Hubei Key Laboratory of Natural Medicinal Chemistry and Resource Evaluation, School of Pharmacy, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, China
Received Date:
24 May 2023 Accepted Date:
05 July 2023 Revised Date:
03 July 2023 Available Online:
15 May 2024
Abstract:
We reported the characterization of a novel brassicicene diterpene biosynthetic gene cluster, which contains a unique α-ketoglutarate-dependent dioxygenase (αKGD) enzyme, AbnI. Our findings revealed that AbnI demonstrates remarkable substrate promiscuity and is capable of activating multiple sites on both 5–8–5 and 5–9–5 brassicicene skeletons, resulting in skeleton modifications and an unexpected ring system rearrangement. These results suggested the potential utility of AbnI as an enzymatic tool for terpene CH functionalization. In addition, the catalytic mechanism of AbnI and its potential ecological implications were discussed.
Microbial metabolites offer a valuable source of natural products with diverse and complex structures [1,2]. Enzymatic modifications, such as C—H functionalization and backbone rearrangement, are encoded within biosynthetic gene clusters (BGCs) that are conserved across strains [3]. However, some strains have acquired unique genes during evolution, enabling them to carry out unexpected structural modifications [4]. Comparative analysis of homologous gene clusters can identify novel enzymes [5–7], expanding our understanding of biosynthetic pathways and offering potential opportunities for enzyme catalysis and synthetic biology design.
Our research focuses on brassicicenes, which are a group of diterpenes that have been discovered in Alternaria brassicicola, a fungus that causes dark leaf spots in Brassica plants [8]. The ring system of some brassicicenes, such as brassicicene A (Fig. S1 in Supporting information), is fused 5–8–5 and is similar to other diterpenes such as fusicoccin A and cotylenin A (Fig. S1) [9], which demonstrate phytohormone-like activity. Although all brassicicenes were initially believed to have 5–8–5 ring systems, our group has recently revised the structures of brassicicenes D–K (Fig. S1). They contain a tricyclo[9.2.1.0]tetradecane core skeleton with a 5–9–5 ring system and a bridgehead double bond [10].
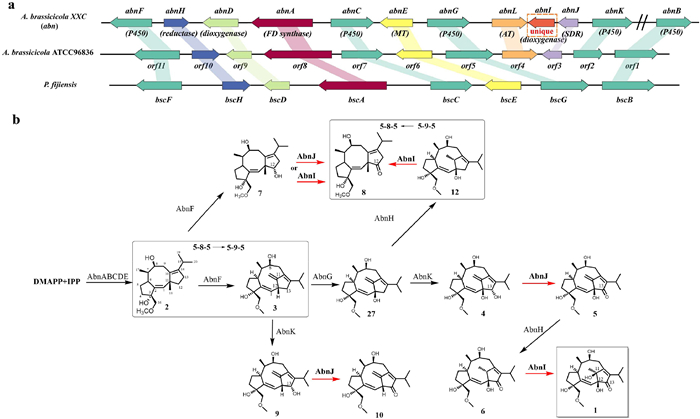
The BGCs for both ring systems of brassicicenes were identified in two fungal species, including A. brassicicola ATCC 96836 and Pseudocercospora fijiensis [11] (Fig. 1a). Oikawa et al. reported on the complete biosynthesis of brassicicene D and found that the enzyme responsible for the ring system rearrangement is cytochrome P450 BscF (Fig. 1b). This skeletal rearrangement, followed by various oxidative tailings, is remarkable as it significantly expands the chemical diversity of brassicicene diterpenes.
Figure 1
Figure 1.
(a) Brassicicene gene cluster (abn) from this study and its comparison with the other two BGCs from the previous works [10,12]. MT, methyltransferase; AT, acetyltransferase. Slash line means the gene location outside the BGC. (b) Biosynthetic pathway for brassicicene M (1) and other derivatives. The black arrows represent the steps that were characterized by Oikawa et al., whereas the red arrows correspond to the steps that were characterized in this study. DMAPP, dimethylallyl diphosphate; IPP, isopentenyl diphosphate.
Of particular note is the observation that the reported brassicicene products from our producer strain A. brassicicola XXC exhibits a greater degree of structural diversity than that obtained from the heterologous expression strain reported by Oikawa et al. (Fig. S1) [12–15]. This observation suggests that there may be other tailoring enzymes yet to be discovered. Here we report on the identification and characterization of AbnI, a cluster-specific enzyme found in the XXC strain that is capable of activating multiple sites on both 5–8–5 and 5–9–5 brassicicene skeletons, resulting in skeleton modifications and an unexpected ring system rearrangement.
To begin, the genome of the XXC strain was sequenced, revealing a terpene BGC that is highly homologous to the two reported BGCs. After naming the new BGC as "abn", a comparative bioinformatic analysis was performed on this cluster (Fig. 1a, Table S4 in Supporting information). Notably, with the exception of AbnI, all of the enzymes found in the abn cluster are present in one or both of the previously reported BGCs, and their functions can be predicted based on the known pathway of brassicicenes [9]. This comparison highlighted the unique nature of AbnI and motivated us to investigate its functions. We analyzed its protein sequence using BLAST and found that it shares 29.93% sequence identity with GA4 desaturase (an αKGD) [16], indicating that AbnI belongs to the family of αKGDs.
To characterize the abn cluster, and in particular, to determine the function of AbnI, we utilized heterologous expression in Aspergillus oryzae NSAR1 (AO). In the previous study, Oikawa et al. had characterized the function of BscA, B, C, D, E, F G, H and Orf2 in a heterologous expression system, but not Orf3. Based on their findings, we were able to predict the functions of most of the enzymes in the abn cluster and transferred them into AO. As expected, the transformant AO-abnABCDE yielded compound 2, the structure of which was confirmed by 1H nuclear magnetic resonance (NMR) (Fig. S19 in Supporting information). Subsequently, we constructed AO-abnFGKJ, AO-abnFGKHJ, and AO-abnFGKHJI for feeding experiments. Specifically, feeding 2 to AO-abnFGKJ confirmed the production of compounds 5, 8, and 10 by comparing them to standards (Fig. S3 in Supporting information). Feeding 2 to AO-abnFGKHJ confirmed the production of compound 6 (Fig. S3). Finally, feeding 2 to AO-abnFGKHJI resulted in the production of a new peak at m/z 403.2091, which was identified as brassicicene M (1) through comparison with the standard (Fig. 2). These results suggested several important findings: AbnJ (Orf3 homologue) may catalyze the oxidation of hydroxyl groups at C-12 of the 5–8–5 ring and C-13 of the 5–9–5 ring, resulting in the production of compounds 5, 8, and 10. Moreover, our most significant finding was that AbnI catalyzed the C—H bond activation at the C-11 position of 6, leading to the production of 1. This is the first report of enzymatic C—H functionalization at this site.
Figure 2
Figure 2.
LC−MS profiles of the metabolites produced by AO heterologous expression of the abn genes.
To gain more insight into reactions catalyzed by AbnJ and AbnI, we performed in vitro activity studies. Both proteins were soluble when fused with N-terminal His-tag (Fig. S4 in Supporting information). AbnJ was co-incubated with compounds 7 and 9 based on its in vivo activity. It was found that in the presence of nicotinamide adenine dinucleotide phosphate (NADP+), AbnJ converted the α–hydroxyl group at the C-13 position of 7 to a carbonyl group. Surprisingly, compound 9 underwent spontaneous oxidation at the C-13 α–hydroxyl group in the buffer, which was accelerated by AbnJ (Fig. 3, Fig. S6b in Supporting information). This observation indicates that AbnJ exhibits substrate promiscuity with respect to the skeleton structure (5–8–5 vs. 5–9–5). We also observed that AbnJ exhibits stereospecificity to its substrates, as it was unable to react with compounds 19 and 20 (Fig. S7 in Supporting information), the β–hydroxyl counterparts of 7 and 9 previously isolated from the XXC strain.
Figure 3
Figure 3.
Extracted ion chromatography (EIC) of in vitro enzyme reaction of AbnJ with 7 and 9.
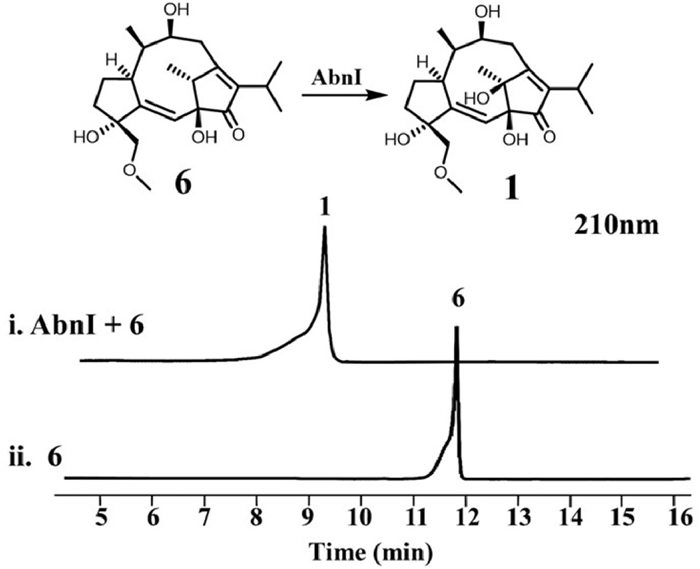
We next performed in vitro assay on AbnI. Although the Pfam analysis of its sequence did not give any known motif, BlastP analysis offered a homologous enzyme GA4 desaturase characterized as an αKGD [19]. So we tried to add Fe2+, alpha-KG, ascorbate and substrate 6 to reconstitute its activity. Indeed 6 was converted to brassicicene M (1) with AbnI catalyzing hydroxylation at C-11 (Fig. 4, lanes ⅰ and ⅱ). Lack of alpha-KG and ascorbate abolished AbnI's activity (Fig. S5 in Supporting information, lanes ⅲ and ⅳ). When not adding Fe2+, activity partly remained, probably because there was Fe2+ left in the active site after protein purification. When free Fe2+ ion in the system was removed by adding EDTA, the activity disappeared (Fig. S5, lanes v and vi). We also employed Michaelis−Menten kinetic analysis on AbnI, showing its kcat = 0.17 ± 0.02 s−1, kM = 1.26 ± 0.36 µmol/L, kcat/kM = 0.13 L µmol−1 s−1 (Fig. S13 in Supporting information).
Figure 4
Figure 4.
High performance liquid chromatography (HPLC) of in vitro enzyme reaction of AbnI with 6 using alpha-KG, ascorbate and Fe2+ as cofactors.
The experiments above demonstrate that AbnI is an αKGD enzyme which serves as a late-stage tailoring enzyme to produce brassicicene M. Our next step was to elucidate its catalytic mechanism. Upon searching its sequence against the InterPro scan [17], we found that it belongs to the hydroxylase/desaturase AsaB-like family (Fig. S14 in Supporting information). Within this family, there are αKGDs such as NvfI (PDB ID: 7DE2) and CTB9 (PDB ID: 7EUU) whose crystal structures were recently reported [18,19]. Using AlphaFold2, we generated a protein structure model of AbnI, which showed structural similarities to NvfI and CTB9 with a root mean square deviation (RMSD) of 2.412 and 1.141 Å (Fig. S15a in Supporting information).
AbnI possesses a classical double stranded β-helix core (DSBH) fold that includes a His291-His157-Asp159 catalytic triad, which is highly similar to that of NvfI. For the AbnI·FeII·αKG complex, we chelated the iron ion between these three residues along with alpha-KG, based on the active site structure of NvfI. The bidentate alpha-KG fits well in the complex, stabilized by hydrogen bonding interactions with R312 and R306. We further conducted molecular docking of 6, resulting in an AbnI·FeⅡ·αKG·substrate 6 complex with the following possible hydrogen bonding interactions: R109 with C8-OH, K155 with C16-O, and D159 with C12-OH (Fig. 5). Site-directed mutagenesis demonstrated that mutations in these residues abolished or greatly reduced the activity of AbnI (Fig. S12 in Supporting information). Notably, C-11 of 6 is closer to the iron center compared to other hydrogen-containing carbons, offering a structure-based explanation for why the C-11 of 6 undergoes hydroxylation in AbnI. AbnI and NvfI lack a tyrosine residue in close proximity to the iron center in the active site, which is present in COX and FtmOx1-like enzymes that mediate hydrogen atom transfer (HAT). This observation suggests that AbnI does not depend on this residue to mediate the HAT in the catalytic process, unlike the FtmOx1 or COX-like model [18,20–23]. This finding supports the direct hydroxyl rebound mechanism, where the FeIV=O species abstracts the hydrogen at C-11 of compound 6, followed by the direct oxygen rebound between the substrate radical and the ferric–hydroxy species (Fig. S8a in Supporting information).
Figure 5
Figure 5.
Molecular docking of AbnI·FeⅡ·αKG·substrate 6 complex.
Meanwhile, when comparing the active site of AbnI to other αKGDs of the AsaB-like family, we noticed that AbnI has a significantly larger active site volume (Fig. S15b in Supporting information), indicating a potentially flexible substrate scope. To investigate this possibility, we conducted enzymatic assays with various brassicicene derivatives, including 5–8–5 skeleton compounds 7 and 11, 5–9–5 skeleton compounds 12 and 13, and dimer compounds 14 and 15. Surprisingly, AbnI was able to transform all the compounds except for 13 and 15 (Fig. 6 and Fig. S1). We compared the catalytic efficiency of AbnI against these substrates and found that the order of efficiency was 6 > 12 > 11 > 7 (Fig. S18 in Supporting information). Conducting molecular docking of the substrates separately revealed that they all fall within a reasonable reaction distance near the catalytic center (Fig. S16 and Table S7 in Supporting information).
Figure 6
Figure 6.
Enzymatic assays of AbnI with the brassicicene derivatives. The asterisk on 18 indicates that its structure was not fully elucidated due to the limited amount.
Specifically, AbnI was found to activate C-7 of 11 and 14, resulting in the formation of novel compounds 16 and 18, respectively (NMR and structure elucidation results were shown in Figs. S20–S26, S29–S31, Tables S5 and S6 in Supporting information), while C-12 activation of 7 and 11 led to the formation of 8 and 17 respectively (Figs. S27 and S28 in Supporting information). Although AbnJ is also capable of catalysing the conversion from 7 to 8, we propose that the mechanism differs between the two enzymes. AbnJ utilizes a typical hydride transfer mediated by NADP+, whereas AbnI catalyzes a hydroxylation on C-12 followed by dehydration, leading to the formation of the ketone group. The most interesting finding is the C-11 activation of the 5–9–5 skeleton, which can result in at least two outcomes: when C-12 is a hydroxyl group and C-13 is a ketone, as in the case of 6, AbnI catalyzes the hydroxylation on C-11. When C-12 is a hydroxyl group and C-13 is unmodified, as in the case of 12, AbnI catalyzes the Wagner–Meerwein (W-M) rearrangement to form 8 (Fig. 6, lines ⅲ and ⅳ, the reaction mechanism proposed in Scheme S1b in Supporting information). Based on our simulation calculations of bond dissociation energies and molecular docking, it is speculated that the substrate promiscuity resulting from the C-13 carbonyl group may be due to the combined effects of the stability of the radical intermediate and the distance of the radical intermediate from the catalytic center (Scheme S1 and Figs. S32 and S34 for detailed mechanism discussion in Supporting information). Notably, AbnI is the second enzyme in the BGC that can cause the skeleton rearrangement, with the first being the P450 AbnF, which catalyzes the oxidative rearrangement from 5 to 8–5 to 5–9–5. Additionally, a 5–9–4 skeleton product, 24 (Fig. S1), was also found in the XXC strain [24], which could be a rearranged product of compound 1. Upon acid treatment of 1, liquid chromatography–mass spectrometry (LC–MS) analysis revealed the formation of multiple products, one of which corresponded to the standard of 24 (Fig. S9 in Supporting information). Given that compound 1 is the C—H functionalization product of AbnI, it is evident that the rearrangement of the 5–9–5 skeleton induced by AbnI is diverse and intriguing.
In our results, AbnI demonstrated substrate and product promiscuity, which raises an interesting question: what is the ecological significance of introducing AbnI into the brassicicene BGC? The first point is obvious: AbnI brings about chemical diversity, leading to the creation of new structures such as 1 and 16. On the other hand, we observed 8 was generated by three distinct pathways, with one pathway being catalyzed by AbnJ and the other two being catalyzed by AbnI (Fig. 1). To investigate the role of AbnI in the production yield of 8, we conducted a feeding experiment and found that the concentration of 8 was significantly higher in the AO-abnFGKBJI compared to the AO-abnFGKBJ transformants (Fig. S17 in Supporting information). This suggests a possible role for AbnI in increasing the concentration of 8 in the metabolite, and potentially increasing the concentration of other downstream 5–8–5 skeleton compounds. As there have been limited investigations into the bioactivity of 5–8–5 and 5–9–5 brassicicenes [13], it is crucial to conduct further exploration into their ecological functions to determine the potential benefits of obtaining high concentration of compound 8.
In summary, we fully characterized a new brassicicene BGC containing a unique αKGD AbnI. Prior to our investigation, the origin of C-11 hydroxyl group in compound 1 was unclear, with two possible mechanisms: (1) through the addition of water to the C-11 carbocation after W-M rearrangement during 5–9–5 skeleton formation (Fig. S8b in Supporting information); (2) through enzymatic hydroxylation on 6 (Fig. S8a). Our in vivo and in vitro results provided compelling evidence supporting the second mechanism, resolving the ambiguity regarding the formation of the C-11 hydroxyl group. Furthermore, AbnI demonstrated remarkable substrate promiscuity, capable of activating multiple sites on 5–8–5 and 5–9–5 skeletons, indicating its potential utility as a catalytic tool for terpene C—H functionalization. Significantly, AbnI collaborates with AbnF to convert 5–8–5 and 5–9–5 ring systems. Its potential role in increasing the concentration of 8 with 5–8–5 skeleton warrants further investigation into its ecological functions.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We are very grateful to Professor Hideaki Oikawa at Division of Chemistry, Graduate School of Science, Hokkaido University for providing Aspergillus oryzae NSAR1 heterologous expression system and useful suggestions on the work. This work was financially supported by the National Key R & D Program of China (Nos. 2021YFA0910500, 2021YFA0910503), the National Natural Science Foundation of China (Nos. 22277035, 32000045 and 81973205), the Program for Changjiang Scholars of Ministry of Education of the People's Republic of China (No. T2016088), the National Natural Science Foundation for Distinguished Young Scholars (No. 81725021), the National Science and Technology Project of China (No. 2018ZX09201001–001–003), Innovative Research Groups of the National Natural Science Foundation of China (No. 81721005), and the Fundamental Research Funds for the Central Universities (No. 2020kfyXJJS043). We thank the Analytical and Testing Center at HUST for ECD, UV, and IR analyses.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.108788.
Figure 1
(a) Brassicicene gene cluster (abn) from this study and its comparison with the other two BGCs from the previous works [10,12]. MT, methyltransferase; AT, acetyltransferase. Slash line means the gene location outside the BGC. (b) Biosynthetic pathway for brassicicene M (1) and other derivatives. The black arrows represent the steps that were characterized by Oikawa et al., whereas the red arrows correspond to the steps that were characterized in this study. DMAPP, dimethylallyl diphosphate; IPP, isopentenyl diphosphate.
Figure 6
Enzymatic assays of AbnI with the brassicicene derivatives. The asterisk on 18 indicates that its structure was not fully elucidated due to the limited amount.

 DownLoad:
DownLoad:





 下载:
下载:







