, the lower oxidation state of the early transition-metal ions attempts to balance the high electrostatic charges mainly by two approaches [14,15]. The first effective strategy is anchoring additional {MO} units as the caps on the four-square faces of the {M12O36} shell to achieve a great deal of Keggin family extensions and improve the metal adaptabilities [16–18]. Additionally, it has been reported to employ the alcohol-oxygen organic ligands with low negative charges to replace the terminal or bridge oxygen position to form neutral clusters to balance the instability factors brought by charges [19–23].
Figure 1
Figure 1.
Classic Keggin POMs and IVB-Keggin metal-oxo clusters. (a) Illustration of structure characteristics and research plan for IVB-Keggin. (b) The reported non-vacant IVB Keggin structures. H, C and guests are omitted for clarity.
In view of the fascinating structures and widespread application prospects of the POMs, the development and synthesis of highly stable and novel metal-oxo clusters has always attracted many synthetic chemists to carry out long-term researches [24–28]. The group IVB metals (Ti, Zr, Hf) and the component metals of traditional POMs (V, Nb, Ta, Mo, W) are adjacent and their intrinsic properties are more similar [29–34]. In contrast, IVB group transition metals have larger atomic radius and higher coordination number, which make their coordination modes more abundant and have more assembly possibilities [35–38]. Meanwhile, the lower valences of group IVB metals make advantageous to bond to various negatively charged ligands, which is expected to develop a series of highly stable IVB metal-oxo clusters [36,39–41,43]. To our knowledge, only four different IVB-Keggin have been reported, which are Ti17O44, PTi16O44, Zr13O44 and Hf13O44 clusters (Fig. 1b) [44–48]. For the Ti17O44 and PTi16O44, the central tetrahedron {PO4} and {TiO4} anions are regarded as the core, a {Ti12O36} cage as the shell and the four-square faces capped by four additional [TiO]2+ units. The alkoxy groups are coordinated with sixteen Ti atoms as terminal ligands of low negative charge. But different from these two titanium clusters, the center of the Zr13O44 is a cube and coordinates with the shell {Zr12O36} by (μ4–O)8 of the {ZrO8} core. Furthermore, Zr13O44 has no additional caps attached on the four-square faces. Similarly, alkoxy groups are coordinated with sixteen Zr to balance the charges and stabilize the cluster. And the Hf13O44 is isostructural with Zr13O44. Based on the above few cases of IVB-Keggin clusters, we speculate that the stabilization effect of {MO} caps play a crucial role in the synthesis of Ti-based Keggin. For the heavier metals Zr and Hf, the special {MO8} core is the key to the synthesis of Zr/Hf-based Keggin. Therefore, it is very fascinating and important to further understand the internal factors of the formation of IVB-Keggin and to predict the synthesis probability and function of the unknown clusters. Computational chemistry has shown remarkable advantages in modeling analysis, structural analysis, and structural prediction [49–51].
Herein, we have carried out the research on IVB-Keggin layer by layer according to the "core-shell-cap-terminal" onion pattern through DFT theory (Fig. 1a). The stability analysis and reasonable synthesis prediction of IVB-Keggin were conducted by the interaction energy of each part, the change of orbital energy level and the relaxed force constant of concerned M–O bonds. We found that the introduction of the cap site could enhance structural stability by lowering the HOMO orbital energy level of the shell. In addition, the tolerance of {M12O36} shell is different matching with different {XO4} or {XO8} core structures, which has the most important correlation with the stability of the crystal, and affects whether it could be synthesized in the experiment. And terminal organic ligands not only stabilize structures by balancing charges, but also enhances the strength of metal-oxygen bonds at adjacent sites. We hope that these explorations can help us understand the growth factors of IVB-Keggin better, to support the directed synthesis of more crystal more accurately in experiment.
All of the geometry optimizations were performed with (U)B3LYP functional with Gaussian 09 [52] program at 298.15 K and 1 atm in this work. The LanL2DZ basis sets [53] were applied for the Ti, Zr and Hf atoms, while 6–31G(d) basis sets were employed to other atoms. The SMD [54] solvent model with the parameters for methanol (MeOH) was used for the solution phase. Frequencies were evaluated at the same theoretical level to obtain the Hessian matrix and verify the stationary points to be equilibriums states. The Total density of states (TDOS) and partial density of states (PDOS) were performed for the crystal structure using Multiwfn [55] program with Becke broadening function and full width at half maximum (FWHM) of 0.3 eV. The 3D images of the molecular orbitals were printed by VMD 1.9.3 [56].
In order to investigate the interaction within the “core-shell-cap” structures, the rigid scan interaction energy changes were used as the reference to evaluate the stability of shells modified with different amounts of caps. The specific methods are as follows:
(1)
where and are the single-point energies of and {XO4} with the X–O bond length changes of the tetrahedron {XO4} cores at SMD(MeOH)/(U)B3LYP/[6–31G(d), LanL2DZ (Ti, Zr, Hf)] level; is the electron energy of the fully relaxed {M12O36-xcap} cage structure. The scanning range of the X–O bond length is set from the X–O triple bond length to X–O single bond length, unit in Å, step size in 0.01 Å/step (See the Supporting information details for specific scan parameters for different types {XO4} cores).
In this work to evaluate and predict the stability of IVB-Keggin clusters, the relaxed force constant was selected as the general criterion to investigate the covalent bond strength between O atom in {XO4} cores and the M metal in {M12O36-xcap} cages. The relaxed force constant is defined as the reciprocal of diagonal compliance matrix to replace the Hessian matrix, which avoids the dependence on the coordinate system to calculate force constants and ensure the correspondence between the bonds and the compliance constant. The relaxed force constant is obtained by employing the program Compliance developed by Grunenberg [57]. The related model structures are optimized without virtual frequency by the Gaussian 09 program for obtaining cartesian Hessen matrix to calculate the relaxed force constant.
Based on the Keggin “shell” {M12O36}, we considered anchoring x (x = 0, 2, 4, 6) number of identical metal cap {MO} at the four-square faces to construct caped-shell structures {M12O36-xcap} (M = Ti, Zr, Hf; x = 0, 2, 4, 6), respectively. Several classic metal oxygen tetrahedral centers are considered in {XO4} unit, including {AlO4}, {SiO4}, {PO4}, {GaO4}, {GeO4} and {AsO4}. In addition, {TiO4} {ZrO8} and {HfO8} core was also considered for {M12O36-xcap} cages referring to the reported IVB-Keggin mentioned above. Considering that most of Keggin clusters are metal oxides in the highest valence state, the metals and heteroatoms involved in this paper are defined as the highest oxidation state, and the corresponding valence state of oxygen is −2.
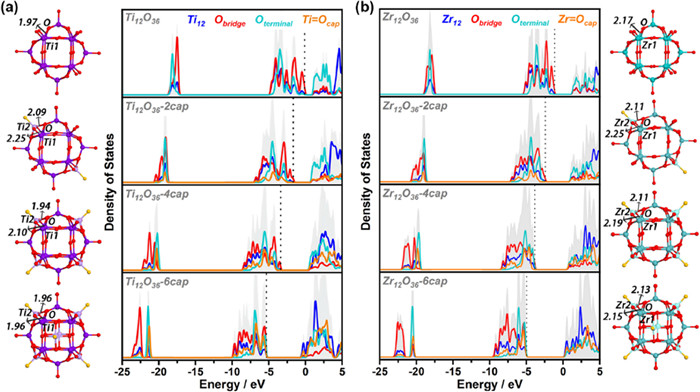
It is well known that caps are important to the stability of Keggin. Therefore, the influence of {MO} cap on {M12O36} shell has been evaluated. Twelve shell structures with the number of 0, 2, 4 and 6 {MO} caps were optimized including Ti12O36, Zr12O36, Hf12O36, etc., respectively, and the total and partial density of states (TDOS and PDOS) were calculated. As shown in Fig. 2a, for the Ti12O36, the unoccupied states are mainly located on the terminal oxygen moieties, while the occupied states are mainly located on the bridge oxygen moieties. As the number of {TiO} caps increase, the contribution of the unoccupied states of terminal oxygen decreases obviously (light green line near 0 eV) and the unoccupied states of shell titanium atoms increase (blue line near 0 eV). This indicates that the introduction of cap realizes the charge-transfer characteristics between shell metal and oxygen ligand regulates the activity of shell. Importantly, the addition of the {TiO} caps could gradually reduce the energy levels of the frontier molecular orbitals and the inner orbitals corresponding to the occupied states, enhancing the stability of shell structures. Moreover, comparing a series of the optimized shell structures, the Ti2–O bond length generated by the addition of {TiO} caps is shorter than the Ti1–O bond length contained in the {Ti12O36} shell, which means the Ti2–O bond energy seems to be more stable. Based on the above, the addition of {TiO} cap cannot only effectively regulate the stability of shell structures, but further adjust the activity of shell metals. Similar results have been found for cap-modified Zr12O36 and Hf12O36 series structures (Fig. 2b and Fig. S1 in Supporting information).
Figure 2
Figure 2.
The total and partial density of states (TDOS and PDOS). (a) The TDOS and PDOS of Ti12O36, Ti12O36–2cap, Ti12O36–4cap and Ti12O36–6cap. (b) The TDOS and PDOS of Zr12O36, Zr12O36–2cap, Zr12O36–4cap and Zr12O36–6cap. The corresponding optimized structure of the ball-and-stick model is also given. Purple, light purple, green, light green, red and yellow spheres represent shell-Ti, cap-Ti, shell-Zr, cap-Zr, bridge oxygen and terminal oxygen, respectively.
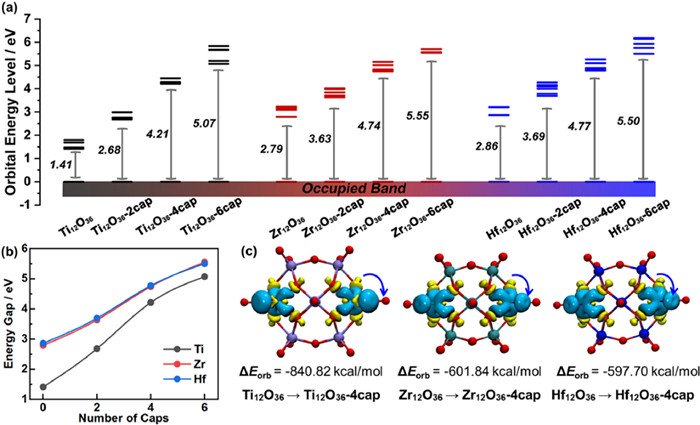
In order to intuitively analyze the IVB-Keggin of different metals, the band gaps of all the optimized {M12O36-xcap} anion cage structures were compared. As shown in Fig. 3a, series shells of Ti, Zr and Hf metal have similar band gap changes, which the band gap gradually increases with the increase of the number of {MO} caps. The results show that the stability of the cage can be effectively improved by adding metal caps to the four-square faces, and the band gap of the structure can be adjusted by controlling the amount of metal caps. In addition, the statistical graph of HOMO-LUMO gap shows that, for Zr and Hf series, the gap values of isomorphic structures are very close, and the corresponding trend of change is almost consistent (Fig. 3b). However, for the shell structures of Ti-GPCs series, the value of Ti12O36 without anchored {TiO} caps are only 1.41 eV, which is much smaller than that of Zr12O36 and Hf12O36 (Egap = 2.79 eV, Egap = 2.86 eV). As the number of {TiO} caps follow 0 → 2 → 4, there is a significant increase in HOMO-LUMO gap, gradually approaching the Zr/Hf gaps. This indicates that the structural stability of Ti12O36 is poor, importantly, the stability can be greatly improved by increasing the number of metal caps to four. In order to further understand the internal factors affecting its stability changes, the shell {M12O36} and the cap {MO} were divided into two moieties respectively, and natural orbitals for chemical valence (NOCV) analysis [58–60] was used to explore the orbital interactions among the fragments through the breakdown of the energy term ΔEorb by Multiwfn [55] program. As can be inferred from Fig. 3c, the main contribution in the overall charge transfer corresponds to the transition from O atoms of four-square faces to the {MO}cap. The corresponding contributions to the orbital interaction energy of Ti12O36–4cap shell amounts to ΔEorb = −840.82 kcal/mol. For comparison, the ΔEorb values of Zr2O36–4cap and Hf2O36–4cap are about 240 kcal/mol lower (ΔEorb = −601.84 kcal/mol and −597.70 kcal/mol, respectively). Based on the above results, the stabilization effect of {MO} caps on Ti-based Keggin are more significant.
Figure 3
Figure 3.
Molecular orbital level and fragment orbital analysis. (a) Frontier molecular orbital levels of {M12O36-xcap} (M = Ti, Zr, Hf; x = 0, 2, 4, 6). (b) HOMO-LUMO gap statistical graph of {M12O36-xcap}. (c) NOCV-deformation densities and associated stabilization energies form {M12O36} to {M12O36–4cap}. The charge flow takes place in the direction yellow → blue.
In addition to the cap affecting the stability of Keggin structure, the matching degree between the central core and the shell would directly determine whether the clusters be formed. Therefore, the interaction between the core and shell, and the compatibility among the core, shell and the cap were investigated. Unfortunately, in the process of complete relaxation structure optimization, the convergence of {Ti12O36@MO4} structure without metal cap modification still fails after many attempts (Fig. S2 in Supporting information). Based on above, the Ti12O36@MO4 structure cannot be obtained and the stabilities of series {M12O36-xcap@XO4} Keggin configurations cannot also be evaluated by fragmental orbital interaction analysis. Therefore, it is attempted to use the shell structure of {M12O36-xcap} obtained by relaxed optimization as the host, the core {MO4} (or {MO8}) as the guest, and the rigid scan of M−O bond length as the variable, to estimate the host-guest interaction energy by the interaction energy change (ΔEr).
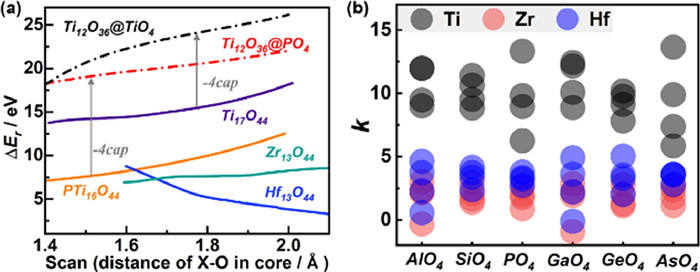
Firstly, to verify the reliability of this evaluation method, we evaluated the ΔEr based on four synthesized structures (Ti17O44, PTi16O44, Zr13O44, Hf13O44) as models. The shell structures Ti12O36–2cap, Zr12O36 and Hf12O36 were employed as the host, and the {TiO4}, {PO4}, {ZrO8} and {HfO8} were matched inside the cage as the guest. The host-guest ΔEr is scanned in the X–O bond length range of 1.3–2.1 A. As shown in Fig. 4a, the ΔEr of Zr13O44, and Hf13O44 are about 5 eV and the ΔEr of Zr12O36 and Hf12O36 are mainly distributed between 8 and 15 eV. For comparison, the ΔE increased by 10 eV when Ti2O36 as the host without four {TiO} caps. The stability results are consistent with experimental conclusions, further proving the reliability of the evaluation method. Notably, all the energy changes are positive, which means the interaction is dominated by the repulsive force between host and guest. This is because when the clusters are fully relaxed, the {M12O36-xcap} hosts will change correspondingly with the change of the guest {XO4} to stabilize the cluster. While the rigid scanning ignores this stabilization energy and overestimates the repulsion between host and guest. However, the relative ΔEr are also of reference value, which reflects the structural stability to a certain extent. And the slope of the curve obtained by this method can also be used as a reference for the tolerance of the {M12O36-xcap} shells.
Figure 4
Figure 4.
Rigid interaction analysis of core-shell. (a) Rigid interaction energy changes (ΔEr) of reported structures Ti17O44, PTi16O44, Zr13O44, Hf13O44 and uncapped Ti12O36@TiO4, Ti12O36@PO4. (b) The statistical graph of slope k of the interaction energy change curve of {M12O36-xcap@XO4} IVB-Keggin structures.
Based on this, we further calculated 72 numbers of ΔEr curves for the {M12O36–6cap@XO4} IVB-Keggin clusters with linear combination of 16 shell hosts and 6 hetero-core guests (Fig. S3 in Supporting information), and obtained the corresponding slope values through linear fitting. In Fig. 4b, the analysis shows the slope ranges of Zr/Hf-based Keggin almost overlap (0–5), and the slope ranges of Ti-based Keggin (6–15) are significantly higher than those of Zr/Hf-based Keggin. This indicates that Zr- and Hf-based Keggin have similar structure and stability, while Ti-based Keggin have worse stability, and {Ti12O36} shell has worse tolerance. The reported Zr/Hf metal-oxo clusters also tend to be more stable [44,46].
Based on the above, the cap’s impact on the shell, as well as the kernel and shell matching have been estimated in general. However, the size effect of {MO4} is not well considered due to the overestimation of the mutual exclusion energy in the interaction energy variation. It is well known that size effect is one of the important factors affecting crystal synthesis. Therefore, in order to better predict the possibility of new IVB-Keggin clusters, we use the relaxed force constant (RFC) as another reference standard for evaluation [57]. It can not only consider the structural relaxation, but also convert the molecular vibration frequency into an index to evaluate the strength of different bond, which is beneficial as a unified reference for clusters containing different kinds of cores and shells.
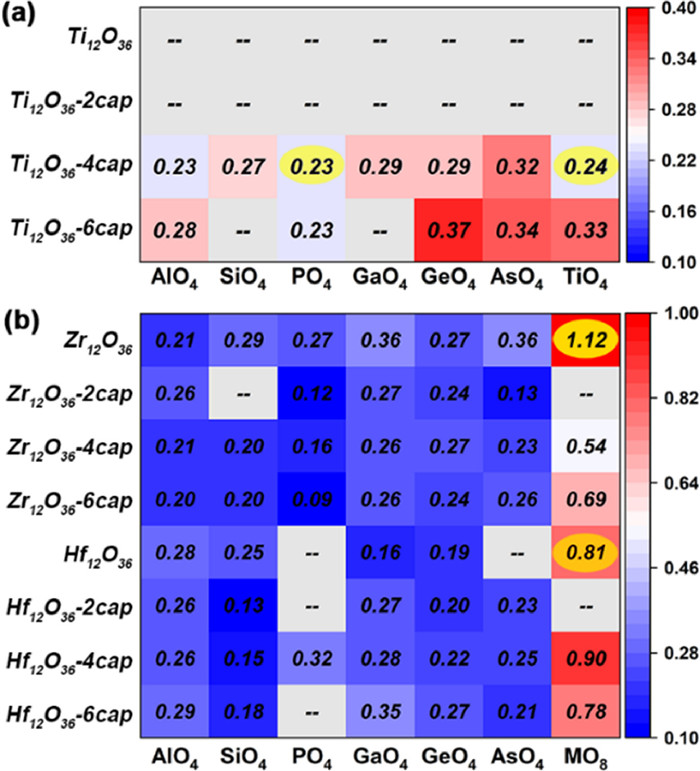
We optimized 84 {M12O36-xcap@XO4/8} IVB-Keggin structures at SMD(MeOH)/(U)B3LYP/[6–31G(d), LanL2DZ (Ti, Zr, Hf)] level. The corresponding RFCs were calculated using the Compliance program. As shown in Fig. 5a, the series structures of {Ti12O36@XO4} and {Ti12O36–2cap@XO4} fail to converge without obtaining relaxation structure after many attempts in optimization process. In addition, the RFCs of Ti12O36–6cap@SiO4 and Ti12O36–6cap@GaO4 cannot be obtained because of the virtual frequency. For most of the {Ti12O36–4cap@XO4} and {Ti12O36–6cap@XO4}, the values of RCF are mainly distributed in the range of 0.2–0.4. The overall level is above or equal to the RCF of Ti17O44 and PTi16O44, which exists the possibility of being successfully synthesized. For the Zr/Hf-based Keggin structures (Fig. 5b), more of structures with virtual frequencies and the low RFC are distributed in the region of SiO4 and PO4, possibly because the radius of Si and P is small, leading to a host-guest mismatch. The RFC of other heteroatomic tetrahedrons as cores also range from 0.2 to 0.4, similar to Ti-based Keggin. However, when the isometallic cube {MO8} as the core, the values of RCF increased by 5–10 times. And the RCF of the synthesized Zr13O44 and Hf13O44 are 1.12 and 0.81, respectively. This means that it is more difficult for Zr and Hf to form {MO4} tetrahedron-centered heterometal-oxo clusters, but easier to form {MO8} cube-centered homometal-oxo clusters.
Figure 5
Figure 5.
Relaxed force constant (RFC) of {M12O36-xcap@XO4} Keggin-type structures. (a) RFC of Ti-based Keggin structures. (b) RFC of Zr/Hf-based Keggin structures. The constant value increases from blue to red. A few structures with virtual frequency and relaxed optimization failure, the corresponding relaxed force constant cannot be calculated and marked with gray.
According to the report, Hf13O44–OMe structure has ultra-high stability against strong acids and bases [45]. The organic terminal methoxy ligand remained intact at 1 mol/L of HCl and NaOH environment. Therefore, the organic ligand of IVB-Keggin plays an important role in stabilizing the Keggin anions besides the reason of balancing charges. Therefore, we have carried out further study on electronic properties of terminal ligands. The geometry optimization of neutral Hf13O44–OMe has been performed at SMD, (MeOH)/(U)B3LYP/[6–31G(d), LanL2DZ] levels. The associated bond lengths obtained after optimization are in good agreement with the experimental value of crystal data (Table S1 in Supporting information). Firstly, the superposition diagram of Hf13O44 and Hf13O44–OMe shows that the Hf13O44–OMe with terminal -OMe group exits the obvious shrinkage (Fig. 6a). And the bond lengths of Hf1−Oa, Hf2−Oa and Hf2−Oc are shortened accordingly, the corresponding bond strength increases further. The natural population analysis (NPA) indicates that negative charges are uniformly distributed within the whole cluster without -OMe groups, resulting in negative charge of shell Hf (NPA(Hf2) ≈ −0.22). When the terminal -OMe is introduced, the -OMe parts will share a certain negative charge inside the cluster, and the charge of Hf2 rises to 2.14 correspondingly, while the charge of terminal O atom decreases to −0.85. Additionally, the electron localization function (ELF) [61] analysis shows that the electrons of the O atom of the bridged oxygen position were significantly localized with the presence of -OMe ligands (Fig. 6b). The electron density of bridged oxygen is located in the direction of Hf···Ob, enhancing the strength of polar covalent bond Hf−Ob, and the stability of Hf13O44–OMe.
Figure 6
Figure 6.
Comparative analysis of Hf13O44 and Hf13O44–OMe. (a) Geometry changes and average NPA charges of Hf13O44 and Hf13O44–OMe. The Oa, Ob, Oc, Hf1 and Hf2 represent cubic oxygen, bridge oxygen, terminal oxygen, core hafnium and shell hafnium atoms, respectively. The methyl (-Me) part is omitted for clarity. Average distances of M–O bond are given in Å, the corresponding standard deviations marked in parentheses. (b) Color-filled map of electron localization function (ELF). The value increases from blue to red. The blue, red, and white spheres represent the Hf, O and C atoms, respectively, and the H atom is omitted for clarity.
In summary, a series of systematic DFT calculations were performed to investigate the role of each core-shell-cap-terminal components of the IVB-Keggin clusters and to predict the possibility of novel structure synthesis. The {MO}caps could enhance structural stability by increasing the HOMO-LUMO gap of the shell. Terminal organic ligands not only stabilize structures by balancing charges, but enhance the strength of metal-oxygen bonds, and the overall stability of IVB-Keggin. Moreover, for Ti-based Keggin, the appropriate number of {MO} caps can enhance the tolerance for {MO4} tetrahedron-core, while the heavier Zr/Hf metals are more favorable to generate homometal-oxo clusters with {MO8} cube as the core. These perspectives might stimulate new perspectives for understand the growth factors of IVB-Keggin better, and provide theoretical support for experimental synthesis.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (NSFC, Nos. 22225109, 22071109, 92061101, 22173016 and 22301084) and the China Postdoctoral Science Foundation (No. 2023M741232).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.108805.
Figure 1
Classic Keggin POMs and IVB-Keggin metal-oxo clusters. (a) Illustration of structure characteristics and research plan for IVB-Keggin. (b) The reported non-vacant IVB Keggin structures. H, C and guests are omitted for clarity.
Figure 2
The total and partial density of states (TDOS and PDOS). (a) The TDOS and PDOS of Ti12O36, Ti12O36–2cap, Ti12O36–4cap and Ti12O36–6cap. (b) The TDOS and PDOS of Zr12O36, Zr12O36–2cap, Zr12O36–4cap and Zr12O36–6cap. The corresponding optimized structure of the ball-and-stick model is also given. Purple, light purple, green, light green, red and yellow spheres represent shell-Ti, cap-Ti, shell-Zr, cap-Zr, bridge oxygen and terminal oxygen, respectively.
Figure 3
Molecular orbital level and fragment orbital analysis. (a) Frontier molecular orbital levels of {M12O36-xcap} (M = Ti, Zr, Hf; x = 0, 2, 4, 6). (b) HOMO-LUMO gap statistical graph of {M12O36-xcap}. (c) NOCV-deformation densities and associated stabilization energies form {M12O36} to {M12O36–4cap}. The charge flow takes place in the direction yellow → blue.
Figure 4
Rigid interaction analysis of core-shell. (a) Rigid interaction energy changes (ΔEr) of reported structures Ti17O44, PTi16O44, Zr13O44, Hf13O44 and uncapped Ti12O36@TiO4, Ti12O36@PO4. (b) The statistical graph of slope k of the interaction energy change curve of {M12O36-xcap@XO4} IVB-Keggin structures.
Figure 5
Relaxed force constant (RFC) of {M12O36-xcap@XO4} Keggin-type structures. (a) RFC of Ti-based Keggin structures. (b) RFC of Zr/Hf-based Keggin structures. The constant value increases from blue to red. A few structures with virtual frequency and relaxed optimization failure, the corresponding relaxed force constant cannot be calculated and marked with gray.
Figure 6
Comparative analysis of Hf13O44 and Hf13O44–OMe. (a) Geometry changes and average NPA charges of Hf13O44 and Hf13O44–OMe. The Oa, Ob, Oc, Hf1 and Hf2 represent cubic oxygen, bridge oxygen, terminal oxygen, core hafnium and shell hafnium atoms, respectively. The methyl (-Me) part is omitted for clarity. Average distances of M–O bond are given in Å, the corresponding standard deviations marked in parentheses. (b) Color-filled map of electron localization function (ELF). The value increases from blue to red. The blue, red, and white spheres represent the Hf, O and C atoms, respectively, and the H atom is omitted for clarity.

 DownLoad:
DownLoad:






 下载:
下载:







