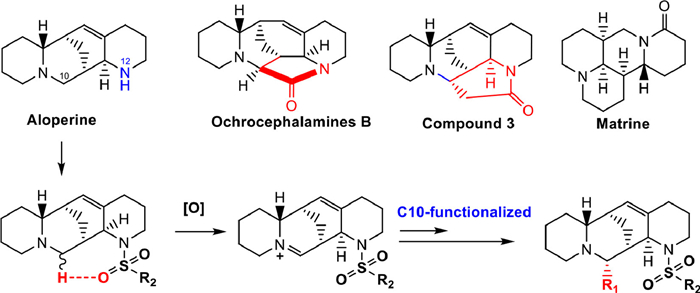
Figure 1.
Structures of the representative alkaloids and construction of target compounds.
Chemical construction and anti-HCoV-OC43 evaluation of novel 10,12-disubstituted aloperine derivatives as dual cofactor inhibitors of TMPRSS2 and SR-B1
Yulong Shi , Fenbei Chen , Mengyuan Wu , Xin Zhang , Runze Meng , Kun Wang , Yan Wang , Yuheng Mei , Qionglu Duan , Yinghong Li , Rongmei Gao , Yuhuan Li , Hongbin Deng , Jiandong Jiang , Yanxiang Wang , Danqing Song

The emergence and spread of fast mutating severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variants including BF.7, BQ.1 and XBB have greatly affected the human health and economy worldwide [1–3]. Antiviral agents acting on host components have advantages of broad-spectrum character and reducing the occurrence of drug resistance, therefore being less vulnerable to viral mutation [4–7]. They also have the potential to be used in combination with the current anti-SARS-CoV-2 drugs to combat the mutation of the SARS-CoV-2 [8].
Natural quinolizidine alkaloids possessing unique chemical scaffolds and druglike features, such as aloperine, ochrocephalamines B and matrine (Fig. 1), exhibited multiple pharmacological activities by multi-target mechanisms [9–12], especially against a variety of different viruses. We have demonstrated endocyclic ring displayed various inhibitory effects on hepatitis C virus (HCV), Ebola virus (EBOV) and Marburg virus (MARV) through the host mechanism of action [13–15]. Additionally, mono-substituted aloperine derivatives have exhibited anti-influenza activity and anti-HIV activity by targeting viral target nucleoprotein [16]. More recently, we found that 12N-p-chlorophenylethyl aloperine showed the broad-spectrum anti-coronavirus effects including SARS-CoV-2, SARS-CoV and Middle East respiratory syndrome coronavirus (MERS-CoV) through blocking viral entry process [17], with a good safety profile. However, the direct target proteins of aloperine derivatives against coronaviruses remain unknown. Also, the chemical modifications have only been focused on the 12N-substituents of aloperine, which greatly limits the structural diversification of this kind of compounds. Therefore, it is very meaningful to construct new synthetic approaches to enrich the structural diversities and biological activities of the quinolizidine alkaloids.
The structure of ochrocephalamines B suggests that aloperine may form a new carbon-carbon bond at the C-10 by activating the α-C–H with the introduction of a hydrogen bond receptor at the 12 N atom [18]. Based on this strategy, the introduction of 12N-substituted sulfonyl in aloperine led to the high activation on the 10-α-C–H bond adjacent to tertiary amine as shown in Fig. 1, and thus 10-iminium ion aloperine derivatives as the key intermediates were subtly formed through selective oxidation [19]. As expected, the novel scaffold 10,12-cycloaloperine 3 with a new seven-membered ring was successfully constructed via nucleophilic substitution and ammonolysis. Meanwhile, the key iminium ion intermediates were respectively reacted with Grignard or zinc nucleophiles to acquire various 10,12-disubstituted aloperine derivatives as a new family of compounds. In the present study, 31 novel 10,12-disubstituted aloperine derivatives were prepared and evaluated for their antiviral activities against human coronavirus OC43 (HCoV-OC43) [20], as well as analyzed their structure–activity relationship (SAR). Furthermore, the safety profiling and identification of direct target proteins of the representative compound using chemoproteomic techniques were carried out.
As shown in Scheme 1, upon the treatments of commercially available starting material aloperine using toluenesulfonyl group and oxidizing agent I2, the cyclic tertiary amine could be transformed to the key intermediate 1a with an isolable iminium ion. The iminium was treated with zinc and methyl bromoacetate to introduce a methyl acetate group (2). Then, the sulfonyl was removed by sodium naphthalene ammonolysis to obtain 10,12-cycloaloperine 3. The key intermediates 1a–e aloperine derivatives were reacted with different Grignard or zinc reagents to obtain target compounds of 4–6 series via a nucleophilic addition. This protocol features mild reaction conditions, stereoselectivity and scalability, representing a robust and practical method for broadening the structural diversity of the quinolizidine alkaloids. In addition, an active photoaffinity aloperine probe 10 was designed and prepared through a four-step procedure, including 12N-sulfonyl substitution, ester reduction, hydroxyl amination and functional group coupling reaction. The absolute configuration of target compounds was further confirmed by comparing the calculated and experimental ECD spectra, confirming the newly constructed chiral center at the C10 position having an S configuration.

In view of the similarity between human β-coronavirus and SARS-CoV-2, the HCoV-OC43 in H460 cells model was used for screening and evaluation of the target compounds against HCoV-OC43. The structures and anti-HCoV-OC43 activities of all the target compounds were shown in Table 1, taking ribavirin (RBV) as a positive control. First, compared with RBV, newly constructed compound 3 had no activity against HCoV-OC43. Then, the toluenesulfonyl was retained on the 12N-position, and various groups were respectively introduced on the 10-C position, by which 18 new 10,12-disubstitued aloperine derivatives (4a–r) were obtained and evaluated. Among them, the small alkyl groups were helpful for enhancing activity, and all of them (4a–j) displayed moderate antiviral activities with half maximal inhibitory concentration (IC50) values between 10.7 and 38.5 µmol/L. Compound 4h bearing 10-cyclopropyl had an ideal antiviral activity with an IC50 value of 12.84 µmol/L and an selectivity index (SI) value of 6.2, comparable to that of RBV. Among the other four compounds 4k–n possessing phenyl, p-methoxyphenyl, 4-chlorophenyl and naphthyl, only 4l showed comparable activity with an IC50 value of 12.84 µmol/L. All the analogues (4o–r) with other functional groups like α-substitution ester and 1,3-dioxan-5-ethyl gave decreased activities. Then, different substituted benzyls on the 10-C atom (5a–i) were generated and evaluated. Only compound 5c gave an IC50 value of 12.84 µmol/L and an SI value of 6.2, similar to that of RBV.
 DownLoad:
CSV
DownLoad:
CSV

|
Then, considering the feasibility of di-substituted modification and the yield of reaction, the phenyl was retained at the C-10 position to explore the effect on C-12 modification. Different benzenesulfonyl groups, including 4-(trifluoromethoxy)benzene sulfonyl, naphthosulfonyl, thiophene-2-sulfonyl and 1-methyl-1H-pyrrole-3-ulfonyl, were respectively introduced to produce compounds 6a–d. Compound 6b showed a potent activity with an IC50 value of 5.14 µmol/L, along with an SI value of 9.0, much better than that of RBV. Thus, compound 6b with the best antiviral activity was selected as the representative compound for further investigation.
To assess the safety profile of representative compound 6b, an acute toxicity experiment was performed in Kunming mice. Compound 6b was orally administrated in a single-dosing to the mice at doses of 100, 200, 400 or 800 mg/kg. All animal experiment protocols were approved by the Animal Care and Use Committee at NIFDC, Beijing, China. Animal experiments were approved by the Ethics Committee of the Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China (approval number IBM20221030D1). After monitoring the mice for 7 days, no significant changes in survival status and body weight were observed. As shown in Fig. S1 (Supporting information), there are no obvious damages in hepatic and renal function at all dosages, including aspartate transaminase (AST), alanine transaminase (ALT), urea and serum creatinine (Cre). The median lethal dose value of 6b was over 800 mg/kg, indicating a good safety profile.
To understand which stage of the viral lifecycle was blocked by 6b, we conducted a time-of-addition assay in H460 cells at different time points, before or after virus infection. As shown in Fig. 2A, compound 6b exerted strong inhibitory effects on HCoV-OC43 at 2 h post infection, indicating that it interfered with the viral entry stage through targeting host cell components, basically consistent with the effects observed with compound 12N-p-chlorophenylethyl aloperine [17]. To further determine the exact antiviral effect of compound 6b during the viral entry stage, viral attachment assay and entry assay were conducted. As shown in Fig. 2B, compound 6b exhibited a potent antiviral activity on viral entry with a half maximal effective concentration (EC50) value of 3.7 ± 1.3 µmol/L. And it also showed inhibitory activity on viral attachment with an EC50 value of 14.0 ± 3.7 µmol/L (Fig. 2C). These results further suggested that 6b mainly acted on the viral entry and attachment stage, consistent with the results of the time-of-addition assay. Thus, the capture of direct target proteins of aloperine derivatives against HCoV-OC43 could be carried out in host cells.
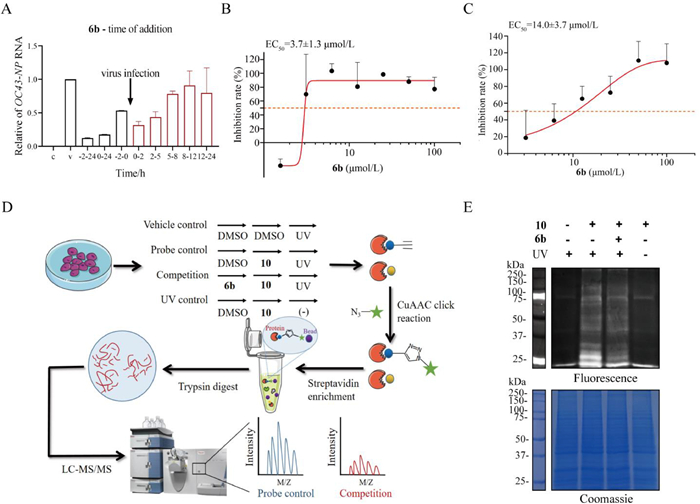
In addition, given the same mechanism of action of the mono- or di-substituted aloperine derivatives, the simple 12N-monosubstituted aloperine was selected as a pharmacophore moiety to construct the active aloperine probes. Also since there is no covalent warhead in aloperine structure, a photoaffinity probe 10 with diazirine and alkyne functional groups was designed and prepared according to our total SAR analysis. As expected, the photoaffinity probe 10 exhibited a comparable potency to that of 6b with IC50 value of 5.14 µmol/L and SI of 6.2, and thus was used as the active photoaffinity probe for capturing direct targets of this kind of compounds using activity-based protein profiling (ABPP) technique (Fig. 2D).
Based on targeting host components against HCoV-OC43 of compound 6b, the host H460 cells were used for fishing direct target proteins using ABPP assay. As shown in Fig. 2D, a total of four sets of experiments were conducted, including competition group and three control groups (vehicle control, probe control and ultraviolet control). The probe-labeled proteomes were conjugated with Cy3-azide or biotin-azide via a click reaction for further fluorescence imaging or biotin/streptavidin enrichment. Fluorescence imaging showed that with 6b pre-treatment, the fluorescence intensity of competitive inhibition group (bands at 25–37 kDa and 50–100 kDa) was significantly weakened compared to that of probe 10 treatment group, indicating a good specificity of the probe (Fig. 2E). The proteomes enriched by streptavidin magnetic beads were analyzed through liquid chromatography tandem mass spectrometry (LC-MS/MS), and proteins with fold change ≥1.5 (both probe/competition group and probe/vehicle control group) were first selected. According to the mechanism of action of aloperine compounds, the proteins related to virus/host interaction, including the transmembrane protease serine 2 (TMPRSS2), scavenger receptor class B member 1 (SR-B1), cathepsin B (Cat B) and transmembrane protein 106B (TMEM106B), were chosen as candidate targets for further validation (Table S1 in Supporting information).
The four candidate target proteins were further verified via pull down assay respectively. As displayed in Fig. 3A, both TMPRSS2 and SR-B1 were successfully identified by their specific antibody, indicating a direct interaction between 6b and these two proteins. Then, cellular thermal shift experiments (CESTA) were carried out in H460 cells to further verify their direct interaction. The thermal stability of TMPRSS2 and SR-B1 was significantly increased in the presence of 6b (50 µmol/L) under the temperature ranging from 40 ℃ to 70 ℃, which also supported possible direct interactions between 6b and the two proteins (Figs. 3B and C and Fig. S2 in Supporting information). Furthermore, surface plasmon resonance (SPR) assay was performed using recombinant proteins to analyze the affinities between 6b and these proteins, as well as the contribution of each potential target. The analyte 6b exhibited a similar dose-independent interaction with TMPRSS2 and SR-B1 (Figs. 3D and E), and a moderate binding affinity with the Kd values of 9.17 and 11.54 µmol/L, respectively. Molecular docking of TMPRSS2 and SR-B1 with 6b was then conducted using Discovery Studio 4.5 software, docking scores were respectively 107 and 108, basically consistent with their affinity forces. As depicted in Figs. 3F and G, the interaction pattern showed that 6b could not bind to the catalytic center of TMPRSS2, but to the hydrophobic pocket outside the active center. Tyr 431 was responsible for the interaction of 6b and SR-B1 through Pi-Pi T-shaped with naphthyl group, and phenyl interacted with Val 440 by Pi-alkyl interaction.

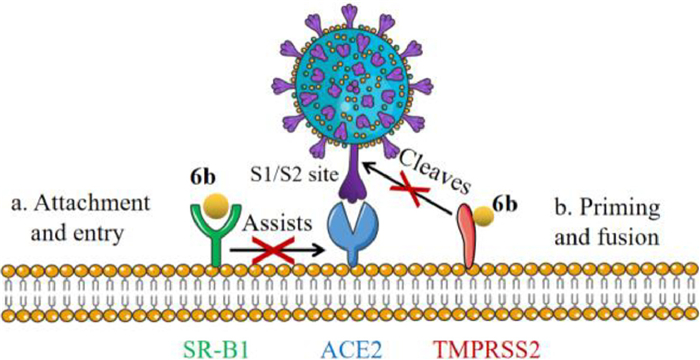
TMPRSS2 plays a critical role in the viral membrane fusion of SARS-CoV-2 [21], thus the inhibitory activity of 6b against TMPRSS2 was evaluated. As shown in Fig. 3H, 6b significantly inhibited the activity of TMPRSS2 with an IC50 value of 1.79 µmol/L, by which the viral fusion and entry were blocked by 6b. These results suggested that compound 6b could deactivate the TMPRSS2 function by binding the hydrophobic pocket adjacent to the active center and changing the protein conformation, rather than directly binding to catalytic center, to suppressing the viral entry and infection.
Meanwhile, SR-B1 is a high-density lipoprotein (HDL) receptor mediating the lipid exchange between cells and bound HDL particles [22], which is also known as a cell receptor of many viruses such as HCV and dengue virus [23,24]. It is recently reported that SR-B1 facilitates the entry of SARS-CoV-2 via angiotensin-converting enzyme 2 (ACE2) in the presence of HDL, which can be strongly inhibited by SR-B1 inhibitors, suggesting that SR-B1 may be a potential therapeutic target against SARS-CoV-2 as an entry cofactor [25,26]. Our results demonstrated that 6b might directly bind to SR-B1 to assist in preventing viral S spite protein from binding to host ACE2, thereby affecting viral attachment and infection. Since SR-B1 is closely related to the transport of HDL, the effect on lipid metabolism of 6b while inhibiting virus entry is actively under investigation.
In order to further verify the contributions of each target proteins, TMPRSS2 and SR-B1 knock-down model was constructed through siRNA interference. As depicted in Fig. 3I and Fig. S3 (Supporting information), no significant difference was found on relative OC43-RdRp RNA level between DMSO and 6b treatment group, when TMPRSS2 or SR-B1 was knocked down. This result further verified the possible interactions between 6b and TMPRSS2/SR-B1.
We subtly developed a robust and practical method for broadening the structural diversity of the quinolizidine alkaloids, with the characteristics of stereoselectivity and scalability. Through selective oxidation on the 10-α-C–H bond induced by sulfonyl and subsequent nucleophilic substitution, 31 new 10,12-disubstituted aloperine derivatives were constructed and evaluated for their antiviral effect against HCoV-OC43. Compound 6b showed the most potent activities and blocked the viral entry stage through a host mechanism of action. Chemoproteomic techniques revealed that compound 6b directly targets both TMPRSS2 and SR-B1 proteins, which act as host cofactors for the virus to bind ACE2 and fuse with the membrane, thereby inhibiting viral entry and infectivity, as displayed in Fig. 4. Interestingly, 6b could deactivate TMPRSS2 function by directly binding to the hydrophobic pocket adjacent to the active center, rather than catalytic center, thereby blocking viral membrane fusion. Our study developed a new class of aloperine scaffold and demonstrated that aloperines act as TMPRSS2 and SR-B1 dual inhibitors to block the HCoV-OC43 entry process, providing the key scientific data for the development of antiviral candidates against human β-coronavirus including SARS-CoV-2 of this kind.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the National Natural Science Foundation of China (No. 81974494) and CAMS Innovation Fund for Medical Sciences (No. 2021-I2M-1–070).
Supplementary material associated with this article can be found, in the online version, at doi:
P. Wang, M.S. Nair, L. Liu, et al., Nature 593 (2021) 130–135. doi: 10.1038/s41586-021-03398-2
D.R. Feikin, M.M. Higdon, L.J. Abu-Raddad, et al., Lancet 399 (2022) 924–944. doi: 10.1016/S0140-6736(22)00152-0
C.H.T. Kwong, J. Mu, S. Li, et al., Chin. Chem. Lett. 32 (2021) 3019–3022. doi: 10.1016/j.cclet.2021.04.008
X. Liu, S. Huuskonen, T. Laitinen, et al., Mol. Syst. Biol. 17 (2021) e10396. doi: 10.15252/msb.202110396
S.M. Heaton, Clin. Transl. Immunol. 8 (2019) e1067. doi: 10.1002/cti2.1067
D.E. Gordon, G.M. Jang, M. Bouhaddou, et al., Nature 583 (2020) 459–468. doi: 10.1038/s41586-020-2286-9
T. Huang, L. Sun, D. Kang, et al., Adv. Exp. Med. Biol. 1322 (2021) 219–260. doi: 10.1007/978-981-16-0267-2_9
S. Xiu, A. Dick, H. Ju, et al., J. Med. Chem. 63 (2020) 12256–12274. doi: 10.1021/acs.jmedchem.0c00502
X. Yuan, J.S. Jiang, Y.A. Yang, et al., Chin. Chem. Lett. 33 (2022) 2923–2927. doi: 10.1016/j.cclet.2021.10.085
R. Wang, X. Deng, Q. Gao, et al., J. Ethnopharmacol. 248 (2020) 112172. doi: 10.1016/j.jep.2019.112172
Y. Li, G. Wang, J. Liu, et al., Eur. J. Med. Chem. 188 (2020) 111972. doi: 10.1016/j.ejmech.2019.111972
X. Yuan, Z.Y. Li, Z.M. Feng, et al., Chin. Chem. Lett. 32 (2021) 4058–4062. doi: 10.1016/j.cclet.2021.04.022
X. Zhang, X.Q. Lv, S. Tang, et al., Eur. J. Med. Chem. 143 (2018) 1053–1065. doi: 10.1016/j.ejmech.2017.12.002
X. Zhang, Q. Liu, N. Zhang, et al., Eur. J. Med. Chem. 149 (2018) 45–55. doi: 10.2298/pac1801045z
X. Zhang, Q. Liu, Q. Li, et al., Acta Pharm. Sin. B 8 (2018) 629–638. doi: 10.2147/ott.s152063
Z. Dang, L. Zhu, L. Xie, et al., Curr. Med. Chem. 28 (2021) 4995–5003. doi: 10.2174/0929867328666201229121802
K. Wang, J.J. Wu, Z. Xin, et al., Bioorg. Chem. 115 (2021) 105196. doi: 10.1016/j.bioorg.2021.105196
K.S. Zhou, P. Yi, T. Yang, et al., Org. Lett. 21 (2019) 5051–5054. doi: 10.1021/acs.orglett.9b01643
T. Maruyama, S. Suga, J.I. Yoshida, Tetrahedron 62 (2006) 6519–6525. doi: 10.1016/j.tet.2006.03.114
N. Keshavarz Valian, B. Pourakbari, K. Asna Ashari, et al., J. Med. Virol. 94 (2022) 1450–1456. doi: 10.1002/jmv.27460
M. Hoffmann, H. Kleine-Weber, S. Schroeder, et al., Cell 181 (2020) e278.
W.J. Shen, S. Azhar, F.B. Kraemer, Annu. Rev. Physiol. 80 (2018) 95–116. doi: 10.1146/annurev-physiol-021317-121550
M. Lavie, S. Sarrazin, R. Montserret, et al., J. Virol. 88 (2014) 10584–10597. doi: 10.1128/JVI.01402-14
A.C. Alcalá, J.L. Maravillas, D. Meza, et al., J. Virol. 96 (2022) e0166421. doi: 10.1128/jvi.01664-21
C. Wei, L. Wan, Q. Yan, et al., Nat. Metab. 2 (2020) 1391–1400. doi: 10.1038/s42255-020-00324-0
G.E.G. Kluck, J.A. Yoo, E.H. Sakarya, et al., Int. J. Mol. Sci. 22 (2021) 10182. doi: 10.3390/ijms221910182

Figure 1 Structures of the representative alkaloids and construction of target compounds.

Scheme 1 The synthesis scheme for the aloperine derivatives. Reaction conditions: (a) K2CO3, N-protecting-reagent, I2, DCM, r.t.; Ag2O, HCl. (b) Zinc powder, methyl bromoacetate (1.2 equiv.), THF, 100 ℃. (c) Grignard reagents, THF. Or zinc powder, THF, 100 ℃. (d) LiAlH4, THF, 0 ℃. (e) PPh3, DEAD, phthalimide, THF, 0℃–r.t. (f) N2H4.H2O, EtOH, reflux. (g) K2CO3, 3-(But-3-yn-1-yl)−3-(2-iodoethyl)−3H-diazirine, CH3CN, 80 ℃. (h) Sodium naphthalene, THF, –20 ℃.

Figure 2 Time of addition assays and target fishing of 6b by ABPP. (A) The time-of-addition experiment. Compound 6b was added at different time points, before or after virus infection. (B). The viral entry assays. Compound 6b solutions of different concentrations were added 2 h after virus infection. (C) The viral attachment assays. Compound 6b solutions of different concentrations and HCoV-OC43 (multiplicity of infection (MOI) = 1) were added at the same time. (D) Overall workflow of the ABPP experiment to identify the direct target proteins of 6b. (E) Labeling of proteins under different conditions.

Figure 3 Validation of the candidate proteins. (A) Pull down assays of TMPRSS2 and SR-B1. (B, C) CETSA assays of TMPRSS2 and SR-B1. (D, E) SPR assays performed on TMRPSS2 (PDB ID: 7MEQ) and SR-B1 (PDB ID: 5KTF). (F, G) 2D modes and solid surface map of TMPRSS2 and SR-B1 interaction pockets with compound 6b. Ligand is colored by element type (N, blue; O, red; C, green; S, yellow), key bonds are indicated by dashed lines between the atoms involved, and the colors of key bonds and residues are shown according to the interaction mode. (H) Inhibitory activity of 6b on TMPRSS2. (I) Constructions of TMPRSS2 and SR-B1 knock-down model, and relative OC43-RdRp RNA level determination.

Figure 4 Cartoon diagram of compound 6b as a dual-target inhibitor against human β-coronavirus.
Table 1. Anti-HCoV-OC43 activity and cytotoxicity of all the target aloperine derivatives.
|
|
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们